
3′-Nitro- and 3′-Aminofluoresceins: Appearance of Previously Missing Dyes

 , , , and
, , , and 
Abstract
1. Introduction
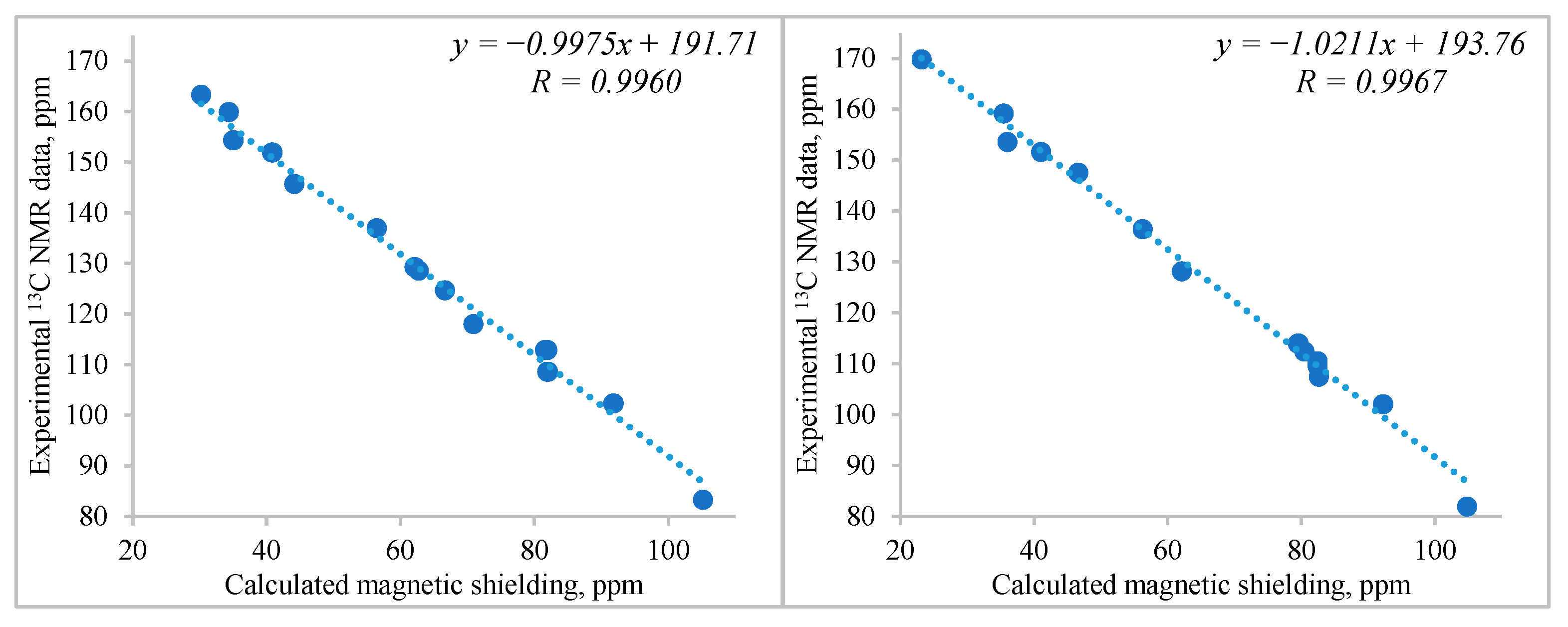
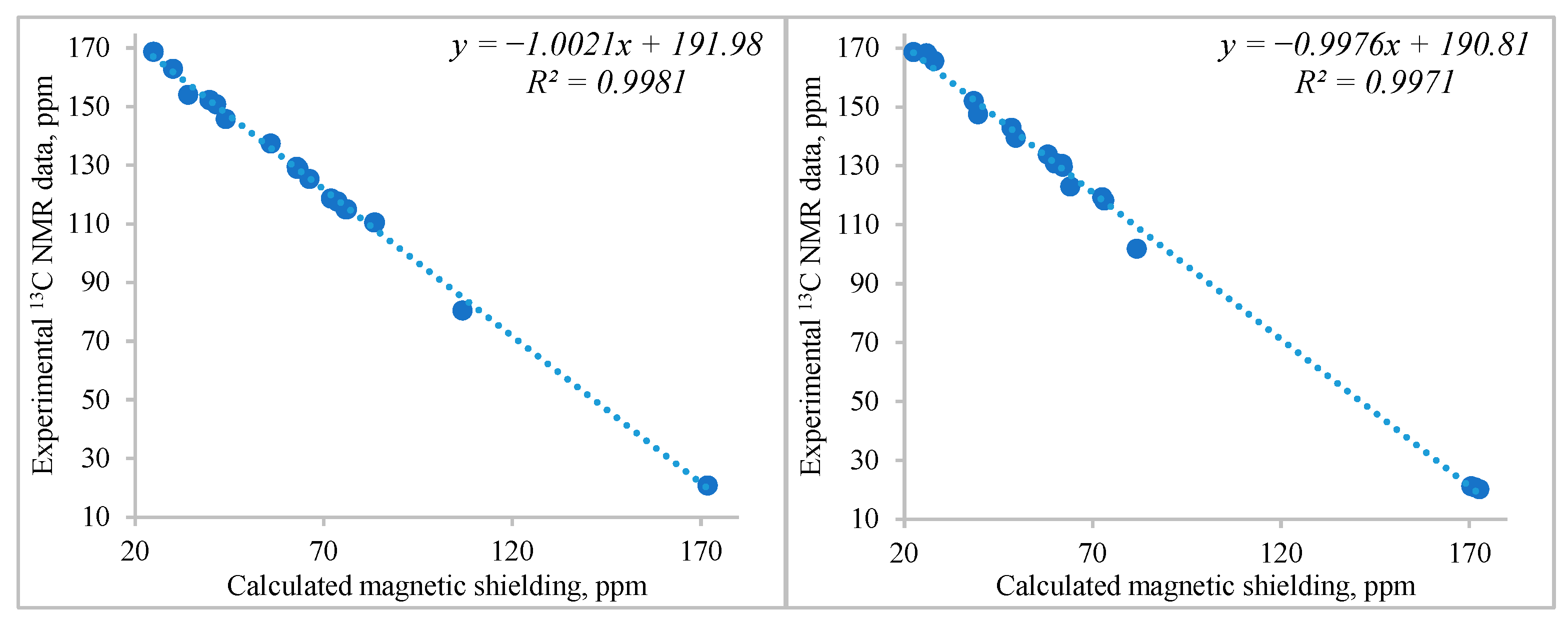
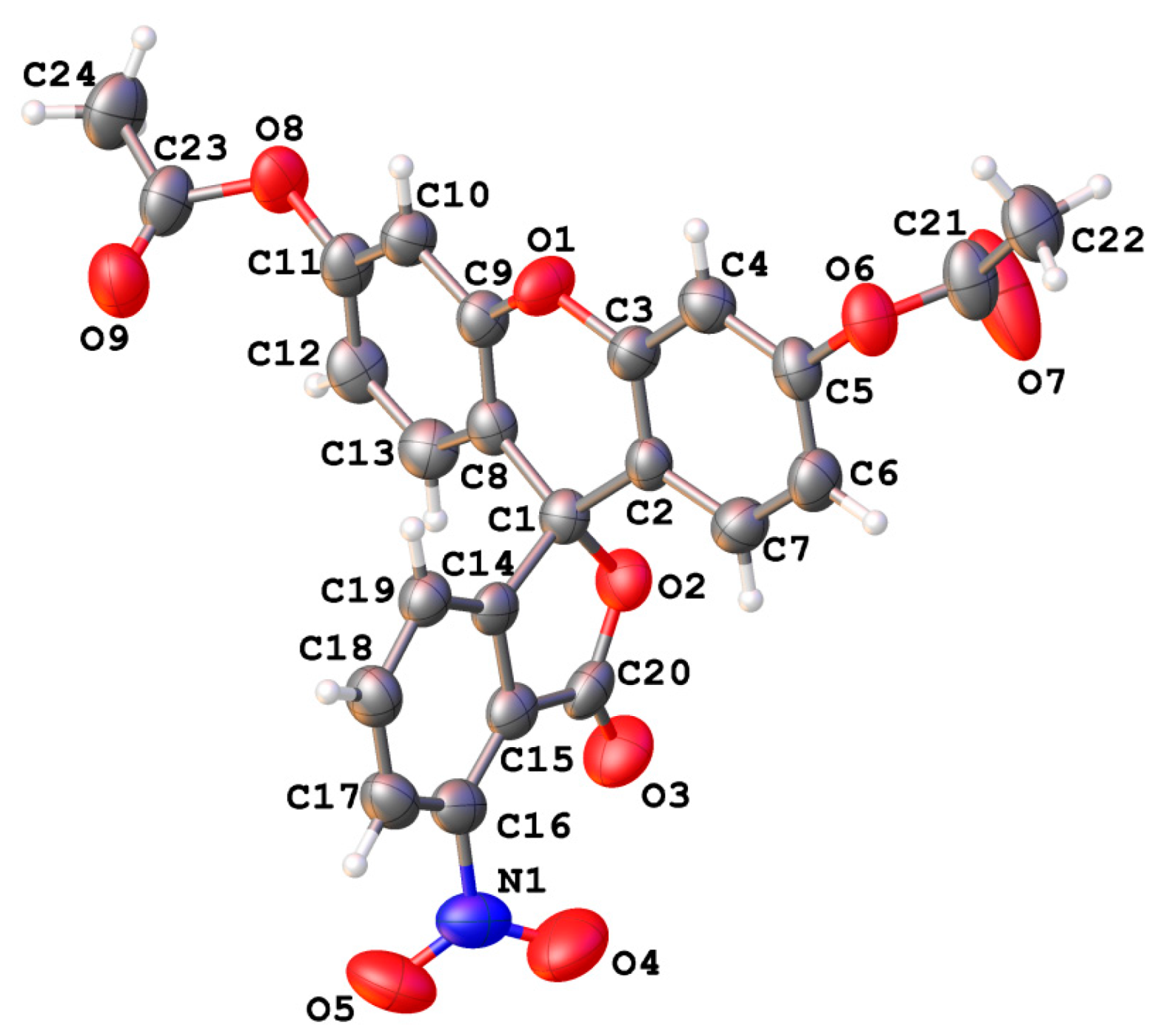
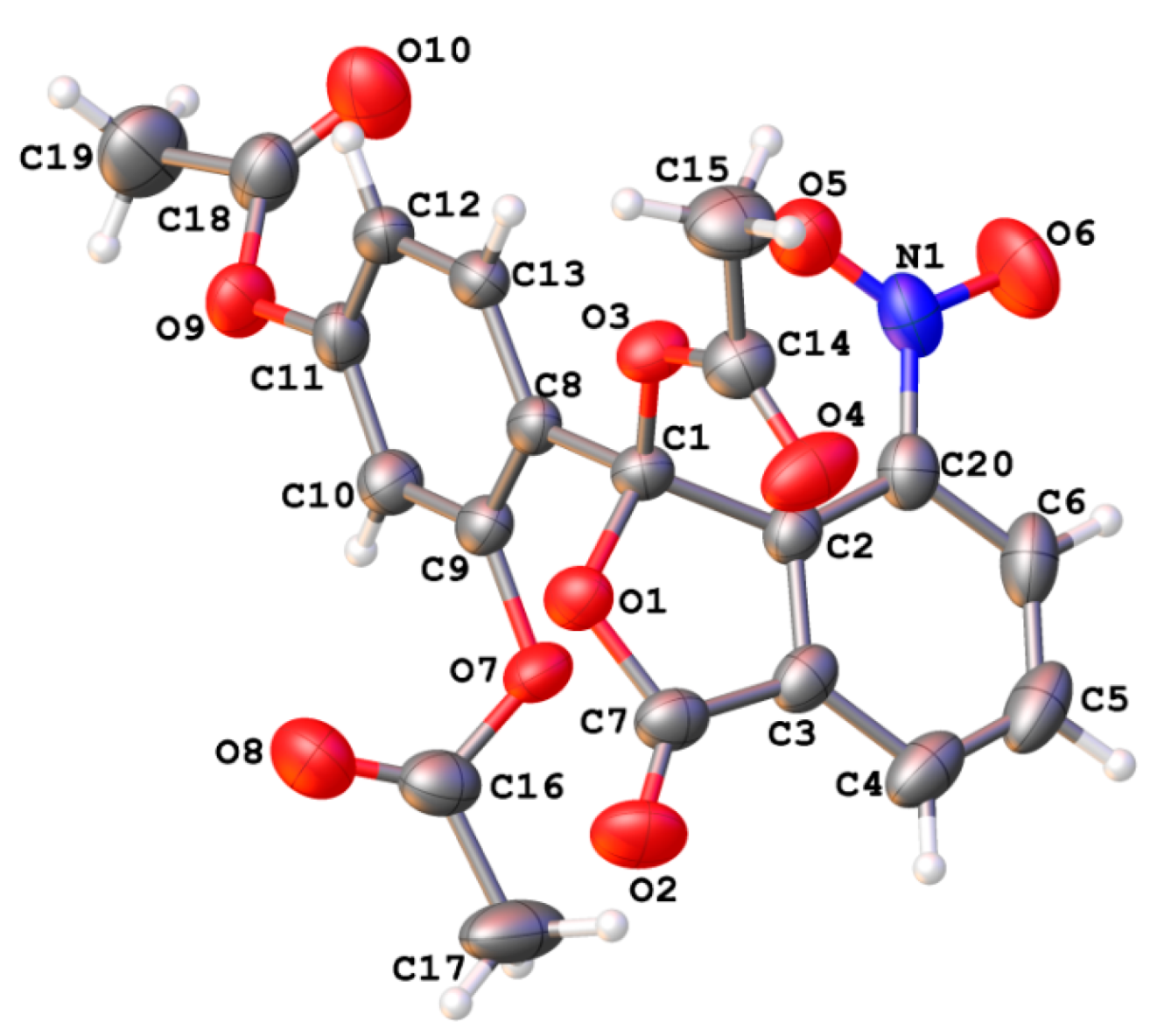
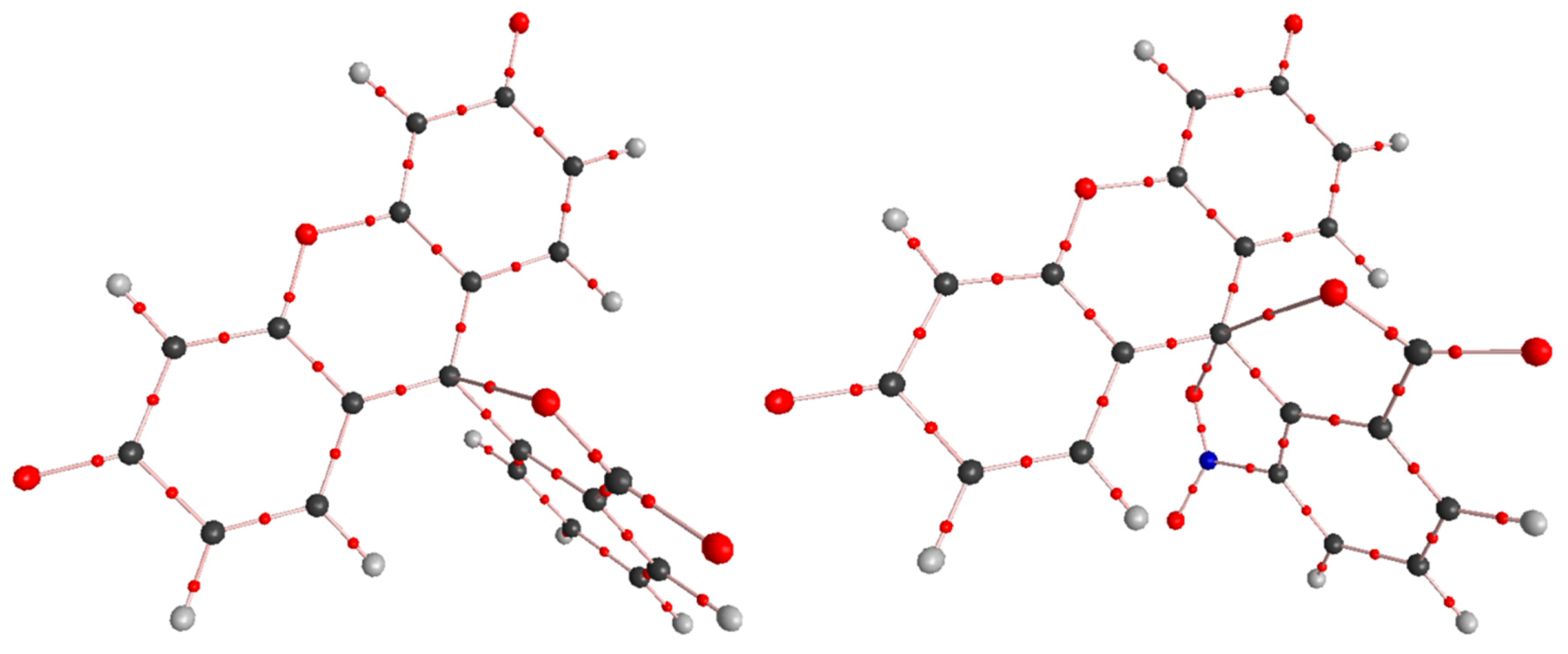
2. Results and Discussion
3. Experimental
3.1. Materials
3.2. Apparatus
3.3. Synthesis of the Dyes
3.3.1. Synthesis, Separation, and Purification of 3′-Nitrofluorescein and 6′-Nitrointermediate
- (i)
- A mixture of 2.00 g 6′-nitrointermediate triacetate, 8 mL of acetic acid, and 1.0 mL of 70% aqueous methanesulfonic acid (MSA) was refluxed for 30 min. The solution was cooled, and the crystallization was initiated by stirring with a glass rod. The solution was left overnight at 6 °C. The next day, small colorless crystals were collected on a glass filter, washed with 5 mL of acetic acid and three portions of water of 10 mL each. The precipitate became heavy with lemon-colored grains. The yield was 1.25 g (88.4%), with m.p. 258–260 °C. 1H NMR (400 MHz, DMSO-d6), δ/ppm: 13.28 (1H, brs, 2′ (COOH)), 11.56 (1H, brs, 5 (PhOH)), 10.58 (1H, brs, 3 (PhOH)), 8.42 (1H, d, J = 7.9 Hz, 5′), 8.32 (1H, d, J = 7.7 Hz, 3′), 7.85 (1H, brm, 4′), 7.11 (1H, brs, 1), 6.29 (2H, m, 2, 4).
- (ii)
- A mixture of 2.00 g 6′-nitrointermediate triacetate, 3 mL of acetic acid, and 1.0 mL of 70% aqueous MSA was refluxed for 15 min. Then 6.0 mL of hot water was added and the solution was slowly cooled. The next day, the lemon-colored crystals were collected and washed with water. The yield was 1.33 g (94.1%).
- (iii)
- A 2.00 g of crude 6′-nitrointermediate was recrystallized from 12.5 mL of acetic acid. The yellow crystals were collected and washed with 4 mL of acetic acid. The yield was 1.44 g (72.0%), with m.p. 256 °C.
3.3.2. Synthesis of 3′-Aminofluorescein
3.3.3. Synthesis of 3′-Nitrofluorescein and 3′-Aminofluorescein Methyl Esters
3.3.4. Some Additional Experiments
- (i)
- A mixture of 0.20 g of 3′-nitrofluorescein and 4.0 g of 50% NaOH was heated at 85–90 °C for 5 h. The reaction course was monitored by measuring the electronic absorption spectra. The solution was then cooled, acidified with 10% HCl to pH 6–7 and filtered. The filtrate was dried at 100–105 °C. The residue was then treated with several portions of acetone (10 mL), and the extract was filtered through a membrane filter and dried. The yield was 0.060 g of dry residue.
- (ii)
- A mixture of 0.20 g of 3′-aminofluorescein and 4.0 g of 50% NaOH was heated at 100–105 °C for 2 h. Next, the product mixture was isolated as described above. A yield of 0.10 g of dry residue was obtained. The NMR spectrum was identical to case (i). To isolate the 3′-aminointermediate M3, the resulting mixture was dissolved in 0.5 mL of acetic acid, filtered and acidified with two drops of 35% HCl. The yellow precipitate was collected, washed with several portions of acetic acid and dissolved on heating in 0.5 mL of 50% aqueous ethanol. To the obtained hot solution, three drops of 35% HCl were added. On cooling, yellow crystals precipitated. The product was collected, washed with several portions of acetonitrile and dried. The yield was 0.030 g of 3′-aminointermediate M3. Monoisotopic mass: 273.06. ESI MS: 274.35 [M + H]+, 272.28 [M − H]−. 1H NMR (400 MHz, DMSO-d6), δ/ppm: 12.34 (1H, brs, PhOH), 10.64 (1H, brs, PhOH), 7.28 (1H, t, J = 7.8 Hz, 5′), 7.04 (1H, d, J = 8.4 Hz, 1), 6.90 (1H, d, J = 7.8 Hz, 6′), 6.40 (1H, d, J = 7.8 Hz, 4′), 6.29 (1H, d, J = 2.1 Hz, 4), 6.28 (1H, dd, J = 8.4 Hz, J = 2.1 Hz, 2).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Coons, A.H.; Kaplan, M.H. Localization of antigen in tissue cells: II. Improvements in a method for the detection of antigen by means of fluorescent antibody. J. Exp. Med. 1950, 91, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sigmund, H.; Pfleiderer, W. Nucleotides. Part LXXI: A new type of labelling of nucleosides and nucleotides. Helv. Chim. Acta 2003, 86, 2299–2334. [Google Scholar] [CrossRef]
- Kuroiwa, T. Photodynamic diagnosis and photodynamic therapy for the brain tumors. Prog. Neuro-Oncol. 2014, 21, 4–21. [Google Scholar] [CrossRef]
- Bharate, G.Y.; Qin, H.; Fang, J. Poly(styrene-co-maleic acid)-conjugated 6-aminofluorescein and rhodamine micelle as macromolecular fluorescent probes for micro-tumors detection and imaging. J. Pers. Med. 2022, 12, 1650. [Google Scholar] [CrossRef]
- Hongrapipat, J.; Kopecková, P.; Liu, J.; Prakongpan, S.; Kopecek, J. Combination chemotherapy and photodynamic therapy with fab’ fragment targeted HPMA copolymer conjugates, in human ovarian carcinoma cells. Mol. Pharm. 2008, 5, 696–709. [Google Scholar] [CrossRef]
- Glushko, V.N.; Blokhina, L.I.; Sadovskaya, N.Y. Studies on the synthesis of fluorescein-5-isothiocyanate: A fluorescent nanomarker for biosensors. Russ. J. Gen. Chem. 2015, 85, 2458–2464. [Google Scholar] [CrossRef]
- Reverdin, F. Ueber einen gelben Farbstoff, welcher vom Dinitrofluoresceïn abstammt. Berichte 1897, 30, 332–334. [Google Scholar] [CrossRef]
- Bogert, M.T.; Wright, R.G. Some experiments on the nitro derivatives of fluorescein. J. Am. Chem. Soc. 1905, 27, 1310–1316. [Google Scholar] [CrossRef][Green Version]
- Moir, J. Colour and chemical constitution, Part II.—The spectra of the mixed phthaleins and of the sulphone-phthaleins. Trans. R. Soc. S. Afr. 1918, 7, 111–116. [Google Scholar] [CrossRef]
- Moir, J. Colour and chemical constitution. Part XV. A systematic study of fluorescein and resorcin-benzein. Trans. R. Soc. S. Afr. 1922, 10, 159–164. [Google Scholar] [CrossRef]
- Underwood, H.W.; Wakeman, R.L. Studies in the 3-nitrophthalic acid series. J. Am. Chem. Soc. 1931, 53, 1839–1842. [Google Scholar] [CrossRef]
- Hanna, C.; Smith, W.T. The absorption spectra of derivatives of phthalic anhydride. Proc. Iowa Acad. Sci. 1951, 58, 251–260. [Google Scholar]
- Klonis, N.; Sawyer, W.H. Spectral properties of the prototropic forms of fluorescein in aqueous solution. J. Fluoresc. 1996, 6, 147–157. [Google Scholar] [CrossRef] [PubMed]
- DeRose, P.C.; Kramer, G.W. Bias in the absorption coefficient determination of a fluorescent dye, standard reference material 1932 fluorescein solution. J. Lumin. 2005, 113, 314–320. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Ruud, K.; Helgaker, T.; Bak, K.L.; Jørgensen, P.; Jensen, H.J.A. Hartree-Fock Limit Magnetizabilities from London Orbitals. J. Chem. Phys. 1993, 99, 3847–3859. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 15–18. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Burgi, H.-B.; Dunitz, J.D. Structure Correlation; VCH: Weinheim, Germany, 1994; Volume 2, pp. 741–784. [Google Scholar]
- Polyakova, I.N.; Starikova, Z.A.; Parusnikov, B.V.; Krasavin, I.A.; Dobryakova, G.M.; Zhadanov, B.V. The lactone form of fluorescein: Crystal structure of the 1:1 molecular complex of fluorescein with methanol. J. Struct. Chem. 1984, 25, 752–757. [Google Scholar] [CrossRef]
- Osborn, R.S.; Rogers, D. The crystal and molecular structure of the 1:1 complex of acetone with the lactoid form of fluorescein. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1975, B31, 359–364. [Google Scholar] [CrossRef]
- Arhangelskis, M.; Eddleston, M.D.; Reid, D.G.; Day, G.M.; Bucar, D.-K.; Morris, A.J.; Jones, W. Rationalization of the color properties of fluorescein in the solid state: A combined computational and experimental study. Chem.-Eur. J. 2016, 22, 10065–10073. [Google Scholar] [CrossRef] [PubMed]
- Mchedlov-Petrossyan, N.O.; Cheipesh, T.A.; Shekhovtsov, S.V.; Ushakova, E.V.; Roshal, A.D.; Omelchenko, I.V. Aminofluoresceins vs fluorescein: Ascertained new unusual features of tautomerism and dissociation of hydroxyxanthene dyes in solution. J. Phys. Chem. A 2019, 123, 8845–8859. [Google Scholar] [CrossRef]
- Mchedlov-Petrossyan, N.O.; Cheipesh, T.A.; Shekhovtsov, S.V.; Redko, A.N.; Rybachenko, V.I.; Omelchenko, I.V.; Shishkin, O.V. Ionization and tautomerism of methyl fluorescein and related dyes. Spectrochim. Acta A 2015, 150, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Suresh Kumar, G.S.; Seethalakshmi, P.G.; Bhuvanesh, N.; Kumaresan, S. Studies on the syntheses, structural characterization, antimicrobial-, and DPPH radical scavenging activity of the cocrystals caffeine:cinnamic acid and caffeine:eosin dihydrate. J. Mol. Struct. 2013, 1050, 88–96. [Google Scholar] [CrossRef]
- Hou, F.; Cheng, J.; Xi, P.; Chen, F.; Huang, L.; Xie, G.; Shi, Y.; Liu, H.; Bai, D.; Zeng, Z. Recognition of copper and hydrogen sulfide in vitro using a fluorescein derivative indicator. Dalton Trans. 2012, 41, 5799–5804. [Google Scholar] [CrossRef]
- He, H.; He, T.; Zhang, Z.; Xu, X.; Yang, H.; Qian, X.; Yang, Y. Ring-restricted N-nitrosated rhodamine as a green-light triggered, orange-emission calibrated and fast-releasing nitric oxide donor. Chin. Chem. Lett. 2018, 29, 1497–1499. [Google Scholar] [CrossRef]
- Mchedlov-Petrossyan, N.O.; Cheipesh, T.A.; Roshal, A.D.; Shekhovtsov, S.V.; Moskaeva, E.G.; Omelchenko, I.V. Aminofluoresceins vs fluorescein: Peculiarity of fluorescence. J. Phys. Chem. A 2019, 123, 8860–8870. [Google Scholar] [CrossRef]
- Mchedlov-Petrossyan, N.O.; Cheipesh, T.A.; Roshal, A.D.; Doroshenko, A.O.; Vodolazkaya, N.A. Fluorescence of aminofluoresceins as an indicative process allowing to distinguish between micelles of cationic surfactants and micelle-like aggregates. Methods Appl. Fluor. 2016, 4, 034002. [Google Scholar] [CrossRef] [PubMed]
- Poronik, Y.M.; Sadowski, B.; Szychta, K.; Quina, F.H.; Vullev, V.I.; Gryko, D.T. Revisiting the non-fluorescence of nitroaromatics: Presumption versus reality. J. Mater. Chem. C 2022, 10, 2870–2904. [Google Scholar] [CrossRef]
- Thomas, A.; Kirilova, E.M.; Nagesh, B.V.; Krishna Chaitanya, G.; Philip, R.; Manohara, S.R.; Sudeeksha, H.C.; Siddlingeshwar, B. Influence of nitro group on solvatochromism, nonlinear optical properties of 3-morpholinobenzanthrone: Experimental and theoretical study. J. Photochem. Photobiol. A Chem. 2023, 437, 114434. [Google Scholar] [CrossRef]
- Mchedlov-Petrossyan, N.O.; Cheipesh, T.A.; Moskaeva, E.G.; Shekhovtsov, S.V.; Ostrovskyi, K.I. Towards understanding of stepwise acid-base dissociation in systems inclined to tautomerism: Nitro derivatives of fluorescein in dimethyl sulfoxide. J. Mol. Liquids 2023, 386, 122540. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhikol, O.A.; Shishkin, O.V. Estimating stacking interaction energy using atom in molecules properties: Homodimers of benzene and pyridine. Int. J. Quant. Chem. 2012, 112, 3008–3017. [Google Scholar] [CrossRef]
- Moskaeva, E.G.; Ostrovskiy, K.I.; Shekhovtsov, S.V.; Mchedlov-Petrossyan, N.O. Transmittance of electronic effects in the fluorescein molecule: Nitro and amino groups in the phthalic acid residue. Kharkiv Univ. Bull. 2021, 36, 24–32. [Google Scholar] [CrossRef]
- Dolomanov, O.; Bourhis, L.; Gildea, R.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Atom | δ, ppm | Atom | δ, ppm | Atom | δ, ppm |
|---|---|---|---|---|---|
| 3′-nitrofluorescein 2 | |||||
| 1 | 129.30 | 9 | 83.30 | 1′ | 159.93 |
| 2 | 108.64 | 2′ | 118.06 | ||
| 3 | 154.37 | 2′a (C=O) | 163.33 | 3′ | 145.74 |
| 4 | 102.34 | 4′ | 124.71 | ||
| 1a | 112.89 | 5′ | 136.98 | ||
| 4a | 151.95 | 6′ | 128.62 | ||
| 3′-aminofluorescein 5 | |||||
| 1 | 128.19 | 9 | 81.97 | 1′ | 153.59 |
| 2 | 110.51 | 2′ | 109.59 | ||
| 3 | 159.22 | 2′a (C=O) | 169.85 | 3′ | 147.58 |
| 4 | 102.08 | 4′ | 112.43 | ||
| 1a | 113.99 | 5′ | 136.47 | ||
| 4a | 151.66 | 6′ | 107.45 | ||

| Atom | δ, ppm | Atom | δ, ppm | Atom | δ, ppm |
|---|---|---|---|---|---|
| 1 | 128.83 | C9 | 80.49 | 2′a(C=O) | 162.97 |
| 2 | 117.65 | 1′ | 154.11 | ||
| 3 | 152.29 | Acetyl groups | 2′ | 118.69 | |
| 4 | 110.55 | C=O | 168.80 | 3′ | 145.85 |
| 1a | 115.03 | 4′ | 125.33 | ||
| 4a | 150.85 | CH3 | 20.83 | 5′ | 137.44 |
| 6′ | 129.52 | ||||

| Atom | δ, ppm | Atom | δ, ppm | Acetyl Groups | δ, ppm | |
|---|---|---|---|---|---|---|
| C=O | CH3 | |||||
| 1′ | 139.54 | 1 | 129.72 | at C3 | 168.28 | 20.24 |
| 2′ | 130.66 | 2 | 116.27 | at C5 | 168.70 | 20.82 |
| 2′a (C=O) | 165.62 | 3 | 152.06 | at C7 | 167.76 | 21.26 |
| 3′ | 130.85 | 4 | 119.36 | |||
| 4′ | 133.86 | 5 | 147.60 | |||
| 5′ | 131.06 | 6 | 123.02 | |||
| 6′ | 142.97 | 7 | 101.89 | |||
| Dye | Solvent | Absorption | Emission | |
|---|---|---|---|---|
| λmax, nm | Molar Absorptivity ×10−3, M−1cm−1 | λmax, nm | ||
| 3′-Nitrofluorescein | Water | 501 | 92.18 | 520 |
| 3′-Nitrofluorescein | DMSO | 530 | 117.48 | 541 |
| 3′-Aminofluorescein | Water | 490 | 86.63 | 520 |
| 3′-Aminofluorescein | DMSO | 518 | 110.35 | 536 |
| Compound | λmax, nm | |
|---|---|---|
| In Water | In DMSO | |
| Fluorescein | 490 | 521 |
| 3′-Nitrofluorescein | 501 | 530 |
| 4′-Nitrofluorescein | 495 | 526 |
| 5′-Nitrofluorescein | 494 | 528 |
| 6′-Nitrofluorescein | 504 | 536 |
| Substituent | Angle between Xanthene and Side Benzene Rings Planes, Degrees | C(9)…O Distance, Å, Weak Non-Covalent Intramolecular Interactions Energy, kcal/mol | Electric Charge on C(9) Atom |
|---|---|---|---|
| H | 90° | 2.67 Å 3.91 kcal/mol | +0.259 |
| 3′-NO2 | 71° | 3.02 Å No bond path found | +0.198 |
| 4′-NO2 | 90° | 2.68 Å 3.82 kcal/mol | +0.257 |
| 5′-NO2 | 90° | 2.68 Å 3.83 kcal/mol | +0.256 |
| 6′-NO2 | 72° | 2.72 Å 3.58 kcal/mol | +0.237 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shekhovtsov, S.V.; Omelchenko, I.V.; Shishkina, S.V.; Doroshenko, A.O.; Vus, K.O.; Vlasenko, H.S.; Mchedlov-Petrossyan, N.O. 3′-Nitro- and 3′-Aminofluoresceins: Appearance of Previously Missing Dyes. Colorants 2023, 2, 500-517. https://doi.org/10.3390/colorants2030024
Shekhovtsov SV, Omelchenko IV, Shishkina SV, Doroshenko AO, Vus KO, Vlasenko HS, Mchedlov-Petrossyan NO. 3′-Nitro- and 3′-Aminofluoresceins: Appearance of Previously Missing Dyes. Colorants. 2023; 2(3):500-517. https://doi.org/10.3390/colorants2030024
Chicago/Turabian StyleShekhovtsov, Sergey V., Iryna V. Omelchenko, Svitlana V. Shishkina, Andrey O. Doroshenko, Kateryna O. Vus, Hanna S. Vlasenko, and Nikolay O. Mchedlov-Petrossyan. 2023. "3′-Nitro- and 3′-Aminofluoresceins: Appearance of Previously Missing Dyes" Colorants 2, no. 3: 500-517. https://doi.org/10.3390/colorants2030024
APA StyleShekhovtsov, S. V., Omelchenko, I. V., Shishkina, S. V., Doroshenko, A. O., Vus, K. O., Vlasenko, H. S., & Mchedlov-Petrossyan, N. O. (2023). 3′-Nitro- and 3′-Aminofluoresceins: Appearance of Previously Missing Dyes. Colorants, 2(3), 500-517. https://doi.org/10.3390/colorants2030024