Improve in CO2 and CH4 Adsorption Capacity on Carbon Microfibers Synthesized by Electrospinning of PAN

 ,
,  , and
, and
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. CMF Preparation
2.3. Characterization Techniques
3. Results and Discussion
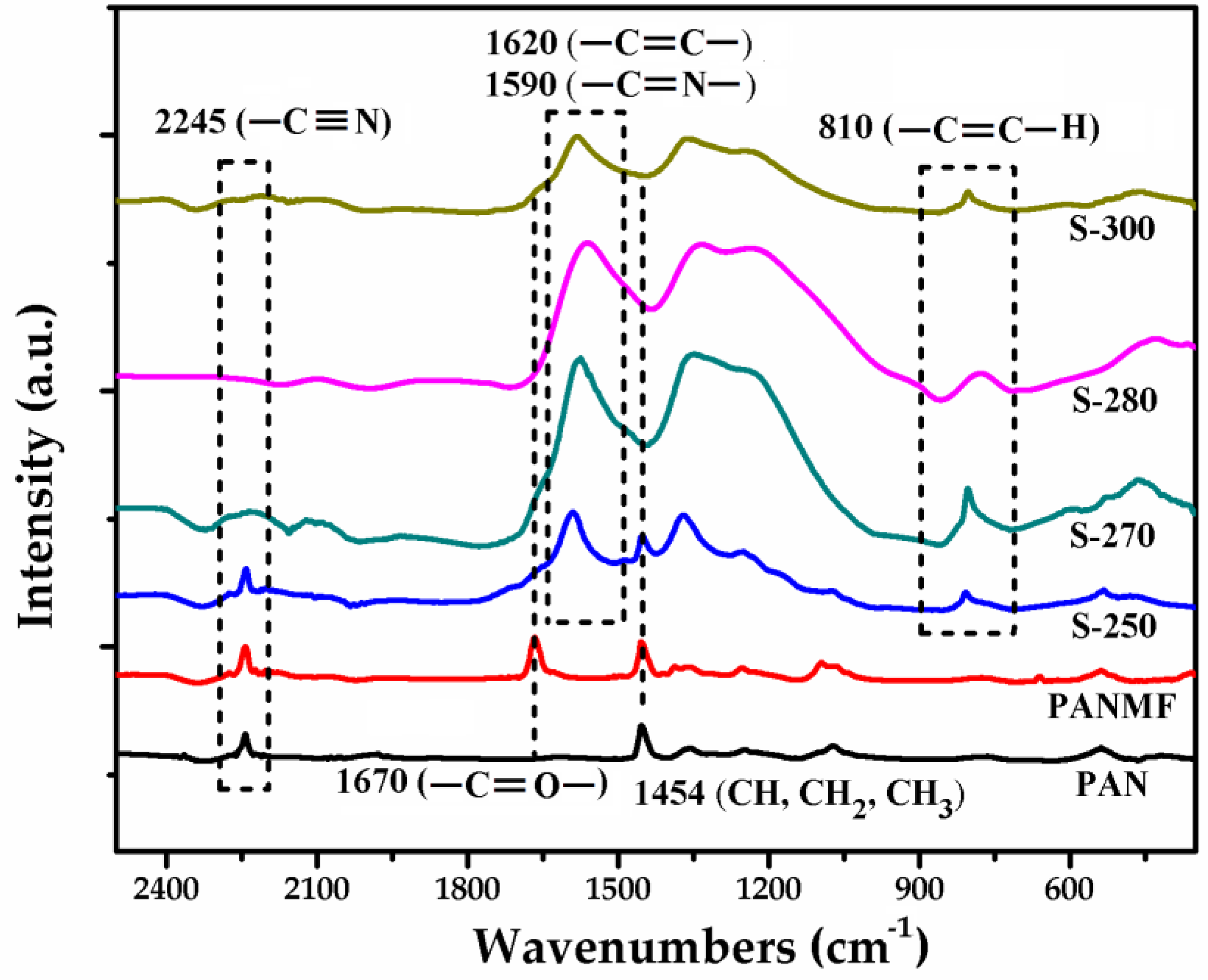
3.1. Chemical Characterization of PANMFs
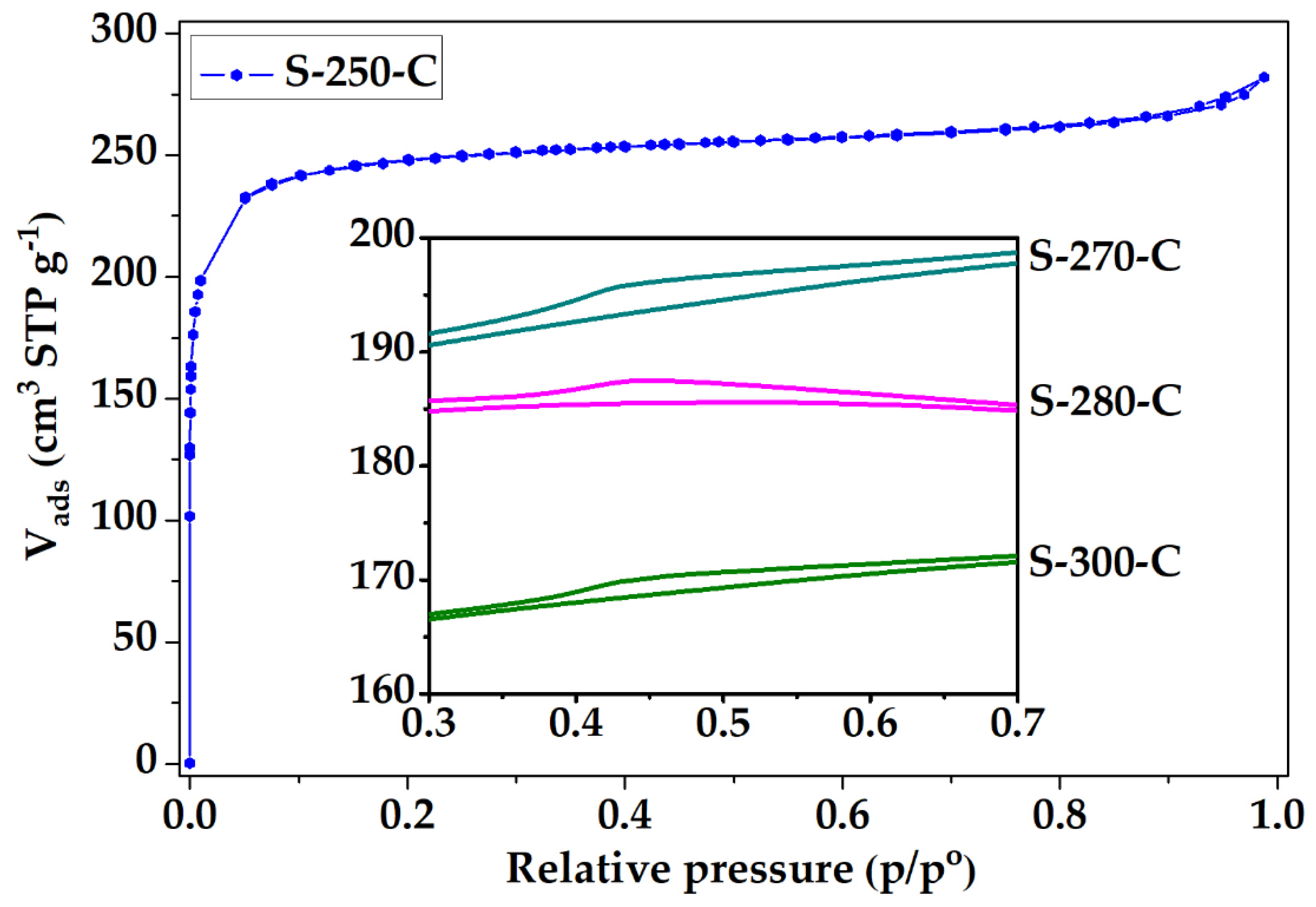
3.2. Structural and Chemical Characterization of CMF
3.2.1. Impact of Stabilization Temperature
3.2.2. Impact of Carbonization Temperature
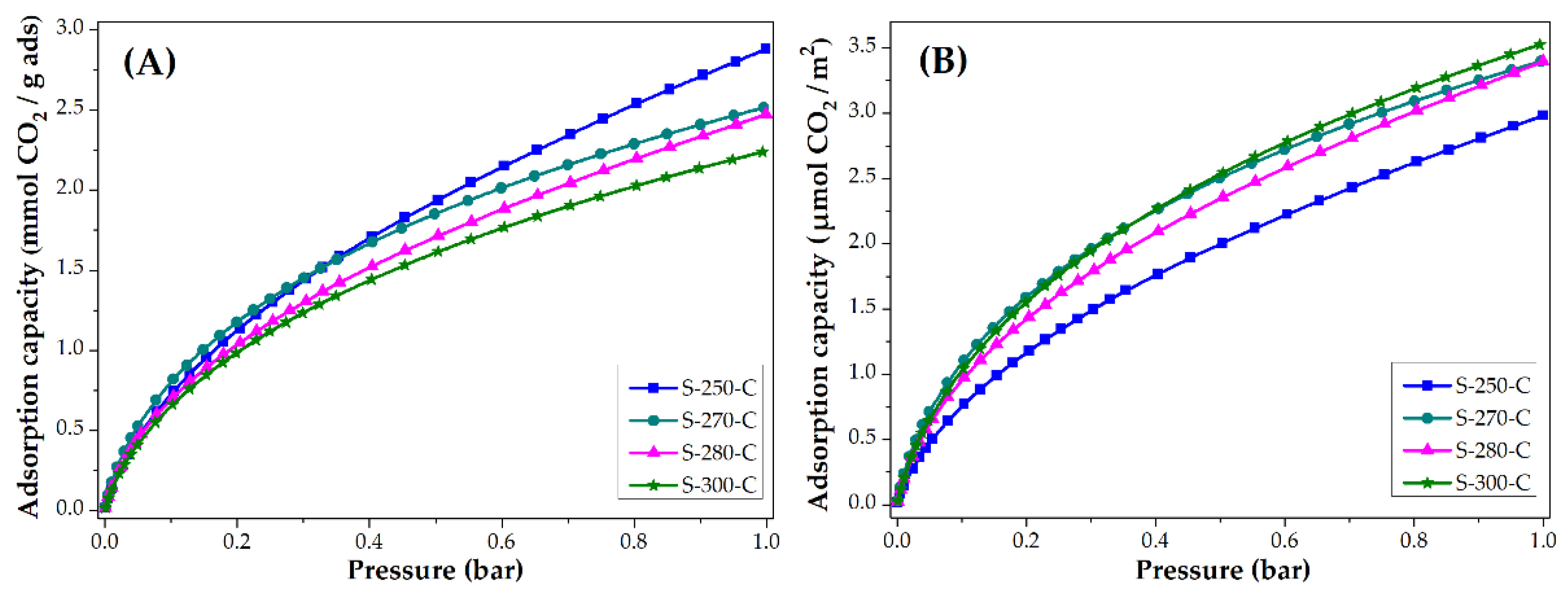
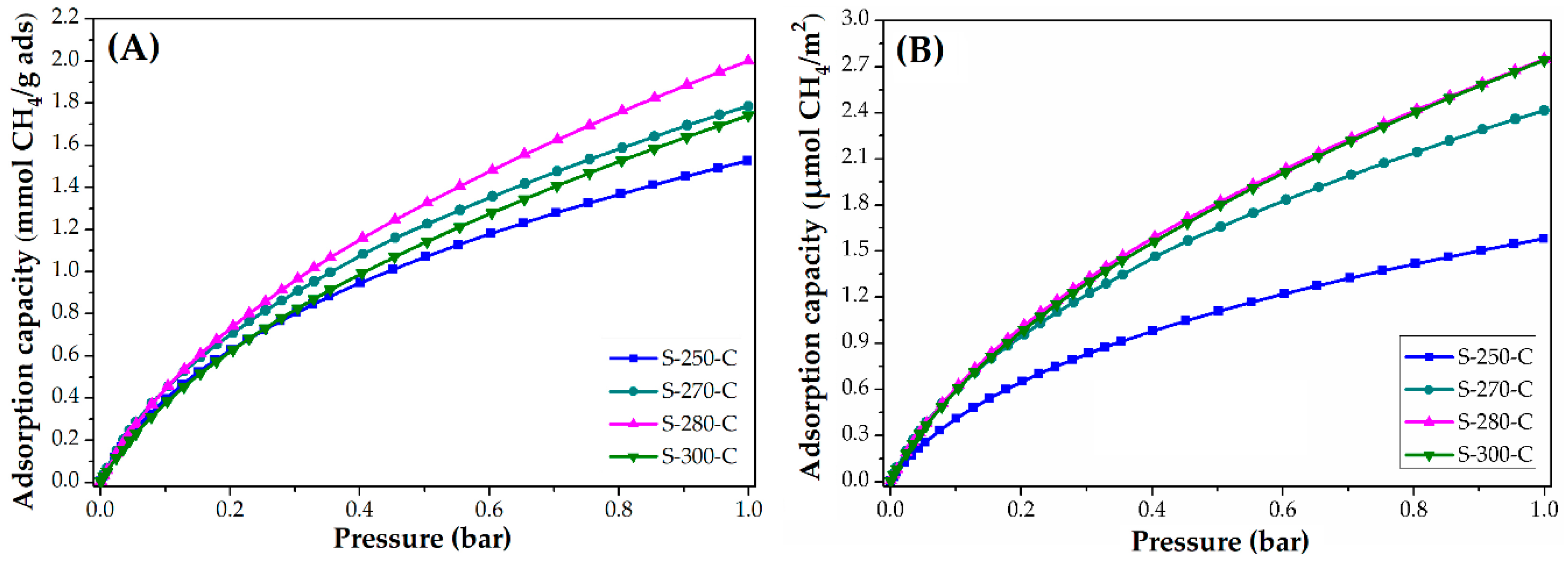
3.3. Applications: CO2 and CH4 Adsorption Capacity
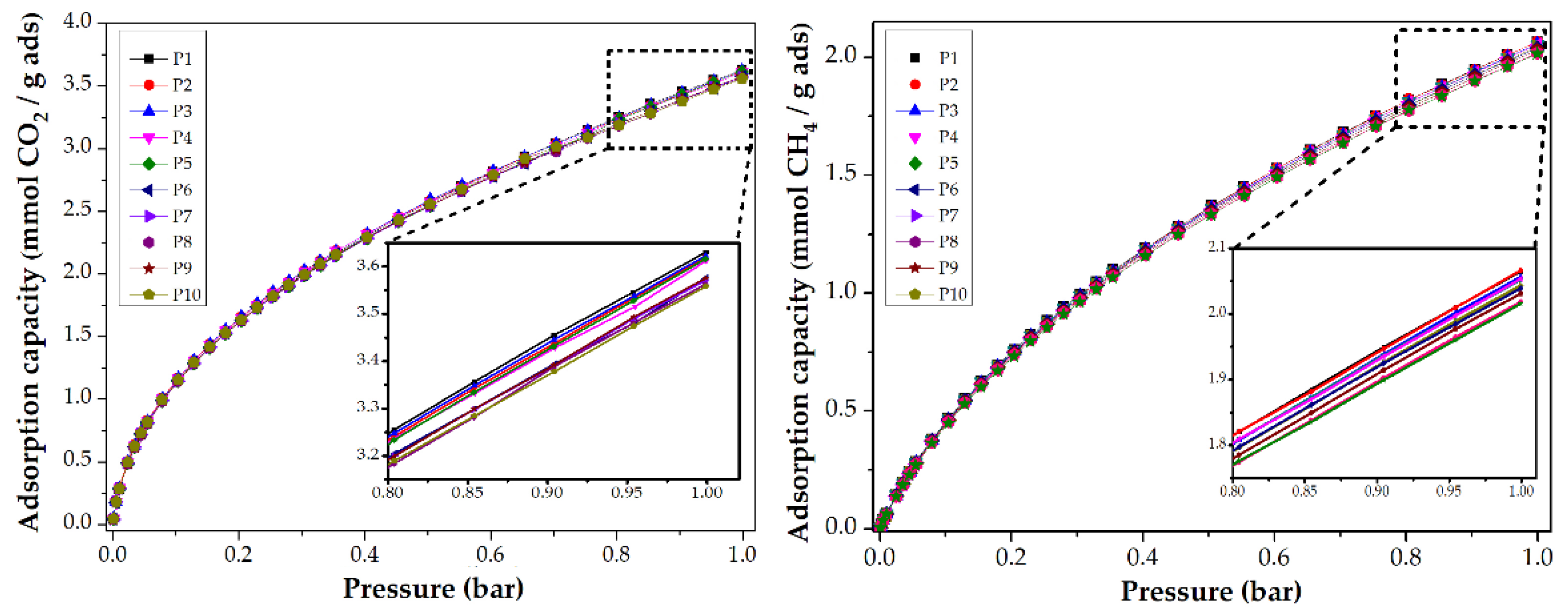
3.3.1. Impact of Stabilization Temperature
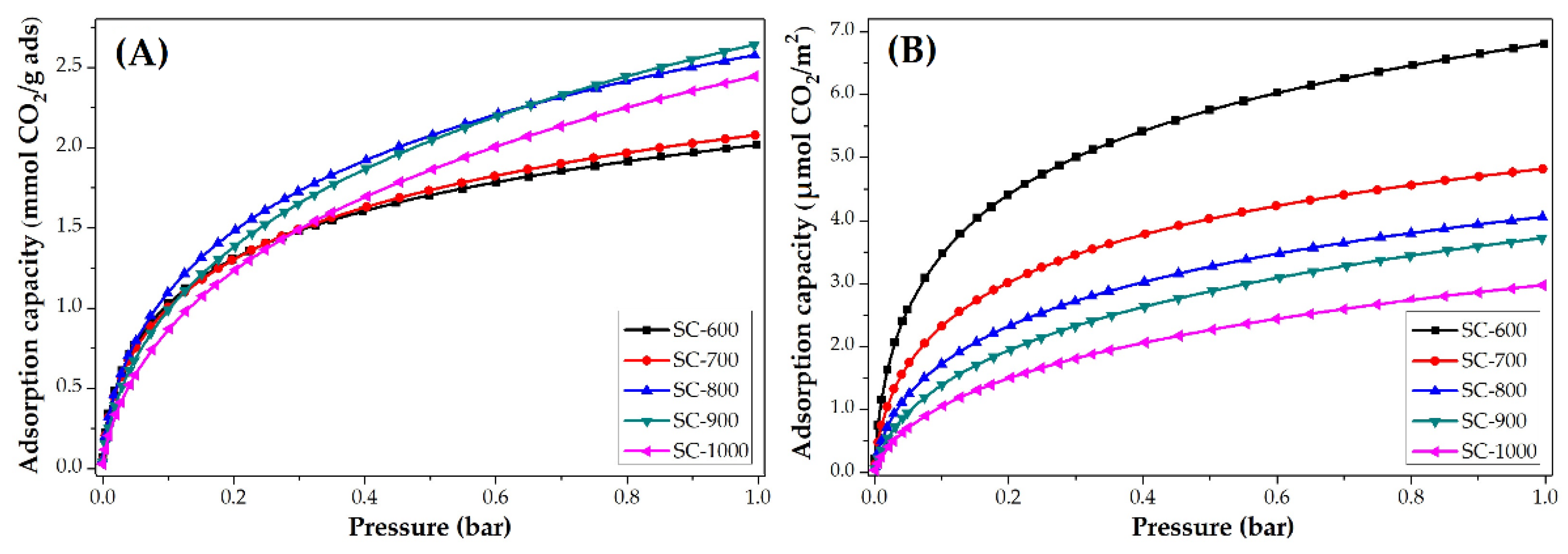
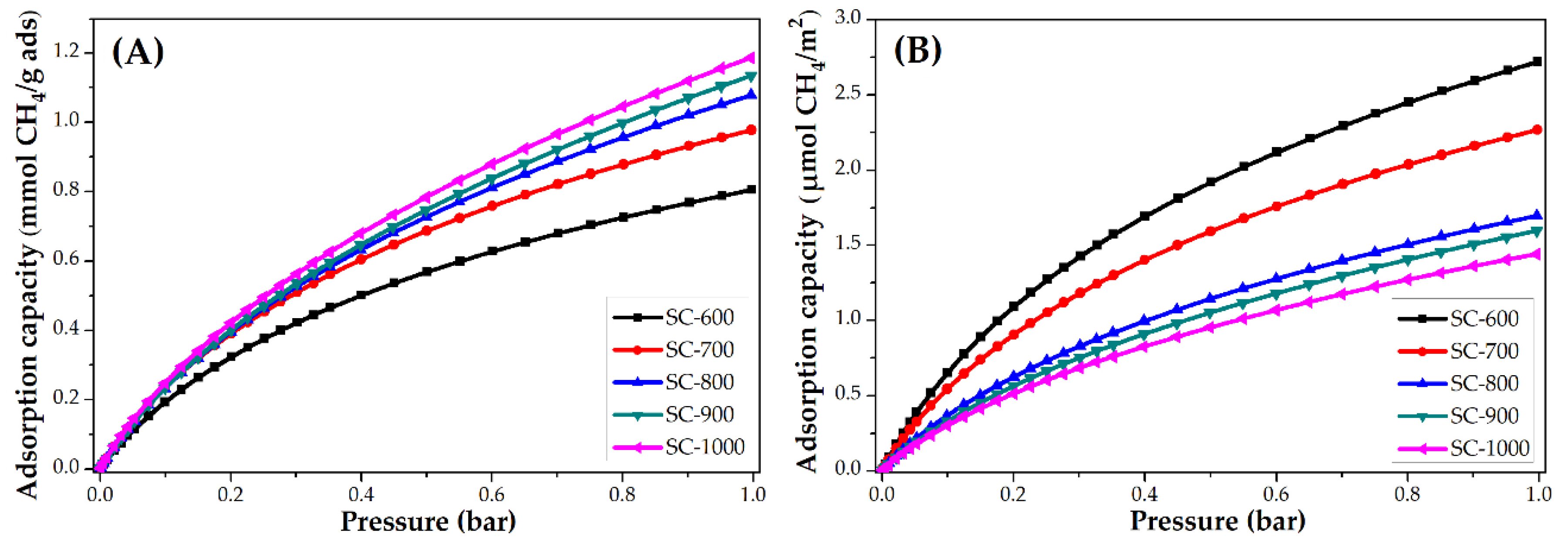
3.3.2. Impact of Carbonization Temperature
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, X.B.; Xu, J. Optimal policies for climate change: A joint consideration of CO2 and methane. Appl. Energy 2018, 211, 1021–1029. [Google Scholar] [CrossRef]
- Meng, L.-Y.; Park, S.-J. Superhydrophobic carbon-based materials: A review of synthesis, structure, and applications. Carbon Lett. 2014, 15, 89–104. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, S.J. A review on solid adsorbents for carbon dioxide capture. J. Ind. Eng. Chem. 2015, 23, 1–11. [Google Scholar] [CrossRef]
- Chaffee, A.L.; Knowles, G.P.; Liang, Z.; Zhang, J.; Xiao, P.; Webley, P.A. CO2 capture by adsorption: Materials and process development. Int. J. Greenh. Gas Control. 2007, 1, 11–18. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Silva, F.W.M.D.; Moura, P.A.S.D.; Sousa, A.G.S.; Vieira, R.S.; Rodriguez-Castellon, E.; Azevedo, D.C.S. CO2 adsorption in amine-grafted zeolite 13X. Appl. Surf. Sci. 2014, 314, 314–321. [Google Scholar] [CrossRef]
- Bao, Z.; Yu, L.; Ren, Q.; Lu, X.; Deng, S. Adsorption of CO2 and CH4 on a magnesium-based metal organic framework. J. Colloid Interface Sci. 2011, 353, 549–556. [Google Scholar] [CrossRef]
- Yong, Z.; Mata, V.; Rodrigues, E. Adsorption of Carbon Dioxide on Basic Alumina at High Temperatures. J. Chem. Eng. Data 2000, 45, 1093–1095. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, L.; Fu, X.; Sun, Y.; Su, W.; Zhou, Y. Adsorption and regeneration study of the mesoporous adsorbent SBA-15 adapted to the capture/separation of CO2 and CH4. Chem. Eng. Sci. 2007, 62, 1101–1110. [Google Scholar] [CrossRef]
- Liu, X.; Li, J.; Zhou, L.; Huang, D.; Zhou, Y. Adsorption of CO2, CH4 and N2 on ordered mesoporous silica molecular sieve. Chem. Phys. Lett. 2005, 415, 198–201. [Google Scholar] [CrossRef]
- Munguía-Cortés, L.; Pérez-Hermosillo, I.; Ojeda-López, R.; Esparza-Schulz, J.M.; Felipe-Mendoza, C.; Cervantes-Uribe, A.; Domínguez-Ortiz, A. APTES-functionalization of SBA-15 using ethanol or toluene: Textural characterization and sorption performance of carbon dioxide. J. Mex. Chem. Soc. 2017, 61, 273–281. [Google Scholar] [CrossRef]
- Ojeda-López, R.; Pérez-Hermosillo, I.J.; Marcos Esparza-Schulz, J.; Cervantes-Uribe, A.; Domínguez-Ortiz, A. SBA-15 materials: Calcination temperature influence on textural properties and total silanol ratio. Adsorption 2015, 21, 659–669. [Google Scholar] [CrossRef]
- Zhang, L.; Aboagye, A.; Kelkar, A.; Lai, C.; Fong, H. A review: Carbon nanofibers from electrospun polyacrylonitrile and their applications. J. Mater. Sci. 2014, 49, 463–480. [Google Scholar] [CrossRef]
- Meng, L.Y.; Park, S.J. Effect of heat treatment on CO2 adsorption of KOH-activated graphite nanofibers. J. Colloid Interface Sci. 2010, 352, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Jung, D.W.; Adelodun, A.A.; Jo, Y.M. Evaluation of CO2 adsorption capacity of electrospun carbon fibers with thermal and chemical activation. J. Appl. Polym. Sci. 2017, 134, 45534–45541. [Google Scholar] [CrossRef]
- Meng, L.Y.; Park, S.J. One-pot synthetic method to prepare highly N-doped nanoporous carbons for CO2 adsorption. Mater. Chem. Phys. 2014, 143, 1158–1163. [Google Scholar] [CrossRef]
- Kim, J.; Maiti, A.; Lin, L.C.; Stolaroff, J.K.; Smit, B.; Aines, R.D. New materials for methane capture from dilute and medium-concentration sources. Nat. Commun. 2013, 4, 1694–1697. [Google Scholar] [CrossRef]
- Golebiowska, M.; Roth, M.; Firlej, L.; Kuchta, B.; Wexler, C. The reversibility of the adsorption of methane-methyl mercaptan mixtures in nanoporous carbon. Carbon 2012, 50, 225–234. [Google Scholar] [CrossRef]
- Park, S.-J. Carbon Fibers; Springer: New York, NY, USA; London, UK, 2015; Volume 210, ISBN 9789401794770. [Google Scholar]
- Ojeda-López, R.; Ramos-Sánchez, G.; Esparza-Schulz, J.M.; Lartundo, L.; Domínguez-Ortiz, A. On site formation of N-doped carbon nanofibers, an efficient electrocatalyst for fuel cell applications. Int. J. Hydrogen Energy 2017, 42, 30339–30348. [Google Scholar] [CrossRef]
- Alarifi, I.M.; Khan, W.S.; Asmatulu, R. Synthesis of electrospun polyacrylonitrile-derived carbon fibers and comparison of properties with bulk form. PLoS ONE 2018, 13, 1–14. [Google Scholar] [CrossRef]
- Yuan, L.; Wei, X.; Martinez, J.P.; Yu, C.; Panahi, N.; Gan, J.B.; Zhang, Y.; Gan, Y.X. Reaction Spinning Titanium Dioxide Particle-Coated Carbon Fiber for Photoelectric Energy Conversion. Fibers 2019, 7, 49. [Google Scholar] [CrossRef]
- Gan, Y.; Yu, C.; Panahi, N.; Gan, J.; Cheng, W. Processing Iron Oxide Nanoparticle-Loaded Composite Carbon Fiber and the Photosensitivity Characterization. Fibers 2019, 7, 25. [Google Scholar] [CrossRef]
- Xue, C.; Wilson, L.D. A Spectroscopic Study of Solid-Phase Chitosan/Cyclodextrin-Based Electrospun Fibers. Fibers 2019, 7, 48. [Google Scholar] [CrossRef]
- Rahaman, M.S.A.; Ismail, A.F.; Mustafa, A.A. Review of heat treatment on polyacrylonitrile fiber. Polym. Degrad. Stab. 2007, 92, 1421–1432. [Google Scholar] [CrossRef]
- Huang, Z.M.; Zhang, Y.Z.; Kotaki, M.; Ramakrishna, S. A review on polymer nanofibers by electrospinning and their applications in nanocomposites. Compos. Sci. Technol. 2003, 63, 2223–2253. [Google Scholar] [CrossRef]
- Mottaghitalab, V.; Haghi, A.K. A study on electrospinning of polyacrylonitrile nanofibers. Korean J. Chem. Eng. 2011, 28, 114–118. [Google Scholar] [CrossRef]
- Paul, P. An Introduction to Electrospinning Process. Man-Made Text. India 2005, 48, 367–371. [Google Scholar]
- Nataraj, S.K.; Yang, K.S.; Aminabhavi, T.M. Polyacrylonitrile-based nanofibers—A state-of-the-art review. Prog. Polym. Sci. 2012, 37, 487–513. [Google Scholar] [CrossRef]
- Wu, M.; Wang, Q.; Li, K.; Wu, Y.; Liu, H. Optimization of stabilization conditions for electrospun polyacrylonitrile nanofibers. Polym. Degrad. Stab. 2012, 97, 1511–1519. [Google Scholar] [CrossRef]
- Dalton, S.; Heatley, F.; Budd, P.M. Thermal stabilization of polyacrylonitrile fibres. Polymer 1999, 40, 5531–5543. [Google Scholar] [CrossRef]
- Xue, Y.; Liu, J.; Liang, J. Correlative study of critical reactions in polyacrylonitrile based carbon fiber precursors during thermal-oxidative stabilization. Polym. Degrad. Stab. 2013, 98, 219–229. [Google Scholar] [CrossRef]
- Ko, T.-H. The influence of pyrolisis properties and microstructure of modified PAN fiber during carbonization. J. Appl. Polym. Sci. 1991, 43, 589–600. [Google Scholar] [CrossRef]
- Wangxi, Z.; Jie, L.; Gang, W. Evolution of structure and properties of PAN precursors during their conversion to carbon fibers. Carbon 2003, 41, 2805–2812. [Google Scholar] [CrossRef]
- Liu, J.; Wang, P.H.; Li, R.Y. Continuous carbonization of polyacrylonitrile-based oxidized fibers: Aspects on mechanical properties and morphological structure. J. Appl. Polym. Sci. 1994, 52, 945–950. [Google Scholar] [CrossRef]
- Laffont, L.; Monthioux, M.; Serin, V.; Mathur, R.B.; Guimon, C.; Guimon, M.F. An EELS study of the structural and chemical transformation of PAN polymer to solid carbon. Carbon 2004, 42, 2485–2494. [Google Scholar] [CrossRef]
- Kumar, A.; Ganguly, A.; Papakonstantinou, P. Thermal stability study of nitrogen functionalities in a graphene network. J. Phys. Condens. Matter 2012, 24, 235503–235508. [Google Scholar] [CrossRef]
- Schierholz, R.; Kröger, D.; Weinrich, H.; Gehring, M.; Tempel, H.; Kungl, H.; Mayer, J.; Eichel, R.A. The carbonization of polyacrylonitrile-derived electrospun carbon nanofibers studied by: In situ transmission electron microscopy. RSC Adv. 2019, 9, 6267–6277. [Google Scholar] [CrossRef]
- Arbab, S.; Teimoury, A.; Mirbaha, H.; Adolphe, D.C.; Noroozi, B.; Nourpanah, P. Optimum stabilization processing parameters for polyacrylonitrile-based carbon nanofibers and their difference with carbon (micro) fibers. Polym. Degrad. Stab. 2017, 142, 198–208. [Google Scholar] [CrossRef]
- Eddib, A.A.; Chung, D.D.L. Electric permittivity of carbon fiber. Carbon 2019, 143, 475–480. [Google Scholar] [CrossRef]
- Zhou, Z.; Lai, C.; Zhang, L.; Qian, Y.; Hou, H.; Reneker, D.H.; Fong, H. Development of carbon nanofibers from aligned electrospun polyacrylonitrile nanofiber bundles and characterization of their microstructural, electrical, and mechanical properties. Polymer 2009, 50, 2999–3006. [Google Scholar] [CrossRef]
- Arshad, S.N.; Naraghi, M.; Chasiotis, I. Strong carbon nanofibers from electrospun polyacrylonitrile. Carbon 2011, 49, 1710–1719. [Google Scholar] [CrossRef]
- Ojeda-López, R.; Pérez-Hermosillo, I.J.; Esparza-Schulz, J.M.; Ramos-Sánchez, G.; Domínguez-Ortiz, A. Controlling Structural and Electrochemical Properties of CNF through Calcination. In Advances in Materials Science Research; Maryann, C., Wythers, E.D., Eds.; Nova Science Publishers: New York, NY, USA, 2019; pp. 79–118. ISBN 6312317269. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Pels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, K.M. Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis. Carbon 1995, 33, 1641–1653. [Google Scholar] [CrossRef]
- Fitzer, E.; Müller, D.J. The influence of oxygen on the chemical reactions during stabilization of pan as carbon fiber precursor. Carbon 1975, 13, 63–69. [Google Scholar] [CrossRef]
- Roldán, L.; Armenise, S.; Marco, Y.; García-Bordejé, E. Control of nitrogen insertion during the growth of nitrogen-containing carbon nanofibers on cordierite monolith walls. Phys. Chem. Chem. Phys. 2012, 14, 3568–3575. [Google Scholar] [CrossRef]
- Shafeeyan, M.S.; Daud, W.M.A.W.; Houshmand, A.; Arami-Niya, A. Ammonia modification of activated carbon to enhance carbon dioxide adsorption: Effect of pre-oxidation. Appl. Surf. Sci. 2011, 257, 3936–3942. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbate | Textural Properties | S-250-C | S-270-C | S-280-C | S-300-C |
|---|---|---|---|---|---|
| N2 | SBET (m2 g−1) | 966.0 | 740.0 | 728.0 | 635.0 |
| DNLDF (nm) | 0.79 | 0.80 | 0.81 | 0.82 | |
| CO2 | DNLDF, X1 (nm) | 0.51 | 0.51 | 0.51 | 0.51 |
| DNLDF, X2 (nm) | 0.59 | 0.59 | 0.59 | 0.59 | |
| DNLDF, X3 (nm) | 0.68 | 0.71 | 0.69 | 0.71 | |
| DNLDF, X4 (nm) | 0.82 | 0.83 | 0.82 | 0.83 |
| PROPERTIES | SC-600 | SC-700 | SC-800 | SC-900 | SC-1000 |
|---|---|---|---|---|---|
| SBET (m2 g−1) | 296 | 431 | 635 | 710 | 822 |
| DNLDF (nm) | 1.15 | 0.80 | 0.80 | 0.83 | 0.83 |
| DNLDF, X1 (nm) | 0.51 | 0.51 | 0.51 | 0.51 | 0.51 |
| DNLDF, X2 (nm) | 0.59 | 0.59 | 0.59 | 0.59 | 0.59 |
| DNLDF, X3 (nm) | 0.73 | 0.72 | 0.72 | 0.70 | 0.72 |
| DNLDF, X4 (nm) | 0.83 | 0.83 | 0.83 | 0.83 | 0.83 |
| C1s | 78 | 82 | 85 | 90 | 91 |
| N1s | 16 | 13 | 9 | 5 | 5 |
| O1s | 6 | 5 | 6 | 5 | 4 |
| Pyridinic | 4.48 | 3.64 | 2.79 | 1.15 | 0.9 |
| C≡N | 3.68 | 1.95 | 0.9 | 0.5 | 0.45 |
| Pyrrolic or pyridine | 4.64 | 3.12 | 1.8 | 0.65 | 0.55 |
| Quaternary or graphite | 2.08 | 2.6 | 2.52 | 2 | 2.1 |
| Pyridinic with oxidized species | 0.64 | 0.78 | 0.63 | 0.5 | 0.5 |
| CMF | CO2 | CH4 | ||||
|---|---|---|---|---|---|---|
| mmol CO2/g ads | mg CO2/g ads | μmol CO2/m2 (Normalized) | mmol CH4/g ads | mg CH4/g ads | μmol CH4/m2 (Normalized) | |
| S-250-C | 2.9 | 127.6 | 3.0 | 1.5 | 24.5 | 1.6 |
| S-270-C | 2.5 | 110.0 | 3.4 | 1.8 | 28.7 | 2.4 |
| S-280-C | 2.5 | 110.0 | 3.4 | 2.0 | 32.1 | 2.8 |
| S-300-C | 2.2 | 96.8 | 3.5 | 1.7 | 27.9 | 2.8 |
| CMF | CO2 | CH4 | ||||
|---|---|---|---|---|---|---|
| mmol CO2/g ads | mg CO2/g ads | μmol CO2/m2 (Normalized) | mmol CH4/g ads | mg CH4/g ads | μmol CH4/m2 (Normalized) | |
| SC-600 | 2.0 | 88.7 | 6.8 | 0.8 | 12.8 | 2.7 |
| SC-700 | 2.1 | 92.4 | 4.8 | 1.0 | 16.0 | 2.3 |
| SC-800 | 2.6 | 114.4 | 4.1 | 1.0 | 16.0 | 1.7 |
| SC-900 | 2.6 | 114.4 | 3.7 | 1.1 | 17.6 | 1.6 |
| SC-1000 | 2.4 | 105.6 | 3.0 | 1.2 | 19.2 | 1.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojeda-López, R.; Esparza-Schulz, J.M.; Pérez-Hermosillo, I.J.; Hernández-Gordillo, A.; Domínguez-Ortiz, A. Improve in CO2 and CH4 Adsorption Capacity on Carbon Microfibers Synthesized by Electrospinning of PAN. Fibers 2019, 7, 81. https://doi.org/10.3390/fib7100081
Ojeda-López R, Esparza-Schulz JM, Pérez-Hermosillo IJ, Hernández-Gordillo A, Domínguez-Ortiz A. Improve in CO2 and CH4 Adsorption Capacity on Carbon Microfibers Synthesized by Electrospinning of PAN. Fibers. 2019; 7(10):81. https://doi.org/10.3390/fib7100081
Chicago/Turabian StyleOjeda-López, Reyna, J. Marcos Esparza-Schulz, Isaac J. Pérez-Hermosillo, Armin Hernández-Gordillo, and Armando Domínguez-Ortiz. 2019. "Improve in CO2 and CH4 Adsorption Capacity on Carbon Microfibers Synthesized by Electrospinning of PAN" Fibers 7, no. 10: 81. https://doi.org/10.3390/fib7100081
APA StyleOjeda-López, R., Esparza-Schulz, J. M., Pérez-Hermosillo, I. J., Hernández-Gordillo, A., & Domínguez-Ortiz, A. (2019). Improve in CO2 and CH4 Adsorption Capacity on Carbon Microfibers Synthesized by Electrospinning of PAN. Fibers, 7(10), 81. https://doi.org/10.3390/fib7100081