Impact of Alternative Stabilization Strategies for the Production of PAN-Based Carbon Fibers with High Performance




Abstract
1. Introduction
1.1. Carbon Fibers Overview
1.2. Chemistry of PAN Precursors towards Carbon Fibers Production
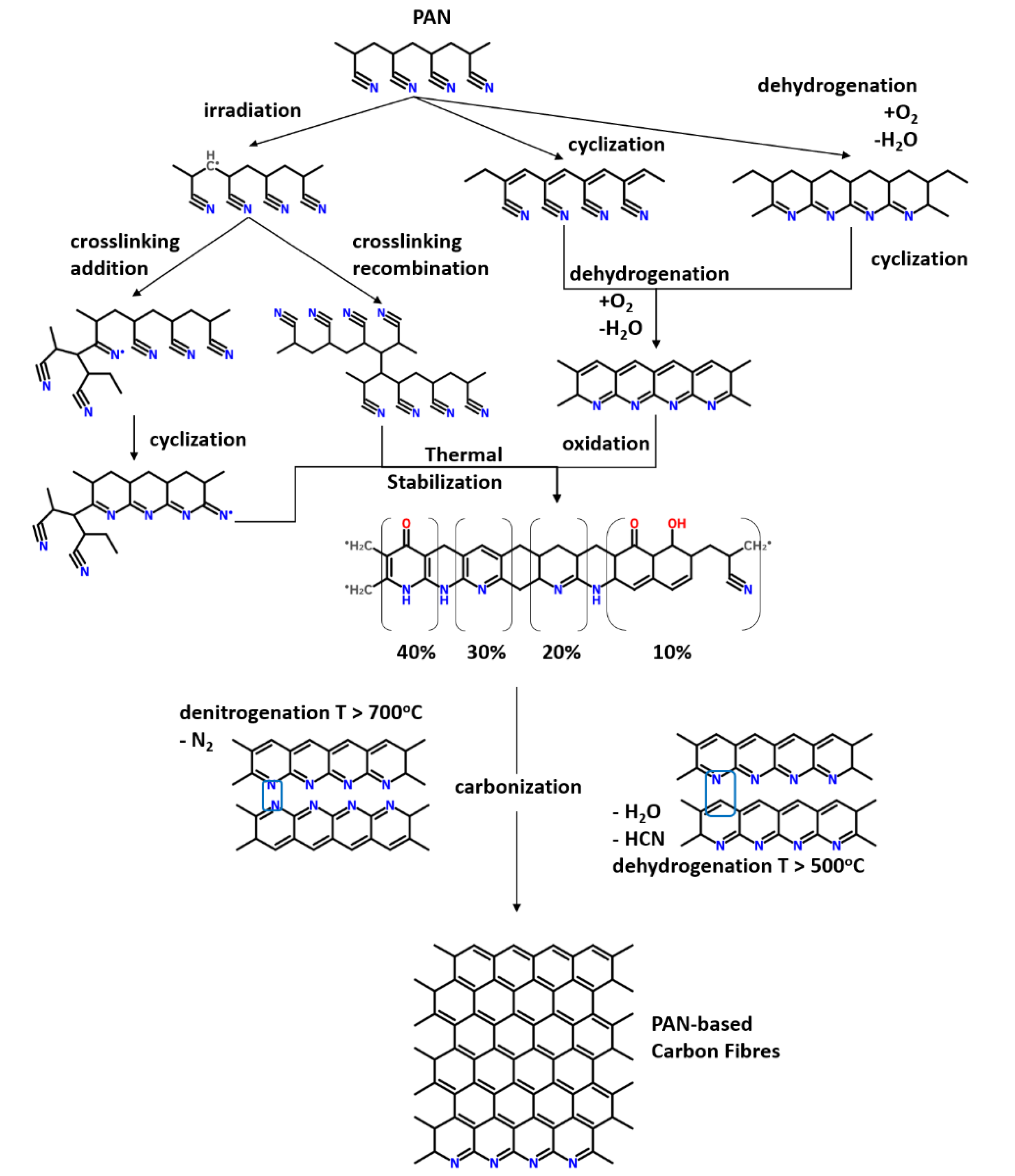
2. Structural Transformations during Stabilization
2.1. Stereochemical Configuration of PAN and Its Effect on the Structure
2.2. Thermomechanical Transformation of PAN
2.3. Outcome of Thermal Treatment: Pre-Oxidized vs. Stabilized PAN Fibers
3. Alternative Pre-Treatments towards High Performance of Carbon Fibers
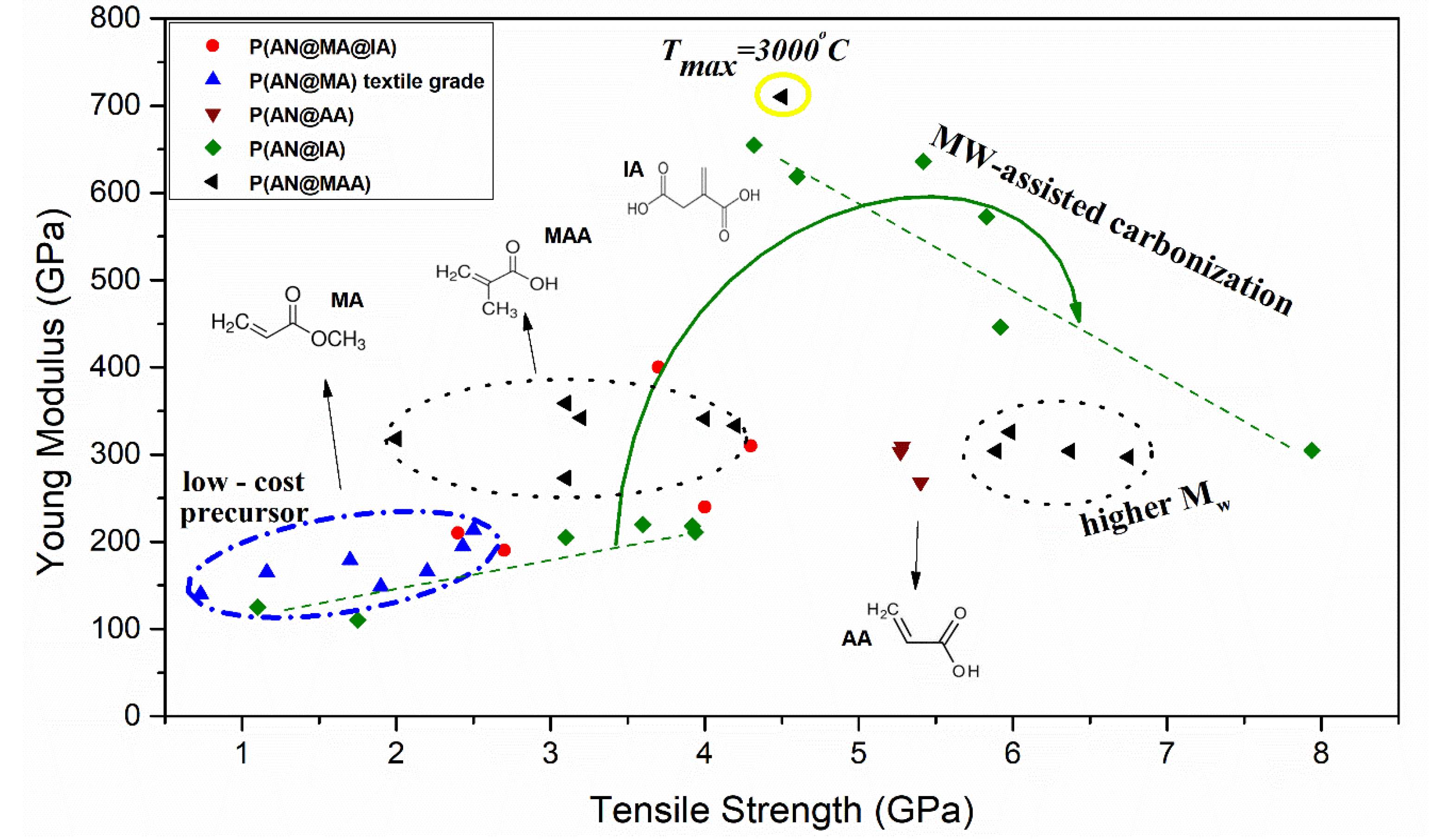
3.1. Carbon Fiber Performance Relation on Chemical Features
3.2. Stabilization Advances to Engineer Optimized Carbon Fiber Performance
3.3. Carbonization Effect on Carbon Fiber Performance
4. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
Appendix A
Physical Structure of PAN Fibers
References
- Bajaj, P.; Roopanwal, A.K. Thermal Stabilization of Acrylic Precursors for the Production of Carbon Fibers: An Overview. J. Macromol. Sci. Part C Polym. Rev. 1997, 37, 97–147. [Google Scholar] [CrossRef]
- Peebles, L.H. Carbon Fibers Formation, Structure, and Properties; Group, T.F., Ed.; CRC Press: 6000 Broken Sound Parkway NW, Boca Raton, Suite 300, FL, USA, 1995. [Google Scholar]
- Manocha, L.M. Carbon Fibers. In Reference Module in Materials Science and Materials Engineering, Encyclopedia of Materials: Science and Technology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2001; pp. 906–916. [Google Scholar] [CrossRef]
- Kaur, J.; Millington, K.; Smith, S. Producing high-quality precursor polymer and fibers to achieve theoretical strength in carbon fibers: A review. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Liu, Y.; Kumar, S. Recent Progress in Fabrication, Structure, and Properties of Carbon Fibers. Polym. Rev. 2012, 52, 234–258. [Google Scholar] [CrossRef]
- Newcomb, B.A. Processing, structure, and properties of carbon fibers. Compos. Part A Appl. Sci. Manuf. 2016, 91, 262–282. [Google Scholar] [CrossRef]
- Sidorina, A.I.; Gunyaeva, A.G. Market for Carbon Fibres and Composites Based on them. A review. Fibre Chem. 2017, 48, 306–310. [Google Scholar] [CrossRef]
- Tanaka, F.; Okabe, T. Historical Review of Processing, Microstructures, and Mechanical Properties of PAN-Based Carbon Fibers. Ref. Modul. Mater. Sci. Mater. Eng. 2018, 66–85. [Google Scholar] [CrossRef]
- Xu, Z.; Gao, C. Graphene fiber: A new trend in carbon fibers. Mater. Today 2015, 18, 480–492. [Google Scholar] [CrossRef]
- Nunna, S.; Naebe, M.; Hameed, N.; Fox, B.L.; Creighton, C. Evolution of radial heterogeneity in polyacrylonitrile fibres during thermal stabilization: An overview. Polym. Degrad. Stab. 2017, 136, 20–30. [Google Scholar] [CrossRef]
- Huang, X. Fabrication and Properties of Carbon Fibers. Materials 2009, 2, 2369–2403. [Google Scholar] [CrossRef]
- Fraczek-Szczypta, A.; Bogun, M.; Blazewicz, S. Carbon fibers modified with carbon nanotubes. J. Mater. Sci. 2009, 44, 4721–4727. [Google Scholar] [CrossRef]
- Jo, A.Y.; Yoo, S.H.; Chung, Y.-S.; Lee, S. Effects of ultraviolet irradiation on stabilization of textile-grade polyacrylonitrile fibers without photo-initiator for preparing carbon fibers. Carbon 2019, 144, 440–448. [Google Scholar] [CrossRef]
- Musiol, P.; Szatkowski, P.; Gubernat, M.; Weselucha-Birczynska, A.; Blazewicz, S. Comparative study of the structure and microstructure of PAN-based nano- and micro-carbon fibers. Ceram. Int. 2016, 42, 11603–11610. [Google Scholar] [CrossRef]
- Santhana Krishnan, G.; Thomas, P.; Naveen, S.; Murali, N. Molecular and thermal studies of carbon fiber precursor polymers with low thermal-oxidative stabilization characteristics. J. Appl. Polym. Sci. 2018, 135, 46381. [Google Scholar] [CrossRef]
- Deurbergue, A.; Oberlin, A. Stabilization and carbonization of pan-based carbon fibers as related to mechanical properties. Carbon 1991, 29, 621–628. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, J.S. Preparation of carbon fibers from linear low density polyethylene. Carbon 2015, 94, 524–530. [Google Scholar] [CrossRef]
- De Palmenaer, A.; Wortberg, G.; Drissen, F.; Seide, G.; Gries, T. Production of Polyethylene Based Carbon Fibres. Chem. Eng. Trans. 2015, 43, 1699–1704. [Google Scholar] [CrossRef]
- Meinl, J.; Schönfeld, K.; Kirsten, M.; Kittler, K.; Michaelis, A.; Cherif, C. Optimization of the temperature program to scale up the stabilization of polyacrylonitrile fibers. Compos. Part A Appl. Sci. Manuf. 2017, 96, 37–45. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Lee, S.; Park, S.; Jo, S.M.; Lee, H.-S.; Joh, H.-I. Continuous and rapid stabilization of polyacrylonitrile fiber bundles assisted by atmospheric pressure plasma for fabricating large-tow carbon fibers. Carbon 2015, 94, 412–416. [Google Scholar] [CrossRef]
- Henrici-Olivé, G.; Olivé, S. The chemistry of carbon fiber formation from polyacrylonitrile. Ind. Dev. 1983, 51, 1–60. [Google Scholar] [CrossRef]
- Peng, G.-Q.; Zhang, X.-H.; Wen, Y.-F.; Yang, Y.-G.; Liu, L. Effect of Coagulation Bath DMSO Concentration on the Structure and Properties of Polyacrylonitrile (PAN) Nascent Fibers during Wet-Spinning. J. Macromol. Sci. Part B 2008, 47, 1130–1141. [Google Scholar] [CrossRef]
- Hao, J.; Liu, Y.; Lu, C. Effect of acrylonitrile sequence distribution on the thermal stabilization reactions and carbon yields of poly(acrylonitrile-co-methyl acrylate). Polym. Degrad. Stab. 2018, 147, 89–96. [Google Scholar] [CrossRef]
- Choi, D.; Kil, H.-S.; Lee, S. Fabrication of low-cost carbon fibers using economical precursors and advanced processing technologies. Carbon 2019, 142, 610–649. [Google Scholar] [CrossRef]
- Cho, D.W.; Ghorpade, R.V.; Hong, S.C. Identifying the role of the acidic comonomer in poly(acrylonitrile- co -itaconic acid) during stabilization process through low temperature electron beam irradiation. Polym. Degrad. Stab. 2018, 153, 220–226. [Google Scholar] [CrossRef]
- Cai, J.Y.; McDonnell, J.; Brackley, C.; O’Brien, L.; Church, J.S.; Millington, K.; Smith, S.; Phair-Sorensen, N. Polyacrylonitrile-based precursors and carbon fibers derived from advanced RAFT technology and conventional methods–The 1st comparative study. Mater. Today Commun. 2016, 9, 22–29. [Google Scholar] [CrossRef]
- Arbab, S.; Zeinolebadi, A. Quantitative analysis of the effects of comonomers and heating conditions on the stabilization reactions of polyacrylonitrile fibers as carbon fiber precursors. Polym. Degrad. Stab. 2017, 139, 107–116. [Google Scholar] [CrossRef]
- Bashir, Z. A critical review of the stabilisation of polyacrylonitrile. Carbon 1991, 29, 1081–1090. [Google Scholar] [CrossRef]
- Guo, X.; Cheng, Y.; Fan, Z.; Feng, Z.; He, L.; Liu, R.; Xu, J. New insights into orientation distribution of high strength polyacrylonitrile-based carbon fibers with skin-core structure. Carbon 2016, 109, 444–452. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, S.; Shen, Z.; Xu, L.; Zhang, L.; Peng, J. Study on the oxidative stabilization of polyacrylonitrile fibers by microwave heating. Polym. Degrad. Stab. 2018, 150, 86–91. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Guo, S.; Xiao, S.; Shen, Z.; Xu, L. Comparison of microwave and conventional heating methods for oxidative stabilization of polyacrylonitrile fibers at different holding time and heating rate. Ceram. Int. 2018, 44, 14377–14385. [Google Scholar] [CrossRef]
- Morris, E.A.; Weisenberger, M.C.; Abdallah, M.G.; Vautard, F.; Grappe, H.; Ozcan, S.; Paulauskas, F.L.; Eberle, C.; Jackson, D.; Mecham, S.J.; et al. High performance carbon fibers from very high molecular weight polyacrylonitrile precursors. Carbon 2016, 101, 245–252. [Google Scholar] [CrossRef]
- Nunna, S.; Naebe, M.; Hameed, N.; Creighton, C.; Naghashian, S.; Jennings, M.J.; Atkiss, S.; Setty, M.; Fox, B.L. Investigation of progress of reactions and evolution of radial heterogeneity in the initial stage of thermal stabilization of PAN precursor fibres. Polym. Degrad. Stab. 2016, 125, 105–114. [Google Scholar] [CrossRef]
- Nunna, S.; Creighton, C.; Hameed, N.; Naebe, M.; Henderson, L.C.; Setty, M.; Fox, B.L. Radial structure and property relationship in the thermal stabilization of PAN precursor fibres. Polym. Test. 2017, 59, 203–211. [Google Scholar] [CrossRef]
- Jain, M.J.; Abhiraman, A.S. Conversion of acrylonitrile-based precursors to carbon fibres. Part 2. Precursor morphology and thermooxidative stabilization. Composites 1987, 18, 263. [Google Scholar] [CrossRef]
- Gupta, A.K.; Paliwal, D.K.; Bajaj, P. Acrylic Precursors for Carbon Fibers. Polym. Rev. 1991, 31, 1–89. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, M.; Zhang, W.; Liu, W.; Yang, C.; Shen, R.; Wu, G. Significantly reduced pre-oxidation period of PAN fibers by continuous electron beam irradiation: Optimization by monitoring radical variation. Polym. Degrad. Stab. 2018, 158, 72–82. [Google Scholar] [CrossRef]
- Chung, D. Carbon Fiber Composites, 1st ed.; Butterworth-Heinmann: Boston, MA, USA, 2012. [Google Scholar]
- Liu, J.; Wang, P.H.; Li, R.Y. Continuous carbonization of polyacrylonitrile-based oxidized fibers: Aspects on mechanical properties and morphological structure. J. Appl. Polym. Sci. 1994, 52, 945–950. [Google Scholar] [CrossRef]
- Park, S.; Kil, H.-S.; Choi, D.; Song, S.-K.; Lee, S. Rapid stabilization of polyacrylonitrile fibers achieved by plasma-assisted thermal treatment on electron-beam irradiated fibers. J. Ind. Eng. Chem. 2019, 69, 449–454. [Google Scholar] [CrossRef]
- Newcomb, B.A.; Giannuzzi, L.A.; Lyons, K.M.; Gulgunje, P.V.; Gupta, K.; Liu, Y.; Kamath, M.; McDonald, K.; Moon, J.; Feng, B.; et al. High resolution transmission electron microscopy study on polyacrylonitrile/carbon nanotube based carbon fibers and the effect of structure development on the thermal and electrical conductivities. Carbon 2015, 93, 502–514. [Google Scholar] [CrossRef]
- Jäger, H.; Cherif, C.; Kirsten, M.; Behnisch, T.; Wolz, D.S.; Böhm, R.; Gude, M. Influence of processing parameters on the properties of carbon fibres-An overview. Mater. Und Werkst. 2016, 47, 1044–1057. [Google Scholar] [CrossRef]
- Chand, S. Review Carbon Fibers for Composites. J. Mater. Sci. 2000, 35, 1303–1313. [Google Scholar] [CrossRef]
- Xue, T.J.; McKinney, M.A.; Wilkie, C.A. The thermal degradation of polyacrylonitrile. Polym. Degrad. Stab. 1997, 58, 193–202. [Google Scholar] [CrossRef]
- Vasishtha, R.; Srivastava, A.K. Polymerization of methacrylic acid and acrylonitrile by p-nitrobenzyl triphenyl phosphonium ylide. Polym. Eng. Sci. 1991, 31, 567–570. [Google Scholar] [CrossRef]
- Acrylonitrile Polymers. In Van Nostrand’s Scientific Encyclopedia; Considine, G.D., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Nuyken, O.; Swift, G. Handbook of Polymer Synthesis, 2nd ed.; Marcel Dekker: New York, NY, USA, 2005. [Google Scholar]
- Ge, H.; Liu, H.; Chen, J.; Wang, C. The skin-core structure of poly(acrylonitrile-itaconic acid) precursor fibers in wet-spinning. J. Appl. Polym. Sci. 2008, 108, 947–952. [Google Scholar] [CrossRef]
- Yu, M.; Wang, C.; Bai, Y.; Zhu, B.; Ji, M.; Xu, Y. Microstructural evolution in polyacrylonitrile fibers during oxidative stabilization. J. Polym. Sci. Part B Polym. Phys. 2008, 46, 759–765. [Google Scholar] [CrossRef]
- Olabisi, O.; Adewale, K. Handbook of Thermoplastics, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Fu, Z.; Liu, B.; Deng, Y.; Ma, J.; Cao, C.; Wang, J.; Ao, Y.; Zhang, H. The suitable itaconic acid content in polyacrylonitrile copolymers used for PAN-based carbon fibers. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Jiang, J.; Lu, X.; Lu, Y. Preparation of polyacrylonitrile with improved isotacticity and low polydispersity. J. Appl. Polym. Sci. 2010. [Google Scholar] [CrossRef]
- Soulis, S.; Anagnou, S.; Milioni, E.; Mpalias, C.; Kartsonakis, I.A.; Kanellopoulou, I.; Markakis, V.; Koumoulos, E.P.; Kontou, E.; Charitidis, C.A. Strategies towards Novel Carbon Fiber Precursors: The Research Results on the Synthesis of PAN Copolymers via AGET ATRP and on Lignin as a Precursor. NanoWorld J. 2015, 1. [Google Scholar] [CrossRef]
- Niu, T.; Jiang, J.; Li, S.; Ni, B.; Liu, X.; Chen, M. Well-Defined High-Molecular-Weight Polyacrylonitrile Formation via Visible-Light-Induced Metal-Free Radical Polymerization. Macromol. Chem. Phys. 2017, 218, 1700169. [Google Scholar] [CrossRef]
- Hao, J.; Li, W.; Suo, X.; Wei, H.; Lu, C.; Liu, Y. Highly isotactic (>60%) polyacrylonitrile-based carbon fiber: Precursor synthesis, fiber spinning, stabilization and carbonization. Polymer 2018, 157, 139–150. [Google Scholar] [CrossRef]
- Krishnan, G.S.; Burkanudeen, A.; Murali, N.; Phadnis, H. Eco-friendly synthesis of carbon fiber precursor polymers and their molecular characteristics. Green Chem. 2012, 14, 1778. [Google Scholar] [CrossRef]
- Ghorpade, R.V.; Cho, D.W.; Hong, S.C. Effect of controlled tacticity of polyacrylonitrile (co)polymers on their thermal oxidative stabilization behaviors and the properties of resulting carbon films. Carbon 2017, 121, 502–511. [Google Scholar] [CrossRef]
- Morris, E.A.; Weisenberger, M.C.; Bradley, S.B.; Abdallah, M.G.; Mecham, S.J.; Pisipati, P.; McGrath, J.E. Synthesis, spinning, and properties of very high molecular weight poly(acrylonitrile- co -methyl acrylate) for high performance precursors for carbon fiber. Polymer 2014, 55, 6471–6482. [Google Scholar] [CrossRef]
- Hatada, K.; Kitayama, T. NMR Spectroscopy of Polymers; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Katsuraya, K.; Hatanaka, K.; Matsuzaki, K.; Minagawa, M. Assignment of finely resolved 13 C NMR spectra of polyacrylonitrile. Polymer 2001, 42, 6323–6326. [Google Scholar] [CrossRef]
- Santhana Krishnan, G.; Burkanudeen, A.; Murali, N.; Phadnis, H. Facile synthesis of stereoregular carbon fiber precursor polymers by template assisted solid phase polymerization. Express Polym. Lett. 2012, 6, 729–738. [Google Scholar] [CrossRef]
- Rwei, S.-P.; Way, T.-F.; Chiang, W.-Y.; Pan, S.-Y. Effect of tacticity on the cyclization of polyacrylonitrile copolymers. Colloid Polym. Sci. 2017, 295, 803–815. [Google Scholar] [CrossRef]
- Sawai, D.; Kanamoto, T.; Yamazaki, H.; Hisatani, K. Dynamic Mechanical Relaxations in Poly(acrylonitrile) with Different Stereoregularities. Macromolecules 2004, 37, 2839–2846. [Google Scholar] [CrossRef]
- Banaie, K.A.; Mirjalili, M.; Eslami-Farsani, R. The study of the correlation between ascending and descending rates of the imposed specific stress and transposition of chemical reactions during PAN precursor fibers stabilization. Polym. Degrad. Stab. 2018, 156, 234–244. [Google Scholar] [CrossRef]
- Spörl, J.M.; Ota, A.; Beyer, R.; Lehr, T.; Müller, A.; Hermanutz, F.; Buchmeiser, M.R. Carbon fibers prepared from tailored reversible-addition-fragmentation transfer copolymerization-derived poly(acrylonitrile)-co-poly(methylmethacrylate). J. Polym. Sci. Part A Polym. Chem. 2014, 52, 1322–1333. [Google Scholar] [CrossRef]
- Hou, C.; Qu, R.; Qun, W.; Ying, L.; Wang, C. Degradation of acrylonitrile-ammonium itaconate copolymers. J. Appl. Polym. Sci. 2005, 98, 1708–1711. [Google Scholar] [CrossRef]
- Wang, Y.X.; Liu, Y.L.; Wang, G.L.; Wang, L.M.; Wang, C.G. Aqueous deposited copolymerization of acrylonitrile with ammonium itaconate under magnetic field. J. Polym. Res. 2011, 18, 1323–1329. [Google Scholar] [CrossRef]
- Krishnan, G.S. Pyrolysis and Thermal Stability of Carbon Fiber Polymer Precursors with Different Microstructures. Polym. Precursor-Deriv. Carbon 2014, 1173, 169–187. [Google Scholar] [CrossRef]
- Kaji, H.; Schmidt-Rohr, K. Conformation and Dynamics of Atactic Poly(acrylonitrile). 3. Characterization of Local Structure by Two-Dimensional2H−13C Solid-State NMR. Macromolecules 2001, 34, 7382–7391. [Google Scholar] [CrossRef]
- Kaji, H.; Schmidt-Rohr, K. Conformation and Dynamics of Atactic Poly(acrylonitrile). 2. Torsion Angle Distributions in Meso Dyads from Two-Dimensional Solid-State Double-Quantum13C NMR. Macromolecules 2001, 34, 7368–7381. [Google Scholar] [CrossRef]
- Coleman, M.M.; Sivy, G.T.; Painter, P.C.; Snyder, R.W.; Gordon, B. Studies of the degradation of acrylonitrile/acrylamide copolymers as a function of composition and temperature. Carbon 1983, 21, 255–267. [Google Scholar] [CrossRef]
- Soulis, S.; Simitzis, J. Thermomechanical behaviour of poly[acrylonitrile-co-(methyl acrylate)] fibres oxidatively treated at temperatures up to 180 °C. Polym. Int. 2005, 54, 1474–1483. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, L.; Wang, M.; Pang, W.; Ge, X. Structural Identification of Polyacrylonitrile during Thermal Treatment by Selective 13C Labeling and Solid-State 13C NMR Spectroscopy. Macromolecules 2014, 47, 3901–3908. [Google Scholar] [CrossRef]
- Liu, X.; Makita, Y.; Hong, Y.-L.; Nishiyama, Y.; Miyoshi, T. Chemical Reactions and Their Kinetics of atactic-Polyacrylonitrile As Revealed by Solid-State 13C NMR. Macromolecules 2016, 50, 244–253. [Google Scholar] [CrossRef]
- Qin, X.; Lu, Y.; Xiao, H.; Hao, Y.; Pan, D. Improving preferred orientation and mechanical properties of PAN-based carbon fibers by pretreating precursor fibers in nitrogen. Carbon 2011, 49, 4598–4600. [Google Scholar] [CrossRef]
- Lei, S.; Wu, S.; Gao, A.; Cao, W.; Li, C.; Xu, L. The formation of conjugated structure and its transformation to pseudo-graphite structure during thermal treatment of polyacrylonitrile. High Perform. Polym. 2016, 29, 1097–1109. [Google Scholar] [CrossRef]
- Watt, W.; Johnson, W. The Effect of Length Changes during the Oxidation of Polyacrylonitrile Fibers on the Young’s Modulus of Carbon Fibers. Appl. Polym. Symp. 1969, 9, 215–227. [Google Scholar]
- Bahl, O.P.; Mathur, R.B. Effect of load on the mechanical properties of carbon fibres from pan precursor. Fibre Sci. Technol. 1979, 12, 31–39. [Google Scholar] [CrossRef]
- Sabet, E.N.; Nourpanah, P.; Arbab, S. A Novel Method for Investigation of Entropic Stress in Prestabilization of PAN-Based Precursor Fibers. Adv. Polym. Technol. 2017, 36, 424–432. [Google Scholar] [CrossRef]
- Fitzer, E.; Frohs, W.; Heine, M. Optimization of stabilization and carbonization treatment of PAN fibres and structural characterization of the resulting carbon fibres. Carbon 1986, 24, 387–395. [Google Scholar] [CrossRef]
- Kalashnik, A.; Panichkina, O.N.; Rudinskaya, G.Y.; Serkov, A.T. Shrinkage Mechanisms in Thermooxidative Stabilization of Acrylic Fibres. Fibre Chem. 2001, 33, 132–139. [Google Scholar] [CrossRef]
- Simitzis, J.; Soulis, S. Correlation of chemical shrinkage of polyacrylonitrile fibres with kinetics of cyclization. Polym. Int. 2008, 57, 99–105. [Google Scholar] [CrossRef]
- Yongping, H.; Tongqing, S.; Haojing, W.; Dong, W. Effect of Heating Rate on the Chemical Reaction during Stabilization of Polyacrylonitrile Fibers. Text. Res. J. 2008, 78, 806–811. [Google Scholar] [CrossRef]
- Hou, Y.; Sun, T.; Wang, H.; Wu, D. A new method for the kinetic study of cyclization reaction during stabilization of polyacrylonitrile fibers. J. Mater. Sci. 2008, 43, 4910–4914. [Google Scholar] [CrossRef]
- Hou, Y.; Sun, T.; Wang, H.; Wu, D. Thermal-shrinkage investigation of the chemical reaction during the stabilization of polyacrylonitrile fibers. J. Appl. Polym. Sci. 2009, 114, 3668–3672. [Google Scholar] [CrossRef]
- Soulis, S.; Dragatogiannis, D.A.; Konstantopoulos, G.; Charitidis, C. Application of PAN fiber length change as the oxidative stabilization process control parameter. Mater. Today Proc. 2018, 5, 27645–27652. [Google Scholar] [CrossRef]
- Sabet, E.N.; Nourpanah, P.; Arbab, S. Quantitative analysis of entropic stress effect on the structural rearrangement during pre-stabilization of PAN precursor fibers. Polymer 2016, 90, 138–146. [Google Scholar] [CrossRef]
- Fazlitdinova, A.G.; Tyumentsev, V.A.; Podkopayev, S.A.; Shveikin, G.P. Changes of polyacrylonitrile fiber fine structure during thermal stabilization. J. Mater. Sci. 2010, 45, 3998–4005. [Google Scholar] [CrossRef]
- Bashir, Z. The Hexagonal Mesophase in Atactic Polyacrylonitrile: A New Interpretation of the Phase Transitions in the Polymer. J. Macromol. Sci. Part B 2001, 40, 41–67. [Google Scholar] [CrossRef]
- Suresh, K.I.; Thomas, K.S.; Rao, B.S.; Nair, C.P.R. Viscoelastic properties of polyacrylonitrile terpolymers during thermo-oxidative stabilization (cyclization). Polym. Adv. Technol. 2008, 19, 831–837. [Google Scholar] [CrossRef]
- Sperling, L.H. Introduction to Physical Polymer Science, 4th ed.; Wiley-Interscience: New York, NY, USA, 2005. [Google Scholar]
- Bashir, Z.; Rastogi, S. The Explanation of the Increase in Slope at the Tg in the Plot of d-Spacing Versus Temperature in Polyacrylonitrile. J. Macromol. Sci. Part B 2004, 44, 55–78. [Google Scholar] [CrossRef]
- Bai, Y.-J.; Wang, C.-G.; Lun, N.; Wang, Y.-X.; Yu, M.-J.; Zhu, B. HRTEM microstructures of PAN precursor fibers. Carbon 2006, 44, 1773–1778. [Google Scholar] [CrossRef]
- He, D.-X.; Wang, C.-G.; Bai, Y.-J.; Lun, N.; Zhu, B.; Wang, Y.-X. Microstructural evolution during thermal stabilization of PAN fibers. J. Mater. Sci. 2007, 42, 7402–7407. [Google Scholar] [CrossRef]
- Nguyen-Thai, N.U.; Hong, S.C. Controlled architectures of poly(acrylonitrile-co-itaconic acid) for efficient structural transformation into carbon materials. Carbon 2014, 69, 571–581. [Google Scholar] [CrossRef]
- Liu, Y.; Choi, Y.H.; Chae, H.G.; Gulgunje, P.; Kumar, S. Temperature dependent tensile behavior of gel-spun polyacrylonitrile and polyacrylonitrile/carbon nanotube composite fibers. Polymer 2013, 54, 4003–4009. [Google Scholar] [CrossRef]
- Ogawa, H.; Saito, K. Oxidation behavior of polyacrylonitrile fibers evaluated by new stabilization index. Carbon 1995, 33, 783–788. [Google Scholar] [CrossRef]
- Qian, X.; Zhi, J.; Chen, L.; Zhong, J.; Wang, X.; Zhang, Y.; Song, S. Evolution of microstructure and electrical property in the conversion of high strength carbon fiber to high modulus and ultrahigh modulus carbon fiber. Compos. Part A Appl. Sci. Manuf. 2018, 112, 111–118. [Google Scholar] [CrossRef]
- Fochler, H.S.; Mooney, J.R.; Ball, L.E.; Boyer, R.D.; Grasselli, J.G. Infrared and NMR spectroscopic studies of the thermal degradation of polyacrylonitrile. Spectrochim. Acta Part A Mol. Spectrosc. 1985, 41, 271–278. [Google Scholar] [CrossRef]
- Usami, T.; Itoh, T.; Ohtani, H.; Tsuge, S. Structural study of polyacrylonitrile fibers during oxidative thermal degradation by pyrolysis-gas chromatography, solid-state carbon-13 NMR, and Fourier-transform infrared spectroscopy. Macromolecules 1990, 23, 2460–2465. [Google Scholar] [CrossRef]
- Bajaj, P.; Sreekumar, T.V.; Sen, K. Thermal behaviour of acrylonitrile copolymers having methacrylic and itaconic acid comonomers. Polymer 2001, 42, 1707–1718. [Google Scholar] [CrossRef]
- Zhu, Y.; Wilding, M.A.; Mukhopadhyay, S.K. Estimation, using infrared spectroscopy, of the cyclization of poly(acrylonitrile) during the stabilization stage of carbon fibre production. J. Mater. Sci. 1996, 31, 3831–3837. [Google Scholar] [CrossRef]
- Krishnan, G.S.; Thomas, P.; Murali, N. Synthesis, characterization, and thermo-mechanical properties of poly(acrylonitrile-co-2,3-dimethyl-1,3-butadiene-co-itaconic acid) as carbon fibre polymer precursors. RSC Adv. 2016, 6, 6182–6190. [Google Scholar] [CrossRef]
- Tsai, J.-S. Comparison of batch and continuous oxidation processes for producing carbon fibre based on PAN fibre. J. Mater. Sci. Lett. 1997, 16, 361–362. [Google Scholar] [CrossRef]
- Tsai, J.-S. Orientation change for polyacrylonitrile precursor during oxidation. J. Mater. Sci. Lett. 1994, 13, 1162–1163. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.K.; Zhu, Y. Structure-Property Relationships of PAN Precursor Fibers During Thermo-oxidative Stabilization. Text. Res. J. 2016, 65, 25–31. [Google Scholar] [CrossRef]
- Gupta, A.K.; Paliwal, D.K.; Bajaj, P. Effect of the nature and mole fraction of acidic comonomer on the stabilization of polyacrylonitrile. J. Appl. Polym. Sci. 1996, 59, 1819–1826. [Google Scholar] [CrossRef]
- Gupta, A.K.; Paliwal, D.K.; Bajaj, P. Effect of an acidic comonomer on thermooxidative stabilization of polyacrylonitrile. J. Appl. Polym. Sci. 1995, 58, 1161–1174. [Google Scholar] [CrossRef]
- Tsai, J.-S.; Lin, C.-H. Effect of comonomer composition on the properties of polyacrylonitrile precursor and resulting carbon fiber. J. Appl. Polym. Sci. 1991, 43, 679–685. [Google Scholar] [CrossRef]
- Bhat, G.S.; Peebles, L.H.; Abhiraman, A.S.; Cook, F.L. Rapid stabilization of acrylic fibers using ammonia: Effect on structure and morphology. J. Appl. Polym. Sci. 1993, 49, 2207–2219. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Zhou, T.; Liu, X.; Yuan, Q.; Zhang, A. New understanding on the reaction pathways of the polyacrylonitrile copolymer fiber pre-oxidation: Online tracking by two-dimensional correlation FTIR spectroscopy. RSC Adv. 2016, 6, 4397–4409. [Google Scholar] [CrossRef]
- Fu, Z.; Ma, J.; Deng, Y.; Wu, G.; Cao, C.; Zhang, H. Structural evolution of poly(acrylonitrile-co-dimethyl itaconate) copolymer during thermal oxidative stabilization. Polym. Adv. Technol. 2015, 26, 322–329. [Google Scholar] [CrossRef]
- Rahaman, M.S.A.; Ismail, A.F.; Mustafa, A. A review of heat treatment on polyacrylonitrile fiber. Polym. Degrad. Stab. 2007, 92, 1421–1432. [Google Scholar] [CrossRef]
- Arbab, S.; Zeinolebadi, A. A procedure for precise determination of thermal stabilization reactions in carbon fiber precursors. Polym. Degrad. Stab. 2013, 98, 2537–2545. [Google Scholar] [CrossRef]
- 115. Farsan, R.E.; Raissi, S.; Shokuhfar, A.; Sedghi, A. FT-IR Study of Stabilized PAN Fibers for Fabrication of Carbon Fibers. World Acad. Sci. Eng. Technol. 2009, 50, 430–433. [Google Scholar]
- Hameed, N.; Sharp, J.; Nunna, S.; Creighton, C.; Magniez, K.; Jyotishkumar, P.; Salim, N.V.; Fox, B. Structural transformation of polyacrylonitrile fibers during stabilization and low temperature carbonization. Polym. Degrad. Stab. 2016, 128, 39–45. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies Tables and Charts, 3rd ed.; John Wiley & Sons, LTD: Middlesex, UK, 2001. [Google Scholar] [CrossRef]
- Gulgunje, P.V.; Newcomb, B.A.; Gupta, K.; Chae, H.G.; Tsotsis, T.K.; Kumar, S. Low-density and high-modulus carbon fibers from polyacrylonitrile with honeycomb structure. Carbon 2015, 95, 710–714. [Google Scholar] [CrossRef]
- Mathur, R.B.; Bahl, O.P.; Mittal, J. Advances in the development of high-performance carbon fibres from pan precursor. Compos. Sci. Technol. 1994, 51, 223–230. [Google Scholar] [CrossRef]
- Chae, H.G.; Newcomb, B.A.; Gulgunje, P.V.; Liu, Y.; Gupta, K.K.; Kamath, M.G.; Lyons, K.M.; Ghoshal, S.; Pramanik, C.; Giannuzzi, L.; et al. High strength and high modulus carbon fibers. Carbon 2015, 93, 81–87. [Google Scholar] [CrossRef]
- Chai, X.; Mi, H.; Zhu, C.; He, C.; Xu, J.; Zhou, X.; Liu, J. Low-temperature thermal stabilization of polyacrylontrile-based precursor fibers towards efficient preparation of carbon fibers with improved mechanical properties. Polymer 2015, 76, 131–139. [Google Scholar] [CrossRef]
- Shin, H.K.; Park, M.; Kang, P.H.; Choi, H.-S.; Park, S.-J. Preparation and characterization of polyacrylonitrile-based carbon fibers produced by electron beam irradiation pretreatment. J. Ind. Eng. Chem. 2014, 20, 3789–3792. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, S.-Y.; Choi, J.; Lee, S.; Jo, S.M.; Joo, J.; Lee, H.-S. Two step microwave plasma carbonization including low plasma power pre-carbonization for polyacrylonitrile based carbon fiber. Polymer 2015, 69, 123–128. [Google Scholar] [CrossRef]
- Sung, M.-G.; Kawabata, Y. Strengthening of carbon fibers by a magnetic field imposed in the stabilization and carbonization process. Mater. Sci. Eng. A 2008, 488, 247–251. [Google Scholar] [CrossRef]
- Xin, G.; Yao, T.; Sun, H.; Scott, S.M.; Shao, D.; Wang, G.; Lian, J. Highly thermally conductive and mechanically strong graphene fibers. Science 2015, 349, 1083–1087. [Google Scholar] [CrossRef]
- Raghubanshi, H.; Dikio, E.D.; Naidoo, E.B. The properties and applications of helical carbon fibers and related materials: A review. J. Ind. Eng. Chem. 2016, 44, 23–42. [Google Scholar] [CrossRef]
- Wu, G.-P.; Lu, C.-X.; Ling, L.-C.; Lu, Y.-G. Comparative investigation on the thermal degradation and stabilization of carbon fiber precursors. Polym. Bull. 2009, 62, 667–678. [Google Scholar] [CrossRef]
- Xue, Y.; Liu, J.; Lian, F.; Liang, J. Effect of the oxygen-induced modification of polyacrylonitrile fibers during thermal-oxidative stabilization on the radial microcrystalline structure of the resulting carbon fibers. Polym. Degrad. Stab. 2013, 98, 2259–2267. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, S.Y.; Lee, S.; Jo, S.; Im, Y.H.; Lee, H.S. Microwave plasma carbonization for the fabrication of polyacrylonitrile-based carbon fiber. Polymer 2015, 56, 590–595. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Guo, S.; Xu, L.; Xiao, S.; Shen, Z. Microwave treatment of pre-oxidized fibers for improving their structure and mechanical properties. Ceram. Int. 2019, 45, 1379–1384. [Google Scholar] [CrossRef]
- Korte, S. Physical Constants of Poly(acrylonitrile), 4th ed.; Brandrup, J., Immergut, E.H., Grulke, E.A., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 1999. [Google Scholar]
- Ji, B.H. Effect of Coagulation Bath Concentration on the Structure and Properties of Polyacrylonitrile as-Spun Fibers during Wet-Spinning. Adv. Mater. Res. 2011, 287, 1832–1836. [Google Scholar] [CrossRef]
- Ouyang, Q.; Chen, Y.-S.; Zhang, N.; Mo, G.-M.; Li, D.-H.; Yan, Q. Effect of Jet Swell and Jet Stretch on the Structure of Wet-Spun Polyacrylonitrile Fiber. J. Macromol. Sci. Part B 2011, 50, 2417–2427. [Google Scholar] [CrossRef]
- Ji, B.-H.; Wang, C.-G.; Wang, Y.-X. Effect of jet stretch on polyacrylonitrile as-spun fiber formation. J. Appl. Polym. Sci. 2007, 103, 3348–3352. [Google Scholar] [CrossRef]
- Minagawa, M.; Yamada, H.; Yamaguchi, K.; Yoshii, F. Gamma-ray irradiation canal polymerization conditions ensuring highly stereoregular (>80%) polyacrylonitrile. Macromolecules 1992, 25, 503–510. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PAN Precursor | Triad Probability Distribution | Propagation * | Remarks | |||
|---|---|---|---|---|---|---|
| mm (Isotactic) | mr (Atactic) | rr (Syndiotactic) | Pm | S ** | ||
| P(AN@IA@MA) | 0.27 | 0.54 | 0.19 | 0.54 | ND *** | Feed composition: 93% AN, 4% MA, 3% IA Solution-precipitation technique/Redox initiator (K2S2O8 and Na2S2O5) Mw: 140,000 g∙mol−1 |
| Aq-PAN | 0.34 | 0.51 | 0.15 | 0.60 | ND | PAN homopolymer Solution-precipitation technique/K2S2O8 and Na2S2O5 |
| Com-PAN | 0.28 | 0.45 | 0.22 | 0.47 | 0.11 | commercial: textile-grade |
| P(AN@IA) | 0.28 | 0.47 | 0.25 | 0.47 | 0.08 | Feed composition 98.5% AN, 1.5% IA Aqueous slurry technique/Free radical initiator (AIBN) Mw: 18,200 g∙mol−1 [61] |
| 13C PAN | 0.34 | 0.38 | 0.28 | 0.44 | 0.17 | 13C- enriched PAN homopolymer Solution/precipitation technique/K2S2O8 and Na2S2O5 Mw: ~450,000 g∙mol−1 [41] |
| Tol-PAN | 0.26 | 0.50 | 0.23 | 0.51 | 0.01 | PAN homopolymer Solution technique/AIBN [60] |
| ATRP-PAN | 0.23 | 0.46 | 0.31 | 0.43 | 0.06 | PAN homopolymer ATR polymerization: AlCl3/AN molar ratio: 0.01 [52] |
| Iso-PAN | 0.37 | 0.48 | 0.15 | 0.54 | 0.12 | PAN homopolymer Stereospecific polymerization (urea-canal complex, ‒78 °C, isotactic-rich) [28,60] |
| At-PAN Iso-PAN-1 Iso-PAN-2 Iso-PAN-3 | 0.25 0.48 0.58 0.68 | 0.51 0.36 0.29 0.22 | 0.24 0.16 0.13 0.10 | 0.51 0.69 0.74 0.79 | 0.01 0.08 0.10 0.10 | Suspension polymerization Polymerization with organometallic compounds Polymerization with organometallic compounds γ-irradiation (urea-canal complex) [63] |
| PAC/01 | 0.48 | 0.37 | 0.15 | 0.52 | 0.24 | Feed composition: 98.5% AN, 1.5% IA Template assisted (NiCl2, MgCl2) solid phase polymerization/AIBN Mw: 25,700 g∙mol−1 [61] |
| PAC/03 | 0.51 | 0.34 | 0.15 | 0.51 | 0.26 | Feed composition: 98.5% AN, 1.5% acrylamide Template assisted (NiCl2, MgCl2) solid phase polymerization/AIBN Mw: 17,300 g∙mol−1 [61] |
| P(AN@MA) | 0.45 | 0.42 | 0.13 | 0.54 | 0.21 | Feed composition: 91% AN, 9% MA Template assisted (MgCl2) solid phase polymerization/AIBN Mw: 37,900 g∙mol−1 [62] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soulis, S.; Konstantopoulos, G.; Koumoulos, E.P.; Charitidis, C.A. Impact of Alternative Stabilization Strategies for the Production of PAN-Based Carbon Fibers with High Performance. Fibers 2020, 8, 33. https://doi.org/10.3390/fib8060033
Soulis S, Konstantopoulos G, Koumoulos EP, Charitidis CA. Impact of Alternative Stabilization Strategies for the Production of PAN-Based Carbon Fibers with High Performance. Fibers. 2020; 8(6):33. https://doi.org/10.3390/fib8060033
Chicago/Turabian StyleSoulis, Spyridon, George Konstantopoulos, Elias P. Koumoulos, and Costas A. Charitidis. 2020. "Impact of Alternative Stabilization Strategies for the Production of PAN-Based Carbon Fibers with High Performance" Fibers 8, no. 6: 33. https://doi.org/10.3390/fib8060033
APA StyleSoulis, S., Konstantopoulos, G., Koumoulos, E. P., & Charitidis, C. A. (2020). Impact of Alternative Stabilization Strategies for the Production of PAN-Based Carbon Fibers with High Performance. Fibers, 8(6), 33. https://doi.org/10.3390/fib8060033