
ARGEOS: A New Bioinformatic Tool for Detailed Systematics Search in GEO and ArrayExpress

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Implementation
2.2. Obtaining GEO Data
2.3. Obtaining AE Data
2.4. Data Analysis
2.5. Analyzing PubMed Data
2.6. Data Export
2.7. Source Code Availability
3. Results
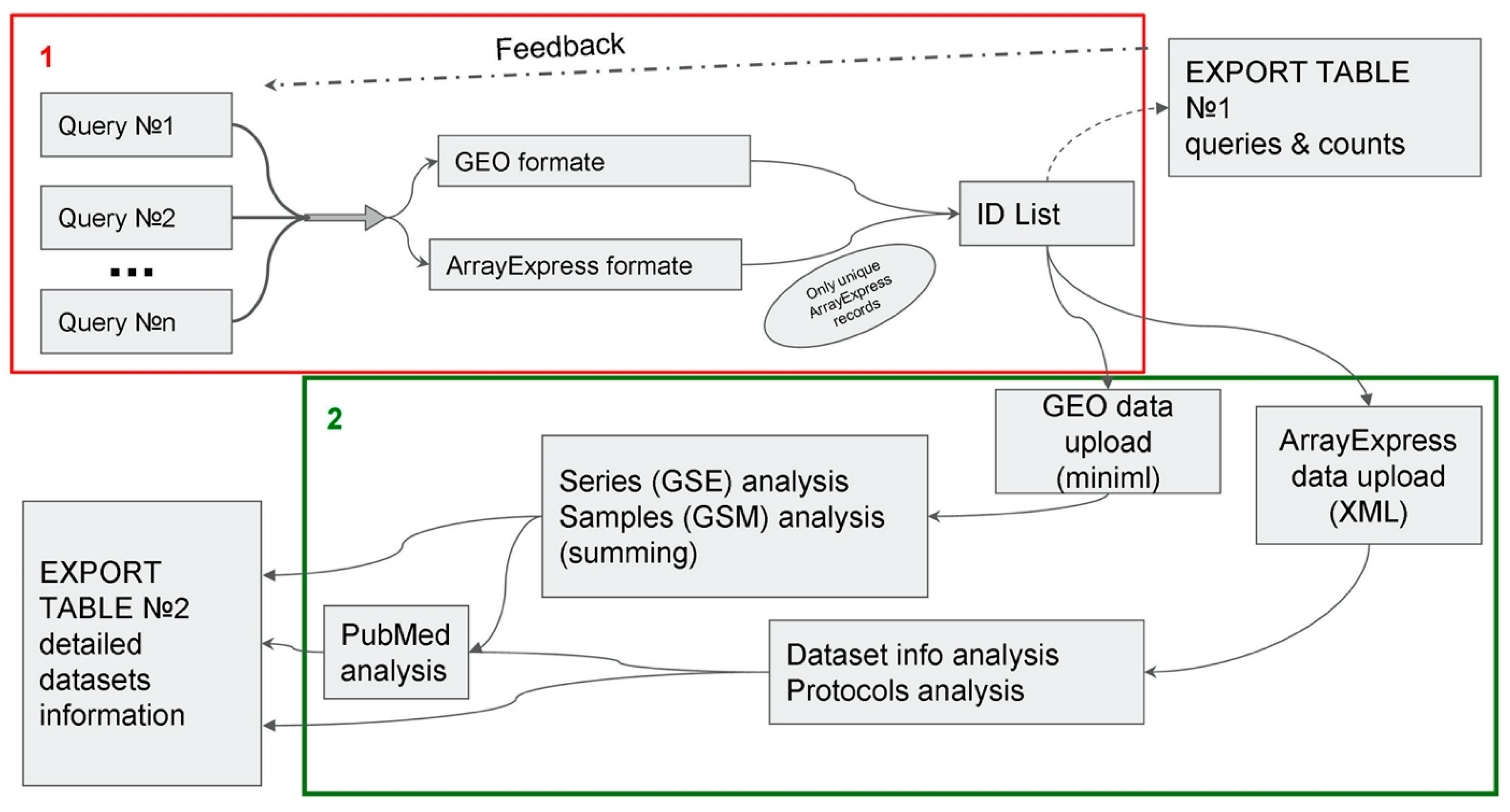
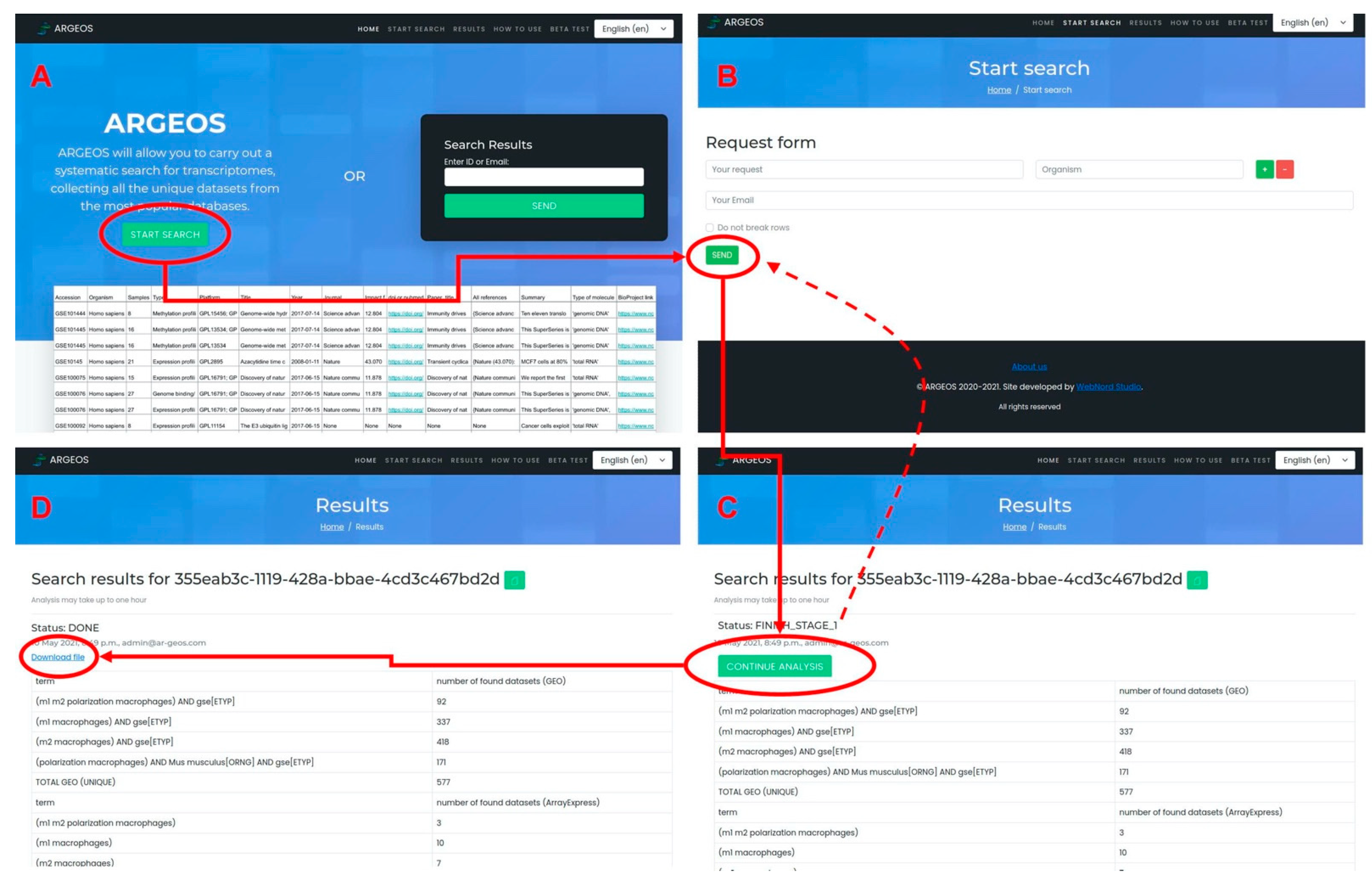
3.1. ARGEOS Work Algorithm
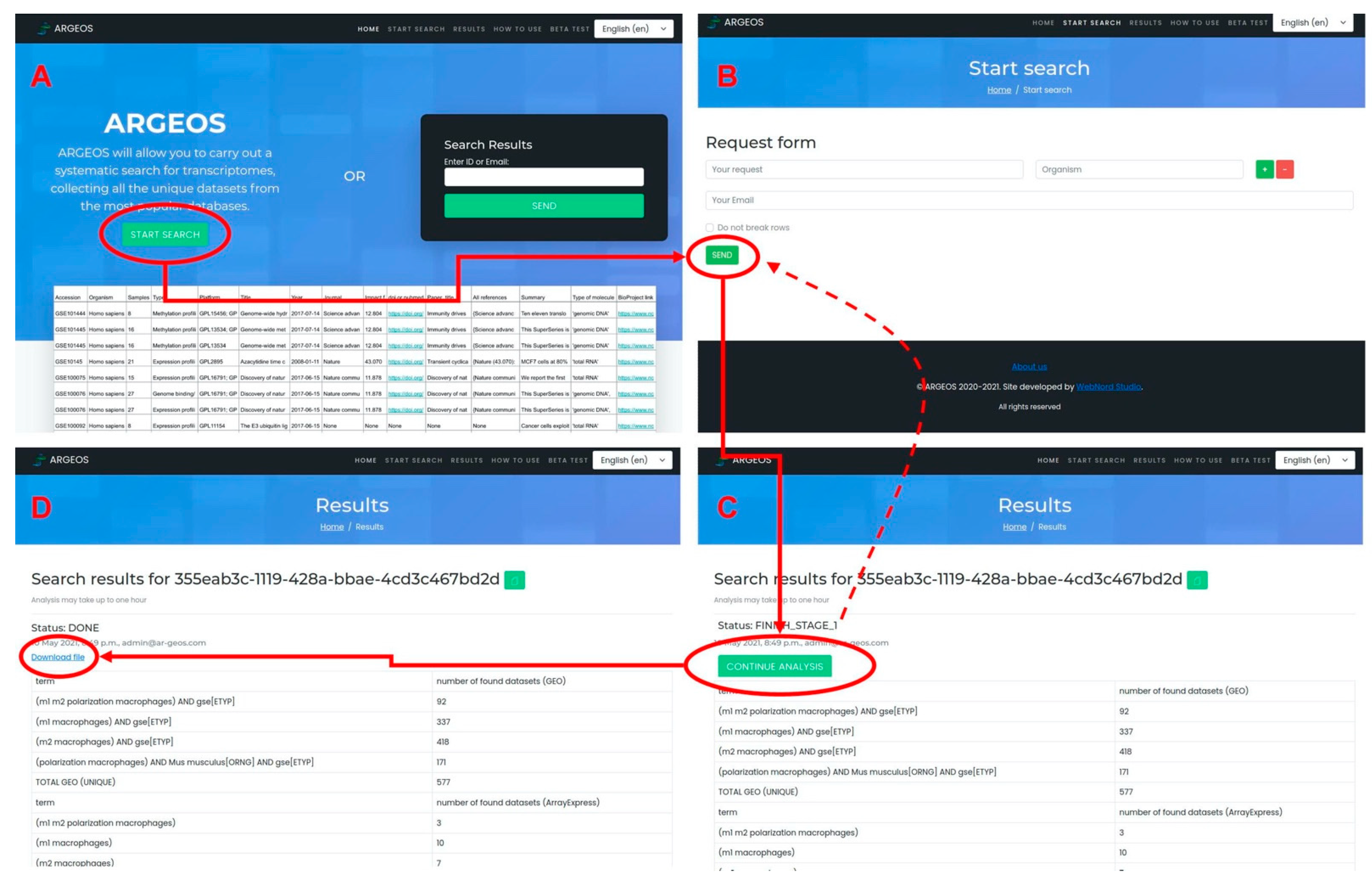
3.2. Graphical Web Interface
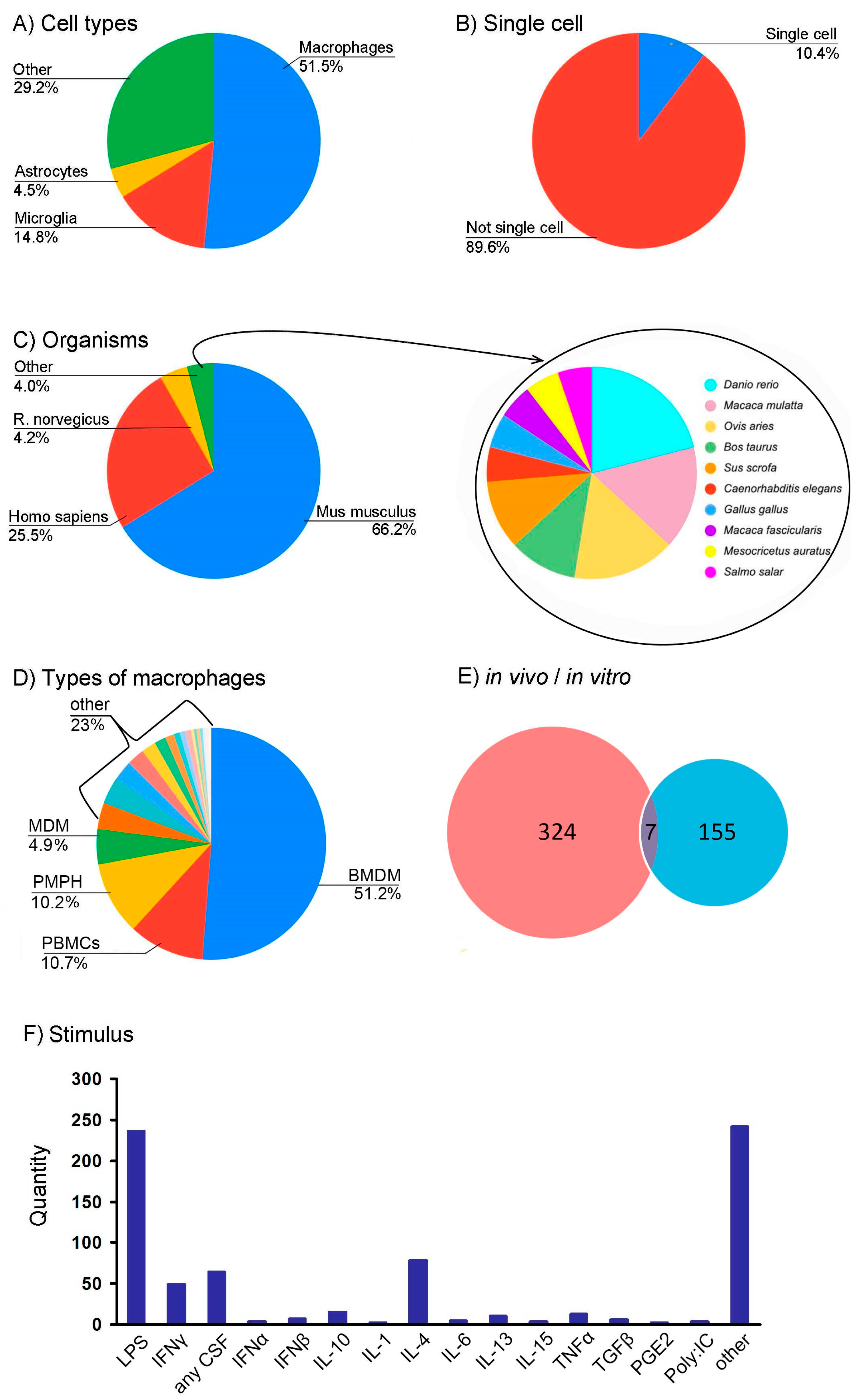
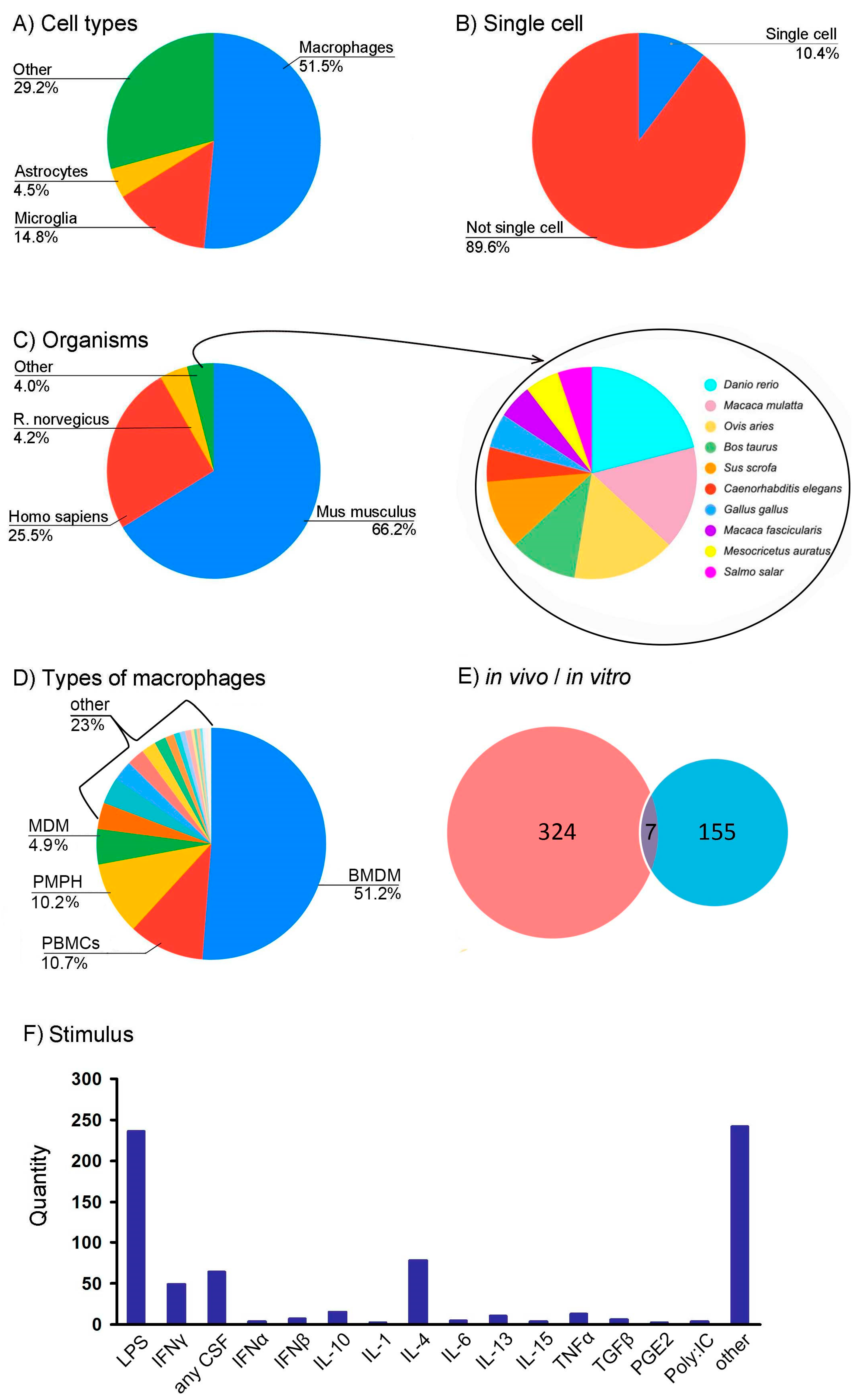
3.3. Creation of RNA-seq Datasets Using ARGEOS to Study the Phenomenon of Changes in Cellular Phenotype
4. Discussion
- Users can send several queries simultaneously, which are supposed to be used for systematic searches.
- It is possible to analyze the number of finds for each query (this allows the user to adjust queries in advanced searches).
- Advanced analysis of information about the dataset is available.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cacheaux, L.P.; Ivens, S.; David, Y.; Lakhter, A.J.; Bar-Klein, G.; Shapira, M.; Heinemann, U.; Friedman, A.; Kaufer, D. Transcriptome profiling reveals TGF-β signaling involvement in epileptogenesis. J. Neurosci. 2009, 29, 8927–8935. [Google Scholar] [CrossRef]
- Stockhammer, O.W.; Zakrzewska, A.; Hegedûs, Z.; Spaink, H.P.; Meijer, A.H. Transcriptome Profiling and functional analyses of the zebrafish embryonic innate immune response to Salmonella infection. J. Immunol. 2009, 182, 5641–5653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Shim, J.K.; Yoon, S.J.; Kim, S.H.; Chang, J.H.; Kang, S.G. Transcriptome profiling-based identification of prognostic subtypes and multi-omics signatures of glioblastoma. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Strunnikova, N.V.; Maminishkis, A.; Barb, J.J.; Wang, F.; Zhi, C.; Sergeev, Y.; Chen, W.; Edwards, A.O.; Stambolian, D.; Abecasis, G.; et al. Transcriptome analysis and molecular signature of human retinal pigment epithelium. Hum. Mol. Genet. 2010, 19, 2468–2486. [Google Scholar] [CrossRef]
- Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome profiling in human diseases: New advances and perspectives. Int. J. Mol. Sci. 2017, 18, 1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Kui, L.; Tang, M.; Li, D.; Wei, K.; Chen, W.; Miao, J.; Dong, Y. High-throughput transcriptome profiling in drug and biomarker discovery. Front. Genet. 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Mashima, J.; Kosuge, T.; Ogasawara, O. DDBJ update: The Genomic Expression Archive (GEA) for functional genomics data. Nucleic Acids Res. 2019, 47, D69–D73. [Google Scholar] [CrossRef]
- Wang, Z.; Lachmann, A.; Ma’ayan, A. Mining data and metadata from the gene expression omnibus. Biophys. Rev. 2019, 11, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Gundersen, G.W.; Jones, M.R.; Rouillard, A.D.; Kou, Y.; Monteiro, C.D.; Feldmann, A.S.; Hu, K.S.; Ma’Ayan, A. GEO2Enrichr: Browser extension and server app to extract gene sets from GEO and analyze them for biological functions. Bioinformatics 2015, 31, 3060–3062. [Google Scholar] [CrossRef]
- Toro-Domínguez, D.; Martorell-Marugán, J.; López-Domínguez, R.; García-Moreno, A.; González-Rumayor, V.; Alarcón-Riquelme, M.E.; Carmona-Sáez, P. ImaGEO: Integrative gene expression meta-analysis from GEO database. Bioinformatics 2019, 35, 880–882. [Google Scholar] [CrossRef]
- Djordjevic, D.; Tang, J.Y.S.; Chen, Y.X.; Kwan, S.L.S.; Ling, R.W.K.; Qian, G.; Woo, C.Y.Y.; Ellis, S.J.; Ho, J.W.K. Discovery of perturbation gene targets via free text metadata mining in Gene Expression Omnibus. Comput. Biol. Chem. 2019, 80, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Mahi, N.; Najafabadi, M.F.; Pilarczyk, M.; Kouril, M.; Medvedovic, M. GREIN: An interactive web platform for re-analyzing GEO RNA-seq data. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Alaimo, S.; Di Maria, A.; Shasha, D.; Ferro, A.; Pulvirenti, A. TACITuS: Transcriptomic data collector, integrator, and selector on big data platform. BMC Bioinform. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivliev, A.E.; ’t Hoen, P.A.C.; Villerius, M.P.; den Dunnen, J.T.; Brandt, B.W. Microarray retriever: A web-based tool for searching and large scale retrieval of public microarray data. Nucleic Acids Res. 2008, 36, W327–W331. [Google Scholar] [CrossRef] [PubMed]
- Bono, H. All of gene expression (AOE): An integrated index for public gene expression databases. PLoS ONE 2020, 15, e0227076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Chistyakov, D.V.; Gavrish, G.E.; Goriainov, S.V.; Chistyakov, V.V.; Astakhova, A.A.; Azbukina, N.V.; Sergeeva, M.G. Oxylipin profiles as functional characteristics of acute inflammatory responses in astrocytes pre-treated with IL-4, IL-10, or LPS. Int. J. Mol. Sci. 2020, 21, 1780. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2006, 34, D173–D180. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, J.P.; Holmes, E.C. Entrezpy: A Python library to dynamically interact with the NCBI Entrez databases. Bioinformatics 2019, 35, 4511–4514. [Google Scholar] [CrossRef] [PubMed]
- Mackaness, G.B. Cellular resistance to infection. J. Exp. Med. 1962, 116, 381–406. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, E.; Lee, S.; Kim, J.H.; Kim, J.H.; Seo, J.W.; Lee, W.H.; Mori, K.; Nakao, K.; Suk, K. Secreted protein lipocalin-2 promotes microglial M1 polarization. FASEB J. 2013, 27, 1176–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, E.; Kim, J.-H.; Lee, S.; Kim, J.-H.; Seo, J.-W.; Jin, M.; Lee, M.-G.; Jang, I.-S.; Lee, W.-H.; Suk, K. Phenotypic polarization of activated astrocytes: The critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. J. Immunol. 2013, 191, 5204–5219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarassishin, L.; Suh, H.S.; Lee, S.C. LPS and IL-1 differentially activate mouse and human astrocytes: Role of CD14. Glia 2014, 62, 999–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Cassol, E.; Poli, G. Macrophage polarization in health and disease. Sci. World J. 2011, 11, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Siddaway, A.P.; Wood, A.M.; Hedges, L.V. How to do a systematic review: A best practice guide for conducting and reporting narrative reviews, meta-analyses, and meta-syntheses. Annu. Rev. Psychol. 2019, 70, 747–770. [Google Scholar] [CrossRef]
- Linares-Espinós, E.; Hernández, V.; Domínguez-Escrig, J.L.; Fernández-Pello, S.; Hevia, V.; Mayor, J.; Padilla-Fernández, B.; Ribal, M.J. Methodology of a systematic review. Actas Urol. Esp. 2018, 42, 499–506. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of microglia and astrocytes in the neurovascular unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Sarkans, U.; Füllgrabe, A.; Ali, A.; Athar, A.; Behrangi, E.; Diaz, N.; Fexova, S.; George, N.; Iqbal, H.; Kurri, S.; et al. From ArrayExpress to BioStudies. Nucleic Acids Res. 2021, 49, D1502–D1506. [Google Scholar] [CrossRef]
- Ivliev, A.E.; ’t Hoen, P.A.C.; Sergeeva, M.G. Coexpression network analysis identifies transcriptional modules related to proastrocytic differentiation and sprouty signaling in glioma. Cancer Res. 2010, 70, 10060–10070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivliev, A.E.; ’t Hoen, P.A.C.; van Roon-Mom, W.M.C.; Peters, D.J.M.; Sergeeva, M.G. Exploring the transcriptome of ciliated cells using in silico dissection of human tissues. PLoS ONE 2012, 7, e35618. [Google Scholar] [CrossRef] [PubMed]
- Ivliev, A.E.; ’t Hoen, P.A.C.; Borisevich, D.; Nikolsky, Y.; Sergeeva, M.G. Drug repositioning through systematic mining of gene coexpression networks in cancer. PLoS ONE 2016, 11, e0165059. [Google Scholar] [CrossRef]
- Liu, J.; Cao, X. Cellular and molecular regulation of innate inflammatory responses. Cell. Mol. Immunol. 2016, 13, 711–721. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavrish, G.E.; Chistyakov, D.V.; Sergeeva, M.G. ARGEOS: A New Bioinformatic Tool for Detailed Systematics Search in GEO and ArrayExpress. Biology 2021, 10, 1026. https://doi.org/10.3390/biology10101026
Gavrish GE, Chistyakov DV, Sergeeva MG. ARGEOS: A New Bioinformatic Tool for Detailed Systematics Search in GEO and ArrayExpress. Biology. 2021; 10(10):1026. https://doi.org/10.3390/biology10101026
Chicago/Turabian StyleGavrish, Gleb E., Dmitry V. Chistyakov, and Marina G. Sergeeva. 2021. "ARGEOS: A New Bioinformatic Tool for Detailed Systematics Search in GEO and ArrayExpress" Biology 10, no. 10: 1026. https://doi.org/10.3390/biology10101026
APA StyleGavrish, G. E., Chistyakov, D. V., & Sergeeva, M. G. (2021). ARGEOS: A New Bioinformatic Tool for Detailed Systematics Search in GEO and ArrayExpress. Biology, 10(10), 1026. https://doi.org/10.3390/biology10101026