
Phylogeographic and SNPs Analyses of Bemisia tabaci B Mitotype Populations Reveal Only Two of Eight Haplotypes Are Invasive



Abstract
:Simple Summary
Abstract
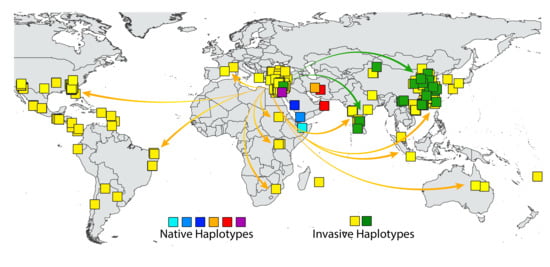
1. Introduction
2. Materials and Methods
2.1. The mtCOI Sequences
2.2. Variant Calling and Population Differentiation
2.3. Phylogeography of Haplotypes
2.4. Network of Haplotypes
2.5. Eco-Geographic Distribution of Haplotypes
3. Results
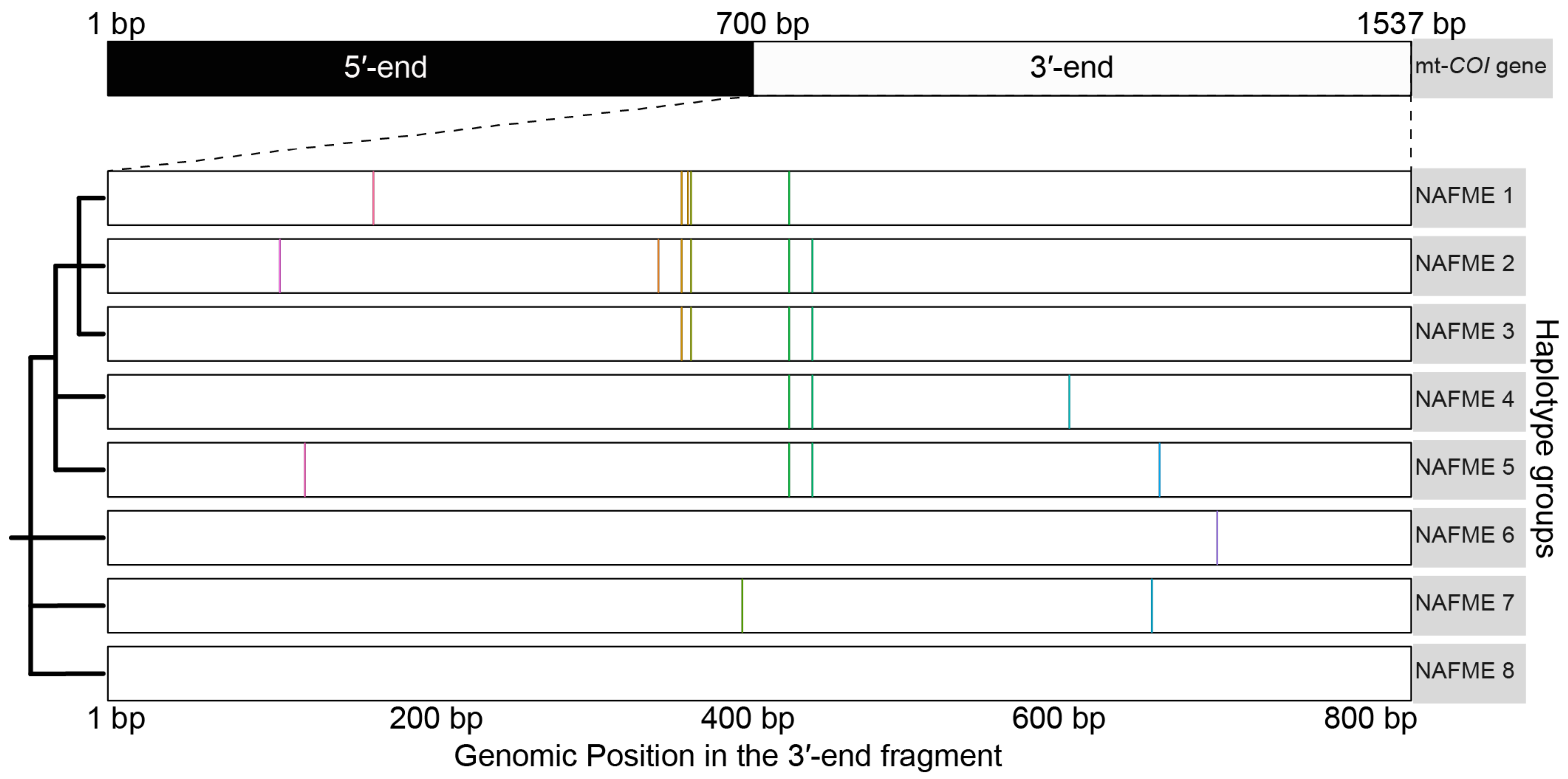
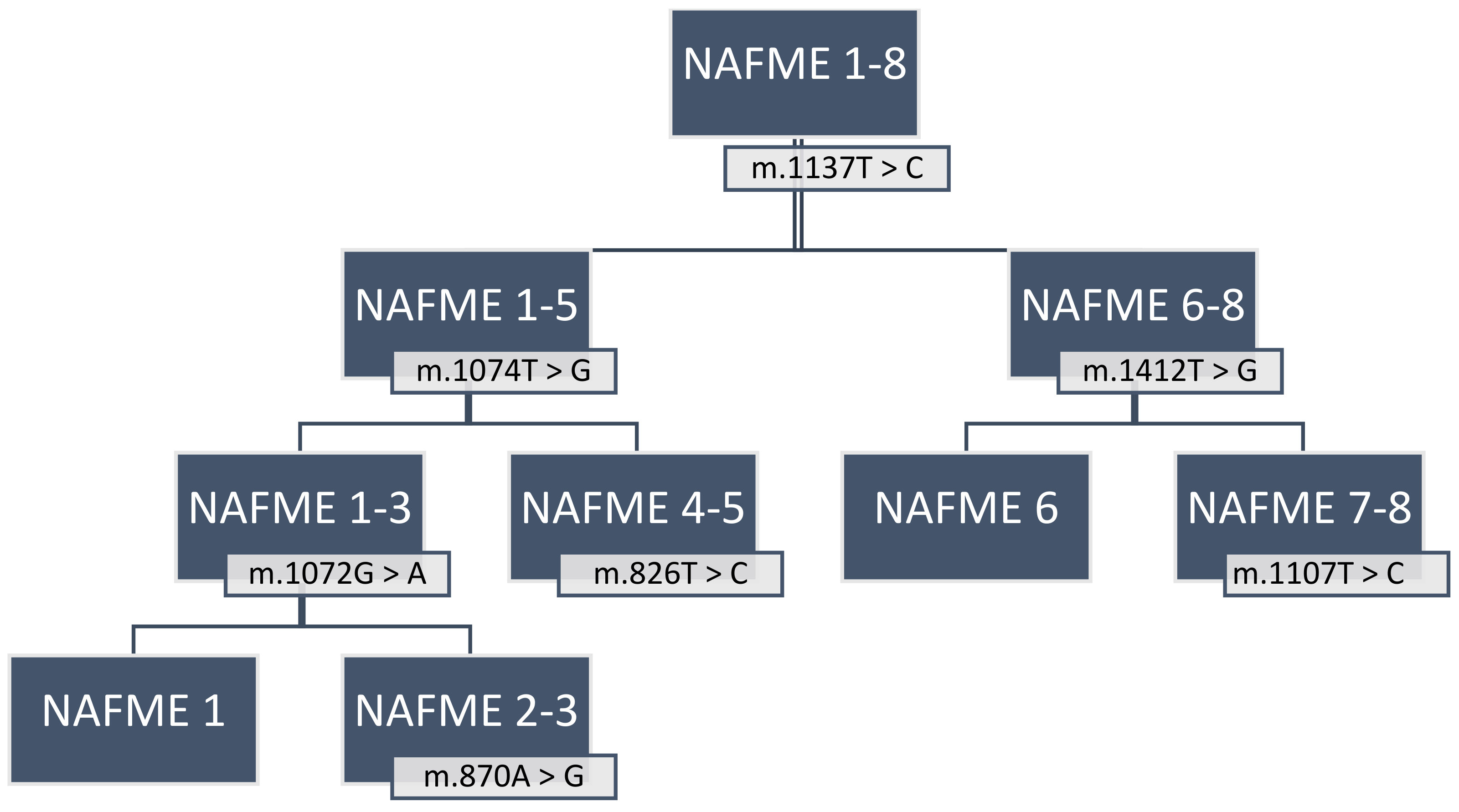
3.1. SNP-Defined Haplotypes of the B Mitotype
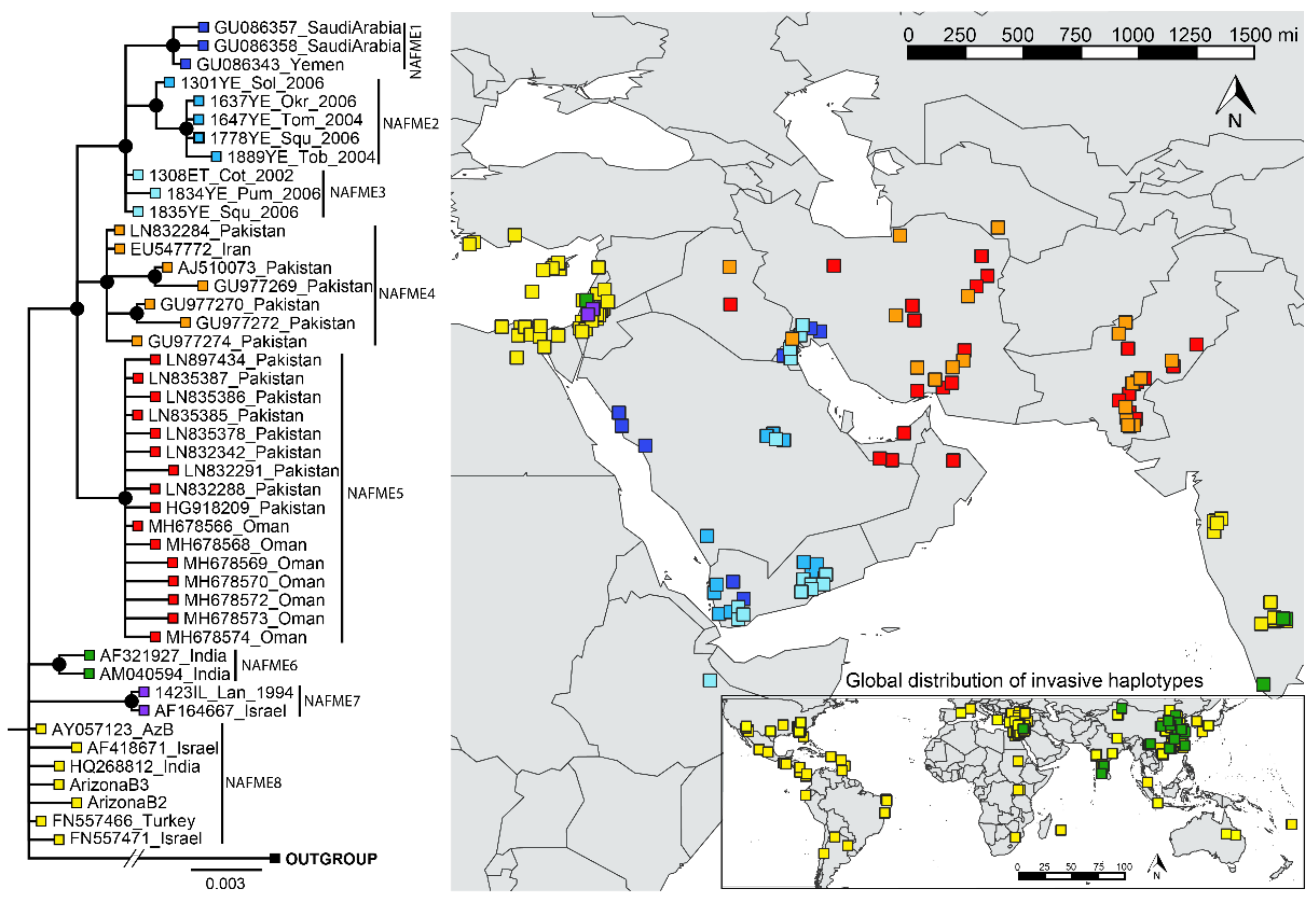
3.2. Phylogenetic Analysis
3.3. Network Analysis and Population Differentiation
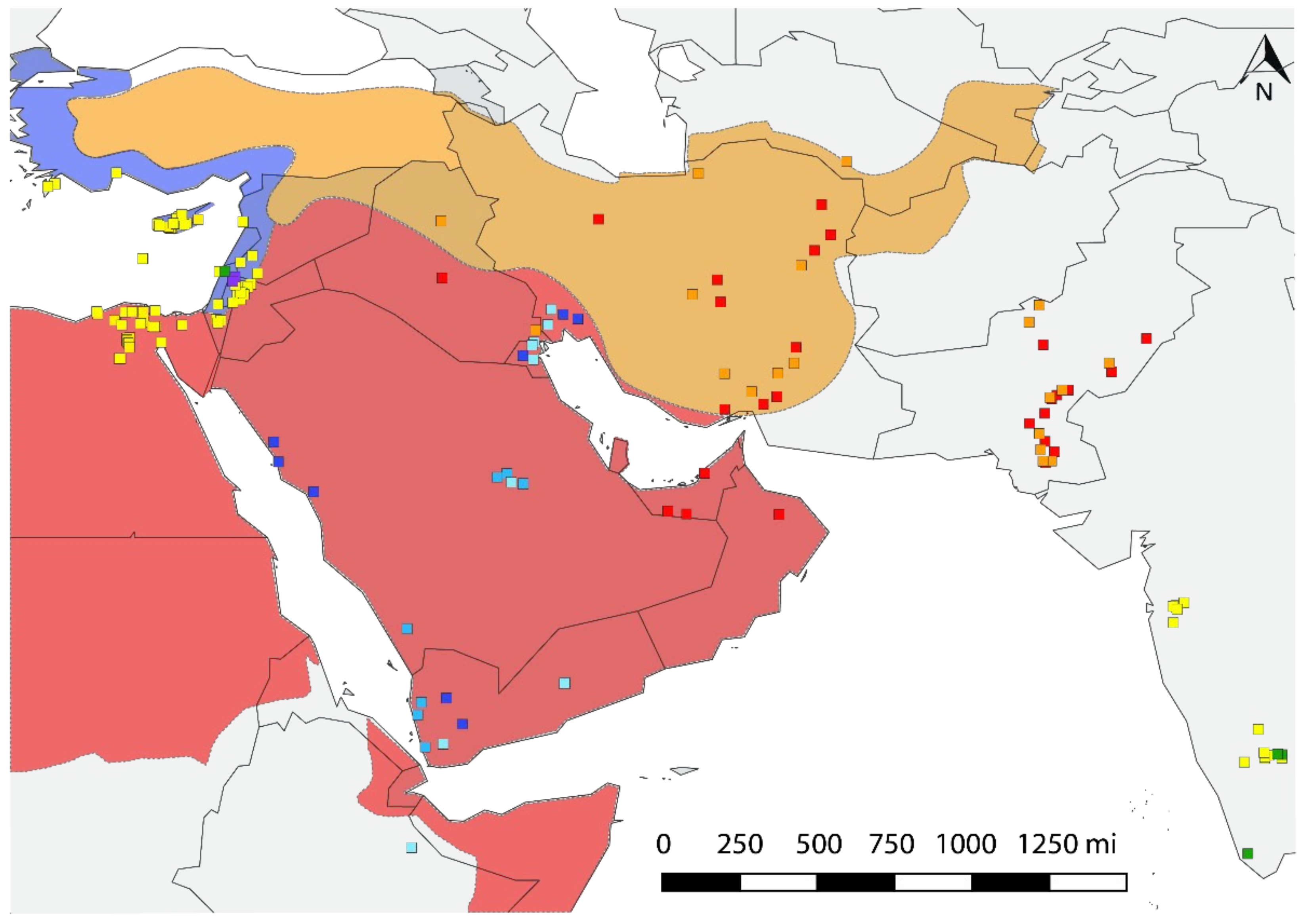
3.4. Eco-Geographic Distribution of Haplotypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, J.K. Phylogenetic biology of the Bemisia tabaci sibling species group. In Bemisia: Bionomics and Management of a Global Pest; Stansly, P.A., Naranjo, S.E., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 31–67. ISBN 978-90-481-2460-2. [Google Scholar]
- De Moya, R.S.; Brown, J.K.; Sweet, A.D.; Walden, K.K.O.; Paredes-Montero, J.R.; Waterhouse, R.M.; Johnson, K.P. Nuclear orthologs derived from whole genome sequencing indicate cryptic diversity in the Bemisia tabaci (Insecta: Aleyrodidae) complex of Whiteflies. Diversity 2019, 11, 151. [Google Scholar] [CrossRef] [Green Version]
- Gill, R.; Brown, J.K. Systematics of Bemisia and Bemisia relatives: Can molecular techniques solve the Bemisia tabaci complex conundrum—A taxonomist’s viewpoint. In Bemisia: Bionomics and Management of a Global Pest; Stansly, P.A., Naranjo, S.E., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 5–29. ISBN 978-90-481-2460-2. [Google Scholar]
- Mound, L.A.; Halsey, S.H. Whitefly of the World: A Systematic Catalogue of the Aleyrodidae (Homoptera) with Host Plant and Natural Enemy Data; British Museum (Natural History) and John Wiley & Sons: Chichester, UK, 1978; ISBN 978-0-471-99634-7. [Google Scholar]
- Cock, M.J.W. (Ed.) Bemisia tabaci–A Literature Survey on the Cotton Whitefly with an Annotated Bibliograph; C.A.B International Institute of Biological Control: Ascot, UK, 1986; ISBN 0-85198-847-4. [Google Scholar]
- Cock, M.J.W. Bemisia tabaci: An Update 1986–1992 on the Cotton Whitefly with an Annotated Bibliography; International Institute of Biological Control: Berkshire, UK, 1993; ISBN 0-85198-847-4. [Google Scholar]
- Brown, J.K.; Frohlich, D.R.; Rosell, R.C. The sweetpotato or silverleaf whiteflies: Biotypes of Bemisia tabaci or a species complex? Annu. Rev. Èntomol. 1995, 40, 511–534. [Google Scholar] [CrossRef]
- Avidov, Z. Bionomics of the tobacco whitefly (Bemisia tabaci Gennad.) in Israel. Ktavim 1956, 7, 25–41. [Google Scholar]
- Brown, J.K. Whitefly-transmitted geminiviruses and associated disorders in the Americas and the Caribbean basin. Plant Dis. 1992, 76, 220. [Google Scholar] [CrossRef]
- Bedford, I.D.; Briddon, R.W.; Brown, J.K.; Rosell, R.C.; Markham, P.G. Geminivirus transmission and biological characterisation of Bemisia tabaci (Gennadius) biotypes from different geographic regions. Ann. Appl. Biol. 1994, 125, 311–325. [Google Scholar] [CrossRef]
- Jones, D.R. Plant viruses transmitted by whiteflies. Eur. J. Plant Pathol. 2003, 109, 195–219. [Google Scholar] [CrossRef]
- Azab, A.K.; Megahed, M.M.; E1-Mirsawi, D.H. On the biology of Bernisia tabaci (Genn.) [Hemiptera—Homoptera: Aleyrodidae]. Bull. Soc. Entomol. Egypt 1971, 4, 305–315. [Google Scholar]
- Horowitz, A.; Podoler, H.; Gerling, D. Life table analysis of the tobacco whitefly Bemisia tabaci (Gennadius) in cotton fields in Israel. Acta Oecologica Oecologia Appl. Fr. 1984, 5, 221–233. [Google Scholar]
- Gerling, D.; Horowitz, A.; Baumgaertner, J. Autecology of Bemisia tabaci. Agric. Ecosyst. Environ. 1986, 17, 5–19. [Google Scholar] [CrossRef]
- Bayhan, E.; Ulusoy, M.R.; Brown, J.K. Host range, distribution, and natural enemies of Bemisia tabaci ‘B biotype’ (Hemiptera: Aleyrodidae) in Turkey. J. Pest Sci. 2006, 79, 233–240. [Google Scholar] [CrossRef]
- Chu, N.; Zhang, Y.-J.; Brown, J.K.; Cong, B.; Xu, B.-Y.; Wu, Q.-J.; Zhu, G.-R.; Society, F.E. The introduction of the exotic q biotype of Bemisia tabaci from the Mediterranean region into China on ornamental crops. Fla. Èntomol. 2006, 89, 168–174. [Google Scholar] [CrossRef]
- Dennehy, T.J.; DeGain, B.; Harpold, G.; Brown, J.K.; Byrne, F.; Morin, S.; Nichols, R. First New World report of Q biotype of Bemisia tabaci (Gennadius) reveals high levels of resistance to insecticides. Resist. Pest Manag. Newsl. 2006, 15, 18–20. [Google Scholar]
- Papayiannis, L.C.; Brown, J.K.; Hadjistylli, M.; Katis, N.I. Bemisia tabaci biotype B associated with tomato yellow leaf curl disease epidemics on Rhodes Island, Greece. Phytoparasitica 2008, 36, 20–22. [Google Scholar] [CrossRef]
- Paredes-Montero, J.R.; Ibarra, M.A.; Arias-Zambrano, M.; Peralta, E.L.; Brown, J.K. Phylo-biogeographical distribution of whitefly Bemisia tabaci (Insecta: Aleyrodidae) mitotypes in Ecuador. Ecosphere 2020, 11, 1–20. [Google Scholar] [CrossRef]
- Ueda, S.; Brown, J.K. First report of the Q biotype of Bemisia tabaci in Japan by mitochondrial cytochrome oxidase I sequence analysis. Phytoparasitica 2006, 34, 405–411. [Google Scholar] [CrossRef]
- Shaurub, E.S.H.; Paredes-Montero, J.R.; Brown, J.K.; Zein, H.S.; Mohamed, A.A. Metabolic resistance to organophosphate insecticides in natural populations of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae) in Egypt and molecular identification of mitotypes. Phytoparasitica 2021, 49, 443–457. [Google Scholar] [CrossRef]
- Bird, J. A Whitefly-transmitted mosaic of Jatropha gossypiifolia. Agr. Exp. Sta. Tech. Pap. 1957, 22, 80–149. [Google Scholar]
- Banks, G.K.; Colvin, J.; Reddy, R.V.C.; Maruthi, M.N.; Muniyappa, V.; Venkatesh, H.M.; Kumar, M.K.; Padmaja, A.S.; Beitia, F.J.; Seal, S.E. First report of the Bemisia tabaci B biotype in India and an associated tomato leaf curl virus disease epidemic. Plant Dis. 2001, 85, 231. [Google Scholar] [CrossRef]
- Lisha, V.S.; Antony, B.; Palaniswami, M.S.; Henneberry, T.J. Bemisia tabaci (Homoptera: Aleyrodidae) biotypes in India. J. Econ. Èntomol. 2003, 96, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Rekha, A.; Maruthi, M.; Muniyappa, V.; Colvin, J. Occurrence of three genotypic clusters of Bemisia tabaci and the rapid spread of the B biotype in south India. Èntomol. Exp. Appl. 2005, 117, 221–233. [Google Scholar] [CrossRef]
- Simón, B.; Cenis, J.L.; Beitia, F.; Khalid, S.; Moreno, I.M.; Fraile, A.; García-Arenal, F. Genetic Structure of field populations of begomoviruses and of their vector Bemisia tabaci in Pakistan. Phytopathology 2003, 93, 1422–1429. [Google Scholar] [CrossRef]
- Burban, C.; Fishpool, L.D.C.; Fauquet, C.; Fargette, D.; Thouvenel, J.-C. Host-associated biotypes within West African populations of the whitefly Bemisia tabaci (Genn.), (Hom., Aleyrodidae). J. Appl. Èntomol. 1992, 113, 416–423. [Google Scholar] [CrossRef]
- Legg, J.P.; Sseruwagi, P.; Boniface, S.; Okao-Okuja, G.; Shirima, R.; Bigirimana, S.; Gashaka, G.; Herrmann, H.-W.; Jeremiah, S.; Obiero, H.; et al. Spatio-temporal patterns of genetic change amongst populations of cassava Bemisia tabaci whiteflies driving virus pandemics in East and Central Africa. Virus Res. 2014, 186, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Sseruwagi, P.; Legg, J.; Maruthi, M.; Colvin, J.; Rey, M.; Brown, J. Genetic diversity of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) populations and presence of the B biotype and a non-B biotype that can induce silverleaf symptoms in squash, in Uganda. Ann. Appl. Biol. 2005, 147, 253–265. [Google Scholar] [CrossRef]
- Byrne, D.N.; Miller, W.B. Carbohydrate and amino acid composition of phloem sap and honeydew produced by Bemisia tabaci. J. Insect Physiol. 1990, 36, 433–439. [Google Scholar] [CrossRef]
- Costa, H.S.; Brown, J.K. Variation in biological characteristics and esterase patterns among populations of Bemisia tabaci, and the association of one population with silverleaf symptom induction. Èntomol. Exp. Appl. 1991, 61, 211–219. [Google Scholar] [CrossRef]
- Hamon, A.B.; Salguero, V. Bemisia tabaci, sweetpotato whitefly. Entomol. Circ. 1987, 292. [Google Scholar]
- Brown, J.K.; Poulos, B.T.; Bird, J. Differential detection of whitefly-transmitted geminiviruses in weed species from Puerto Rico by hybridization analysis with non-radioactive probes. In Proceedings of the American Phytopathology Society-Caribbean Division meeting, Mayaguez, Puerto Rico, 27–31 May 1991. [Google Scholar]
- Brown, J.K.; Coats, S.A.; Bedford, I.D.; Markham, P.G.; Bird, J.; Frohlich, D.R. Characterization and distribution of esterase electromorphs in the whitefly, Bemisia tabaci (Genn.) (Homoptera: Aleyrodidae). Biochem. Genet. 1995, 33, 205–214. [Google Scholar] [CrossRef]
- Costa, H.S.; Brown, J.K. Variability in biological characteristics, isozyme patterns and virus transmission among populations of Bemisia tabaci in Arizona. Phytopathology 1990, 80, 888. [Google Scholar]
- Prabhaker, N.; Coudriet, D.L.; Meyerdirk, D.E. Discrimination of three whitefly species (Homoptera, Aleyrodidae) by electrophoresis of non-specific esterases. J. Appl. Èntomol. 1987, 103, 447–451. [Google Scholar] [CrossRef]
- Wool, D.; Gerling, D.; Nolt, B.L.; Constantino, L.M.; Bellotti, A.C.; Morales, F.J. The use of electrophoresis for identification of adult whiteflies (Homoptera, Aleyrodidae) in Israel and Colombia. J. Appl. Èntomol. 1989, 107, 344–350. [Google Scholar] [CrossRef]
- Wool, D.; Gerling, D.; Bellotti, A.; Morales, F.; Nolt, B. Spatial and temporal genetic variation in populations of the whitefly Bemisia tabaci (Genn.) in Israel and Colombia: An interim report. Int. J. Trop. Insect Sci. 1991, 12, 225–230. [Google Scholar] [CrossRef]
- Costa, H.S.; Brown, J.K.; Sivasupramaniam, S.; Bird, J. Regional distribution, insecticide resistance, and reciprocal crosses between the A and B biotypes of Bemisia tabaci. Int. J. Trop. Insect Sci. 1993, 14, 255–266. [Google Scholar] [CrossRef]
- Simmons, A.M.; Gould, J.; Hoelmer, K.; Goolsby, J. Classical biological control of Bemisia tabaci in the United States—A review of interagency research and implementation. J. Econ. Èntomol. 2009, 102, 1724–1725. [Google Scholar] [CrossRef]
- Brown, J.K. The Bemisia tabaci complex: Genetic and phenotypic variation and relevance to TYLCV-vector interactions. In Tomato Yellow Leaf Curl Virus Disease: Management, Molecular Biology, Breeding for Resistance; Czosnek, H., Ed.; Springer: Dordrecht, The Netherlands, 2007; pp. 25–56. ISBN 978-1-4020-4769-5. [Google Scholar]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phyloge-ography: The mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Castro, J.A.; Picornell, A.; Ramon, M. Mitochondrial DNA: A tool for populational genetics studies. Int. Microbiol. 1998, 1, 327–332. [Google Scholar]
- Harrison, R.G. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. 1989, 4, 6–11. [Google Scholar] [CrossRef]
- Delatte, H.; Reynaud, B.; Granier, M.; Thornary, L.; Lett, J.; Goldbach, R.; Peterschmitt, M. A new silverleaf-inducing biotype Ms of Bemisia tabaci (Hemiptera: Aleyrodidae) indigenous to the islands of the south-west Indian Ocean. Bull. Èntomol. Res. 2005, 95, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohlich, D.R.; Torres-Jerez, I.; Bedford, I.D.; Markham, P.G.; Brown, J.K. A phylogeographical analysis of the Bemisia tabaci species complex based on mitochondrial DNA markers. Mol. Ecol. 1999, 8, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Legg, J.P.; French, R.; Rogan, D.; Okao-Okuja, G.; Brown, J.K. A distinct Bemisia tabaci (Gennadius) (Hemiptera: Sternorrhyncha: Aleyrodidae) genotype cluster is associated with the epidemic of severe cassava mosaic virus disease in Uganda. Mol. Ecol. 2002, 11, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.E.; Zimmermann, N.E.; McVicar, T.R.; Vergopolan, N.; Berg, A.; Wood, E.F. Present and future Köppen-Geiger climate classification maps at 1-km resolution. Sci. Data 2018, 5, 180214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kans, J. Entrez direct: E-utilities on the UNIX command line. In Entrez Programming Utilities Help; Bethesda, M.D., Ed.; National Center for Biotechnology Information, U.S. National Library of Medicine: Bethesda, MD, USA, 2020. [Google Scholar]
- Paredes-Montero, J.R.; Hameed, U.; Rehman, M.Z.U.; Rasool, G.; Haider, M.S.; Herrmann, H.-W.; Brown, J.K. Demographic expansion of the predominant Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) mitotypes associated with the Cotton leaf curl virus epidemic in Pakistan. Ann. Èntomol. Soc. Am. 2019, 112, 265–280. [Google Scholar] [CrossRef]
- Güssow, D.; Clackson, T. Direct clone characterization from plaques and colonies by the polymerase chain reaction. Nucleic Acids Res. 1989, 17, 4000. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.; Maddison, D. Mesquite: A modular system for evolutionary analysis. Evolution 2008, 62, 1103–1118. [Google Scholar]
- MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [CrossRef] [PubMed] [Green Version]
- Song, H.; Buhay, J.E.; Whiting, M.F.; Crandall, K.A. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc. Natl. Acad. Sci. USA 2008, 105, 13486–13491. [Google Scholar] [CrossRef] [Green Version]
- De Barro, P.J.; Liu, S.-S.; Boykin, L.; Dinsdale, A.B. Bemisia tabaci: A statement of species status. Annu. Rev. Èntomol. 2011, 56, 1–19. [Google Scholar] [CrossRef]
- Tay, W.T.; Elfekih, S.; Court, L.N.; Gordon, K.H.; Delatte, H.; De Barro, P.J. The trouble with MEAM2: Implications of pseudogenes on species delimitation in the globally invasive Bemisia tabaci (Hemiptera: Aleyrodidae) cryptic species complex. Genome Biol. Evol. 2017, 9, 2732–2738. [Google Scholar] [CrossRef] [Green Version]
- Raymond, M.; Rousset, F. GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Keenan, K. DiveRsity: A Comprehensive, general purpose population genetics analysis package. R Package Version 2017, 1, 90. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T.-A. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Chapman, B.; Chang, J. Biopython: Python tools for computational biology. ACM Sigbio. Newsl. 2000, 20, 15–19. [Google Scholar] [CrossRef]
- QGIS Development Team. QGIS Geographic Information System. Open Source Geospatial Foundation. 2021. Available online: http://www.qgis.org (accessed on 15 March 2021).
- Peel, M.C.; Finlayson, B.L.; McMahon, T.A. Updated world map of the Köppen-Geiger climate classification. Hydrol. Earth Syst. Sci. 2007, 11, 1633–1644. [Google Scholar] [CrossRef] [Green Version]
- Manafzadeh, S.; Salvo, G.; Conti, E. A tale of migrations from east to west: The Irano-Turanian floristic region as a source of Mediterranean xerophytes. J. Biogeogr. 2014, 41, 366–379. [Google Scholar] [CrossRef]
- Leray, M.; Knowlton, N.; Ho, S.-L.; Nguyen, B.N.; Machida, R.J. GenBank is a reliable resource for 21st century biodiversity research. Proc. Natl. Acad. Sci. USA 2019, 116, 22651–22656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinsdale, A.; Cook, L.G.; Riginos, C.; Buckley, Y.M.; De Barro, P. Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome qxidase 1 to identify species level genetic boundaries. Ann. Entomol. Soc. Am. 2010, 103, 196–208. [Google Scholar] [CrossRef]
- Paredes-Montero, J.R.; Brown, J.K. The NUMTs in the Nuclear Genomes of Bemisia tabaci (Hemiptera: Aleyrodidae) and Its Implications in the Demarcation of Molecular Species; ESA: St. Louis, MO, USA, 2019. [Google Scholar]
- Keohavong, P.; Thilly, W.G. Fidelity of DNA polymerases in DNA amplification. Proc. Natl. Acad. Sci. USA 1989, 86, 9253–9257. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Rannala, B. Molecular phylogenetics: Principles and practice. Nat. Rev. Genet. 2012, 13, 303–314. [Google Scholar] [CrossRef]
- Liu, S.-S.; De Barro, P.J.; Xu, J.; Luan, J.-B.; Zang, L.-S.; Ruan, Y.-M.; Wan, F.-H. Asymmetric mating interactions drive widespread invasion and displacement in a whitefly. Science 2007, 318, 1769–1772. [Google Scholar] [CrossRef]
- Masood, M.; Amin, I.; Hassan, I.; Mansoor, S.; Brown, J.K.; Briddon, R.W. Diversity and distribution of cryptic species of the Bemisia tabaci (Hemiptera: Aleyrodidae) complex in Pakistan. J. Econ. Èntomol. 2017, 110, 2295–2300. [Google Scholar] [CrossRef]
- Ahmed, M.Z.; Ren, S.-X.; Mandour, N.S.; Maruthi, M.N.; Naveed, M.; Qiu, B.-L. Phylogenetic analysis of Bemisia tabaci (Hemiptera: Aleyrodidae) populations from cotton plants in Pakistan, China, and Egypt. J. Pest Sci. 2010, 83, 135–141. [Google Scholar] [CrossRef]
- Djamali, M.; Baumel, A.; Brewer, S.; Jackson, S.; Kadereit, J.W.; López-Vinyallonga, S.; Mehregan, I.; Shabanian, E.; Simakova, A. Ecological implications of Cousinia Cass. (Asteraceae) persistence through the last two glacial–interglacial cycles in the continental Middle East for the Irano-Turanian flora. Rev. Palaeobot. Palynol. 2012, 172, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Shakun, J.D.; Clark, P.U.; He, F.; Marcott, S.A.; Mix, A.C.; Liu, Z.; Otto-Bliesner, B.; Schmittner, A.; Bard, E. Global warming preceded by increasing carbon dioxide concentrations during the last deglaciation. Nature 2012, 484, 49–54. [Google Scholar] [CrossRef]
- Moharrek, F.; Sanmartín, I.; Kazempour-Osaloo, S.; Feliner, G.N. Morphological innovations and vast extensions of mountain habitats triggered rapid diversification within the species-rich Irano-Turanian genus Acantholimon (Plumbaginaceae). Front. Genet. 2019, 9, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talebi, K.S.; Sajedi, T.; Pourhashemi, M. Introduction. In Forests of Iran: A Treasure from the Past, a Hope for the Future; Talebi, K.S., Sajedi, T., Pourhashemi, M., Eds.; Plant and Vegetation; Springer: Dordrecht, The Netherlands, 2014; pp. 1–13. ISBN 978-94-007-7371-4. [Google Scholar]
- Fekrat, L.; Shishehbor, P. Some biological features of Cotton whitefly, Bemisia tabaci (Homoptera: Aleyrodidae) on various host Plants. Pak. J. Biol. Sci. 2007, 10, 3180–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saghafipour, A.; Zahraei-Ramazani, A.; Vatandoost, H.; Asadollahi, A.; Fouladi-Fard, R.; Hamta, A.; Hasanwand, A. Relationship between some environmental and climatic factors on outbreak of whiteflies, the human annoying insects. J. Arthropod-Borne Dis. 2020, 14, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TehranTimes. Silverleaf Whiteflies, Unwelcome Guests to the Capital. Available online: https://www.tehrantimes.com/news/413614/Silverleaf-whiteflies-unwelcome-guests-to-the-capital (accessed on 15 March 2021).
- TehranTimes. Tehran Measures to Control Whiteflies Inadequate. Available online: https://financialtribune.com/articles/people-environment/64973/tehran-measures-to-control-whiteflies-inadequate (accessed on 15 March 2021).
- TehranTimes. Whiteflies All Around: Who Is Displaced? Available online: https://www.tehrantimes.com/news/416359/Whiteflies-all-around-Who-is-displaced (accessed on 15 March 2021).
- Hadjistylli, M.; Roderick, G.; Brown, J.K. Global Population structure of a worldwide pest and virus vector: Genetic diversity and population history of the Bemisia tabaci sibling species group. PLoS ONE 2016, 11, e0165105. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-H.; Ko, C.-C.; Chung, C.-H.; Wang, H.-Y. Multilocus approach to clarify species status and the divergence history of the Bemisia tabaci (Hemiptera: Aleyrodidae) species complex. Mol. Phylogenetics Evol. 2014, 76, 172–180. [Google Scholar] [CrossRef]
- Santos-Garcia, D.; Vargas-Chavez, C.; Moya, A.; Latorre, A.; Silva, F.J. Genome evolution in the primary endosymbiont of whiteflies sheds light on their divergence. Genome Biol. Evol. 2015, 7, 873–888. [Google Scholar] [CrossRef] [Green Version]
- Santos-Garcia, D.; Mestre-Rincon, N.; Ouvrard, D.; Zchori-Fein, E.; Morin, S. Portiera gets wild: Genome instability provides insights into the evolution of both whiteflies and their Endosymbionts. Genome Biol. Evol. 2020, 12, 2107–2124. [Google Scholar] [CrossRef]
- Jaenike, J. Host Specialization in phytophagous insects. Annu. Rev. Ecol. Syst. 1990, 21, 243–273. [Google Scholar] [CrossRef]
- Joshi, A.; Thompson, J.N. Trade-offs and the evolution of host specialization. Evol. Ecol. 1995, 9, 82–92. [Google Scholar] [CrossRef]
- Brucker, R.M.; Bordenstein, S.R. Speciation by symbiosis. Trends Ecol. Evol. 2012, 27, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Mittelbach, G.G.; Schemske, D.W.; Cornell, H.V.; Allen, A.; Brown, J.M.; Bush, M.; Harrison, S.P.; Hurlbert, A.; Knowlton, N.; Lessios, H.A.; et al. Evolution and the latitudinal diversity gradient: Speciation, extinction and biogeography. Ecol. Lett. 2007, 10, 315–331. [Google Scholar] [CrossRef]
- Knowles, L.L. Tests of Pleistocene speciation in montane grasshoppers (genus Melanoplus) from the Sky Islands of Western North America. Evolution 2000, 54, 1337–1348. [Google Scholar] [CrossRef]
- Barnes, W.M. The fidelity of Taq polymerase catalyzing PCR is improved by an N-terminal deletion. Gene 1992, 112, 29–35. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NAFME Haplotypes | Country | SNP | Polymorphic Type | Variant Frequency | Variant Coverage | Variant p-Value * |
|---|---|---|---|---|---|---|
| NAFME 1 | Iran, Kuwait, Saudi Arabia, Yemen | m.870A > G | transition | 66.70% | 7 | 1.50 × 10−7 |
| m.1074T > G | transversion | 100.00% | 7 | 1.00 × 10−12 | ||
| m.1068T > C | transition | 100.00% | 7 | 1.00 × 10−12 | ||
| m.1137T > C | transition | 100.00% | 7 | 1.00 × 10−12 | ||
| m.1072G > A | transition | 100.00% | 7 | 1.00 × 10−12 | ||
| NAFME 2 | Saudi Arabia, Yemen | m.1074T > G | transversion | 100.00% | 10 | 1.00 × 10−18 |
| m.810T > C | transition | 88.90% | 10 | 8.90 × 10−12 | ||
| m.1068T > C | transition | 100.00% | 10 | 1.00 × 10−18 | ||
| m.1137T > C | transition | 100.00% | 10 | 1.00 × 10−18 | ||
| m.1152T > C | transition | 100.00% | 10 | 1.00 × 10−18 | ||
| m.1053G > A | transition | 100.00% | 10 | 1.00 × 10−18 | ||
| NAFME 3 | Ethiopia, Iran, Kuwait, Saudi Arabia, Yemen | m.1074T > G | transversion | 100.00% | 14 | 1.00 × 10−26 |
| m.1068T > C | transition | 100.00% | 14 | 1.00 × 10−26 | ||
| m.1137T > C | transition | 84.60% | 14 | 7.70 × 10−21 | ||
| m.1152T > C | transition | 61.50% | 14 | 1.20 × 10−13 | ||
| NAFME 4 | Iran, Iraq, Pakistan, Turkmenistan | m.1137T > C | transition | 100.00% | 16 | 1.00 × 10−30 |
| m.1152T > C | transition | 100.00% | 16 | 1.00 × 10−30 | ||
| m.1317T > C | transition | 100.00% | 16 | 1.00 × 10−30 | ||
| NAFME 5 | Iran, Iraq, Oman, Pakistan, United Arab Emirates | m.1375C > T | transition | 100.00% | 33 | 1.00 × 10−64 |
| m.826T > C | transition | 100.00% | 33 | 1.00 × 10−64 | ||
| m.1137T > C | transition | 93.80% | 33 | 4.90 × 10−58 | ||
| m.1152T > C | transition | 100.00% | 33 | 1.00 × 10−64 | ||
| NAFME 6 | China, India, Israel | m.1412T > G | transversion | 100.00% | 7 | 1.00 × 10−14 |
| NAFME 7 | Israel | m.1383T > C | transition | 100.00% | 7 | 0.0001 |
| m.1107T > C | transition | 100.00% | 7 | 0.0001 | ||
| NAFME 8 ¥ (AzB) | Argentina, Brazil, China, Colombia, Costa Rica, Cyprus, Ecuador, Egypt, Guatemala, India, Israel, Malaysia, Saudi Arabia, South Korea, Trinidad and Tobago, Turkey, USA, and others α | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paredes-Montero, J.R.; Haq, Q.M.I.; Mohamed, A.A.; Brown, J.K. Phylogeographic and SNPs Analyses of Bemisia tabaci B Mitotype Populations Reveal Only Two of Eight Haplotypes Are Invasive. Biology 2021, 10, 1048. https://doi.org/10.3390/biology10101048
Paredes-Montero JR, Haq QMI, Mohamed AA, Brown JK. Phylogeographic and SNPs Analyses of Bemisia tabaci B Mitotype Populations Reveal Only Two of Eight Haplotypes Are Invasive. Biology. 2021; 10(10):1048. https://doi.org/10.3390/biology10101048
Chicago/Turabian StyleParedes-Montero, Jorge R., Q. M. Imranul Haq, Amr A. Mohamed, and Judith K. Brown. 2021. "Phylogeographic and SNPs Analyses of Bemisia tabaci B Mitotype Populations Reveal Only Two of Eight Haplotypes Are Invasive" Biology 10, no. 10: 1048. https://doi.org/10.3390/biology10101048