
Investigation of the Influence of Hexabenzocoronene in Polyacrylonitrile-Based Precursors for Carbon Fibers

 , , ,
, , ,  ,
, 
Abstract
1. Introduction
2. Experimental
2.1. Materials
2.2. Spinning Solution and Wet-Spinning Process of Precursor Fibers
2.3. Stabilization and Carbonization of PAN and HBC/PAN Fibers
2.4. Characterization
3. Results and Discussion
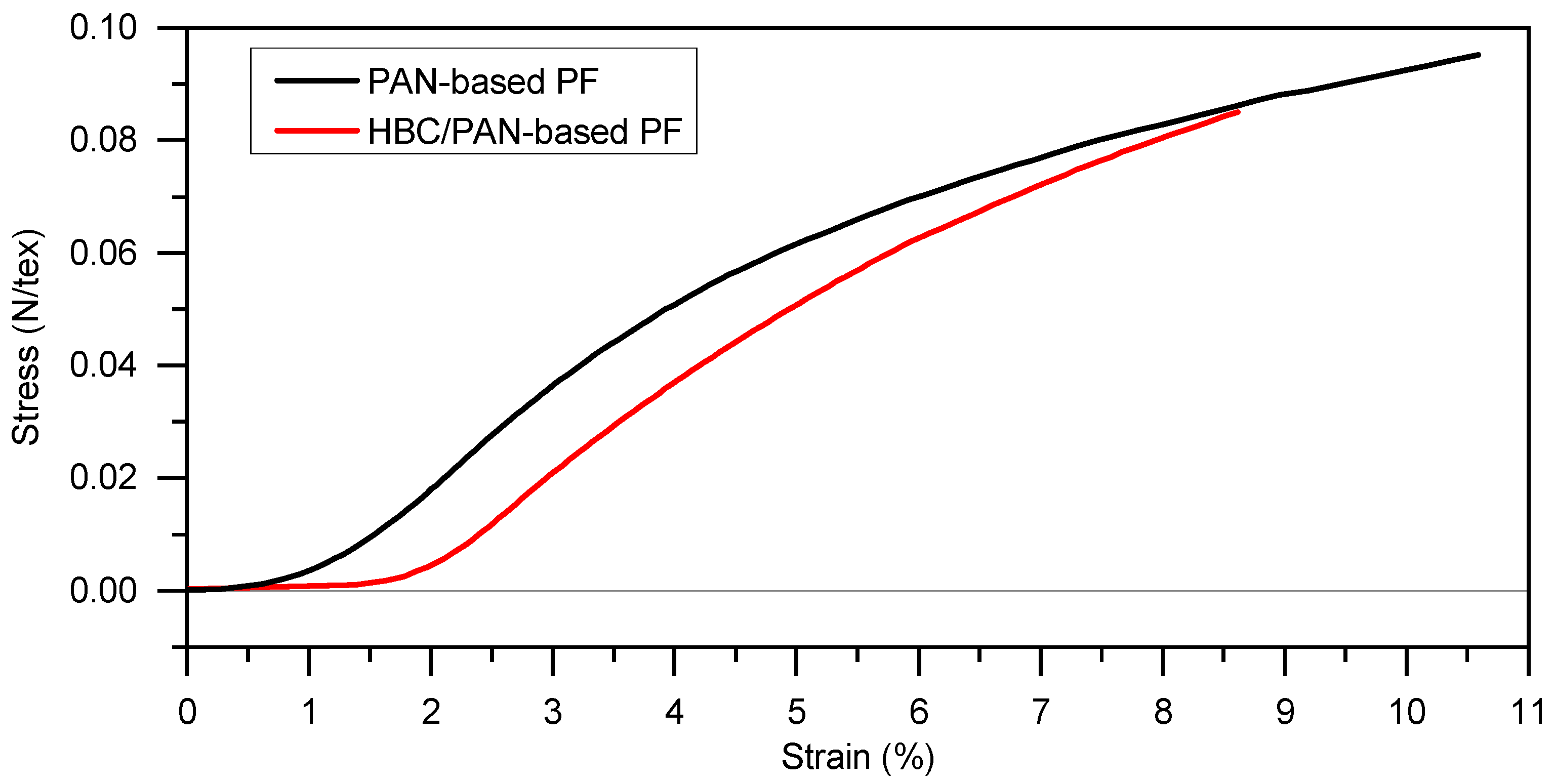
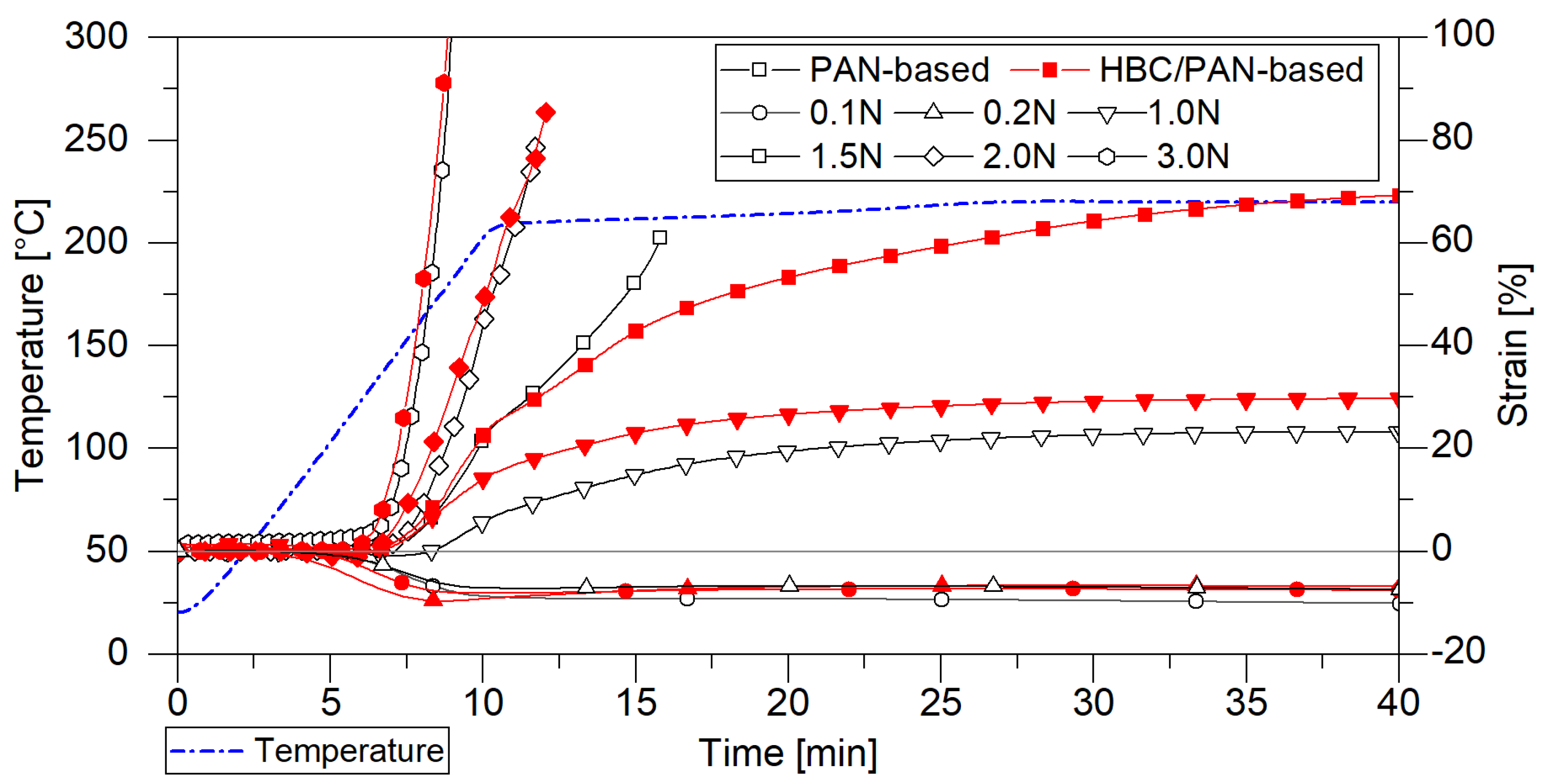
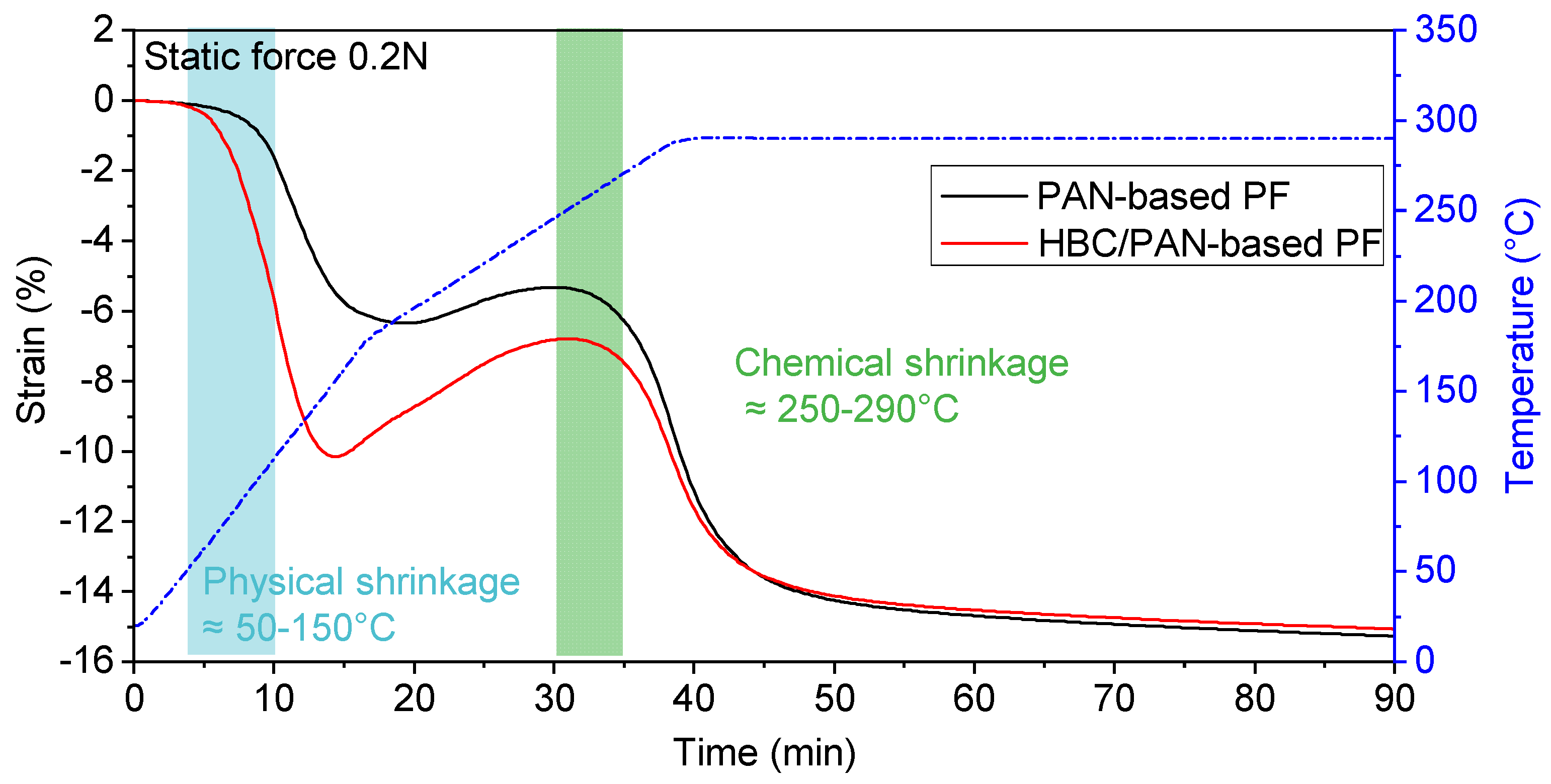
3.1. Mechanical Properties and Thermal Behavior of PAN and HBC/PAN Precursor Fibers and Stabilized Fibers
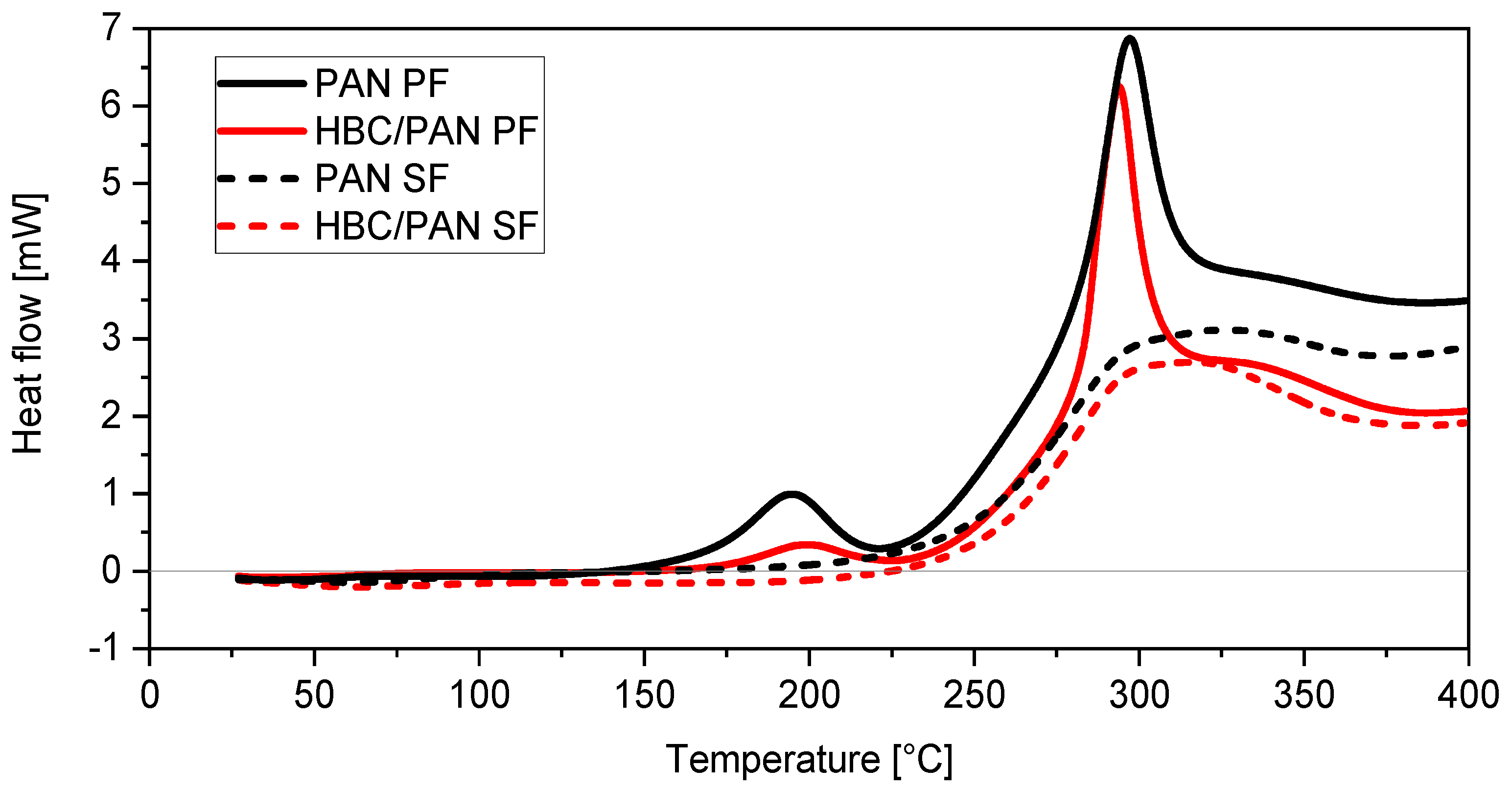
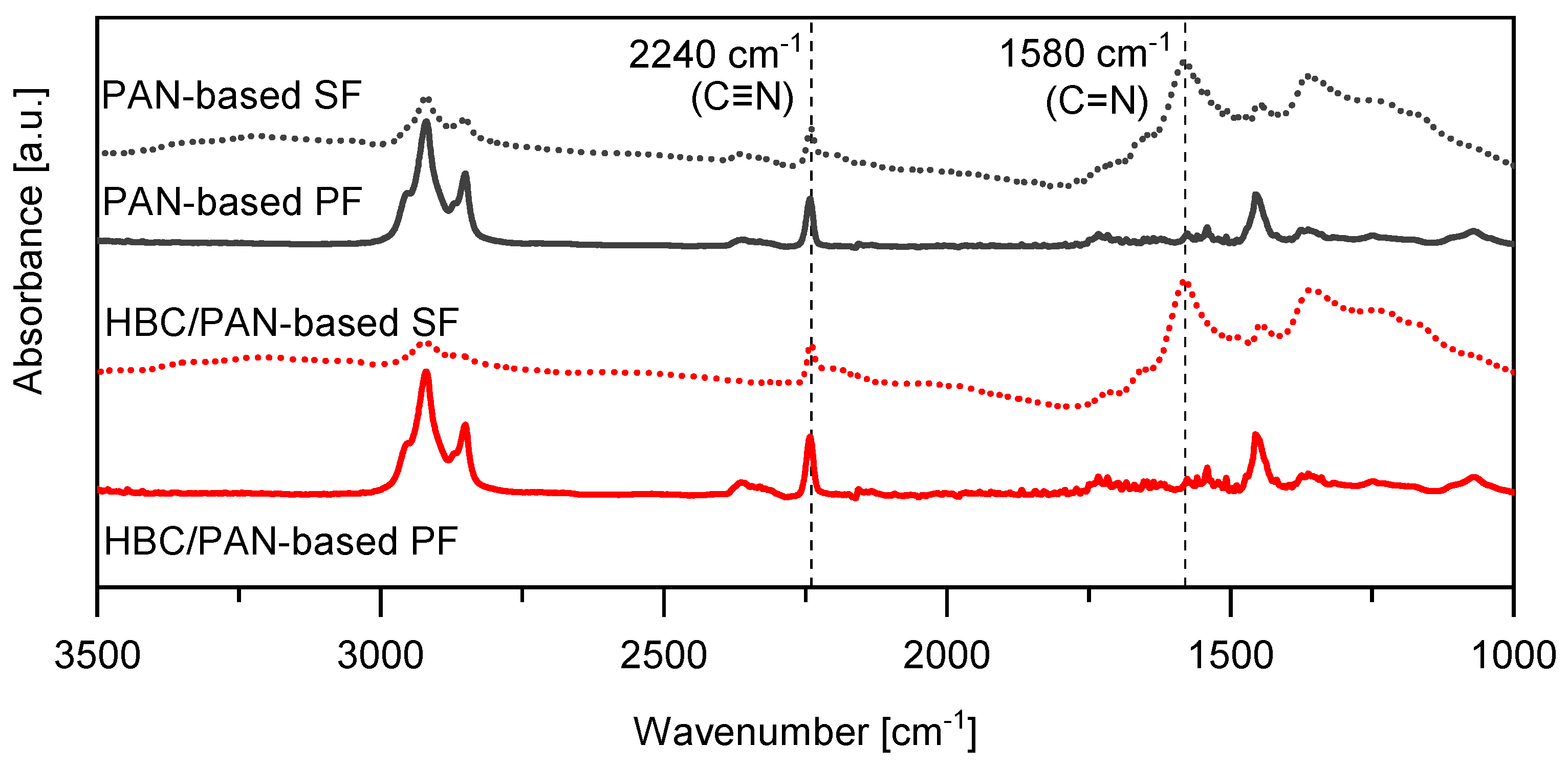
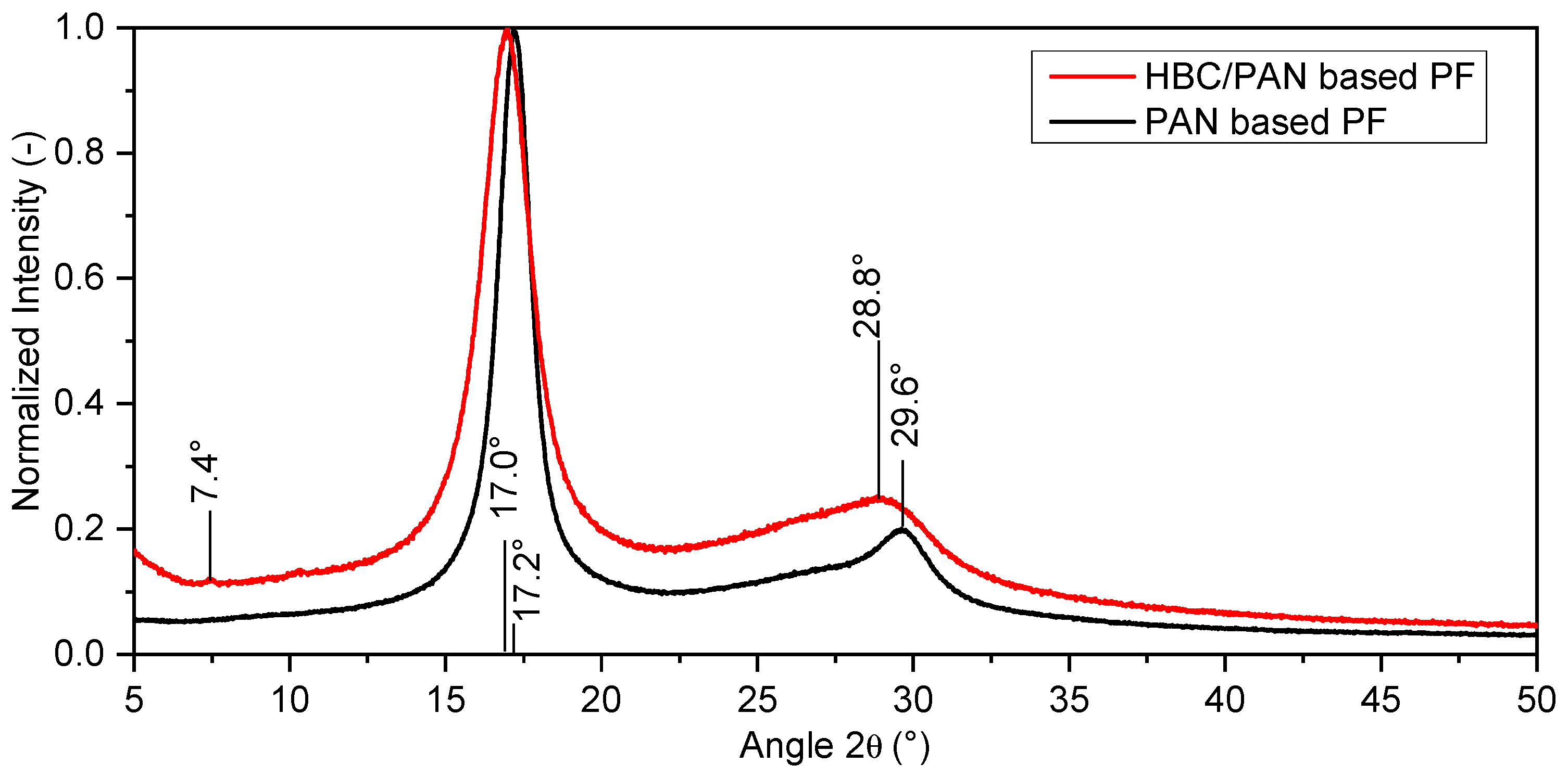
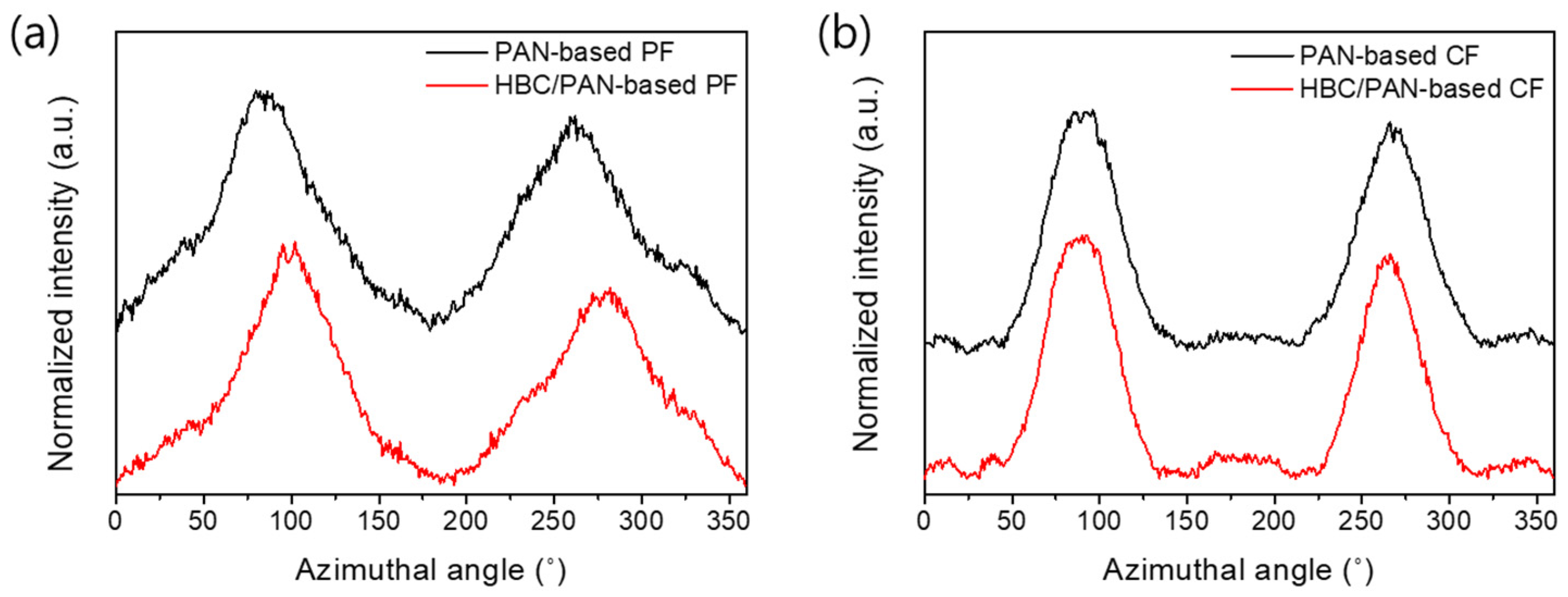
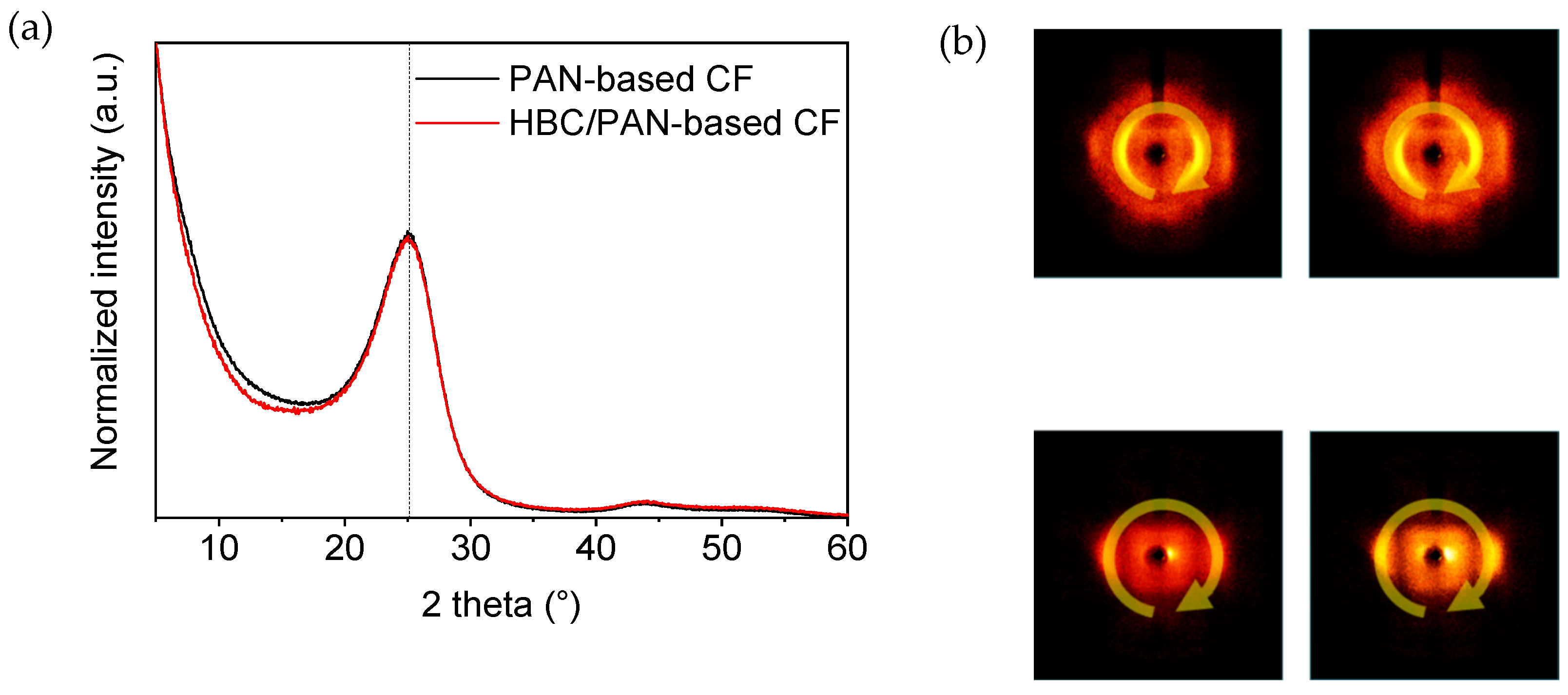
3.2. Structural Analysis of PAN- and HBC/PAN-Based PFs and SFs Using FT-IR and XRD
3.3. Mechanical Properties and Density of PAN- and HBC/PAN-Based Fibers
4. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
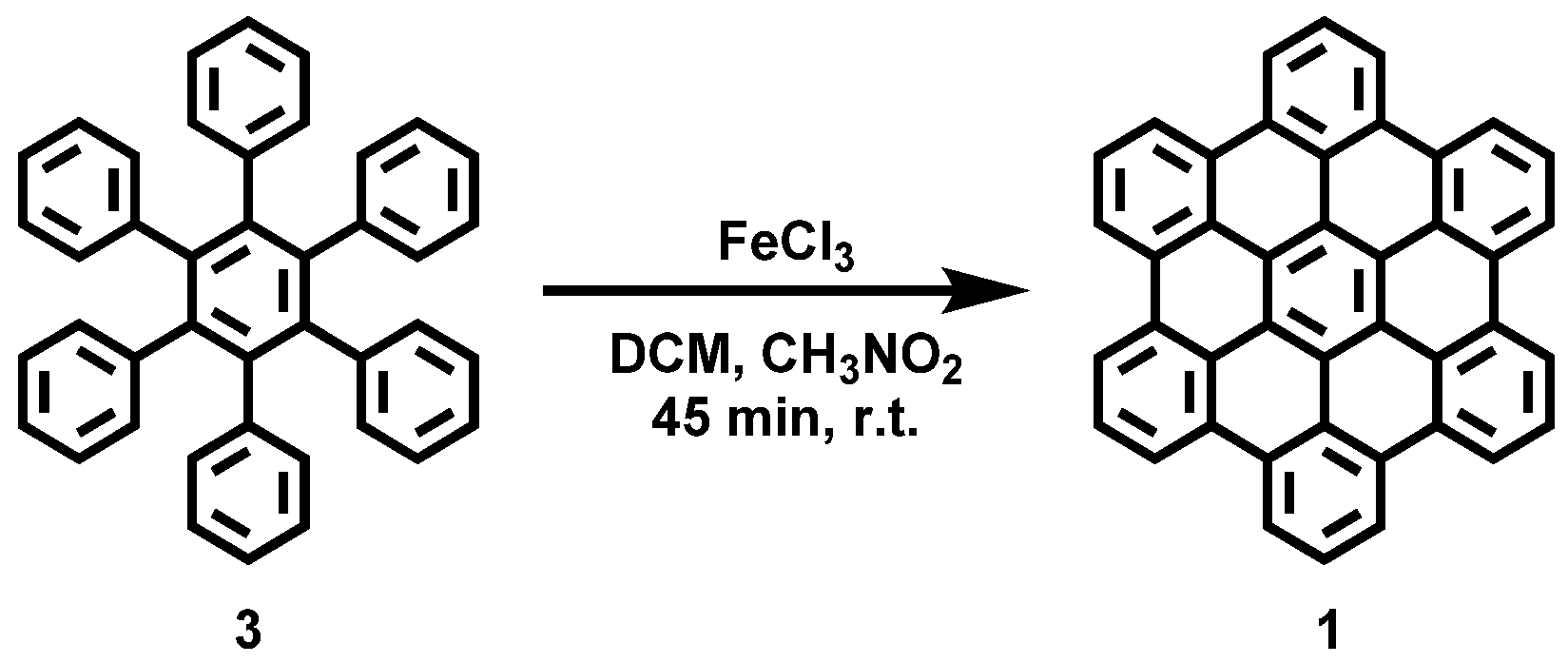
Appendix A.1. Synthesis of HBC (1)


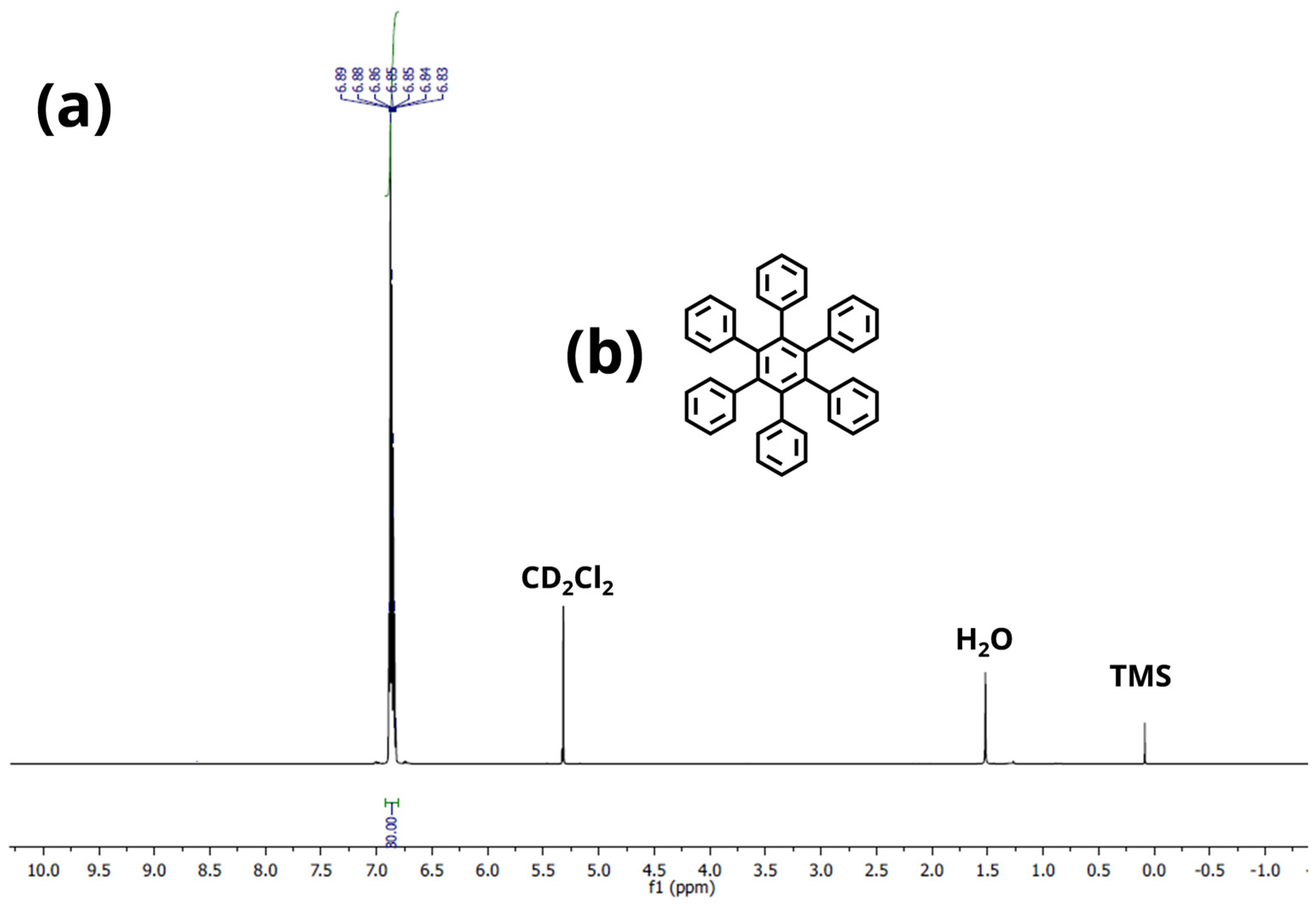
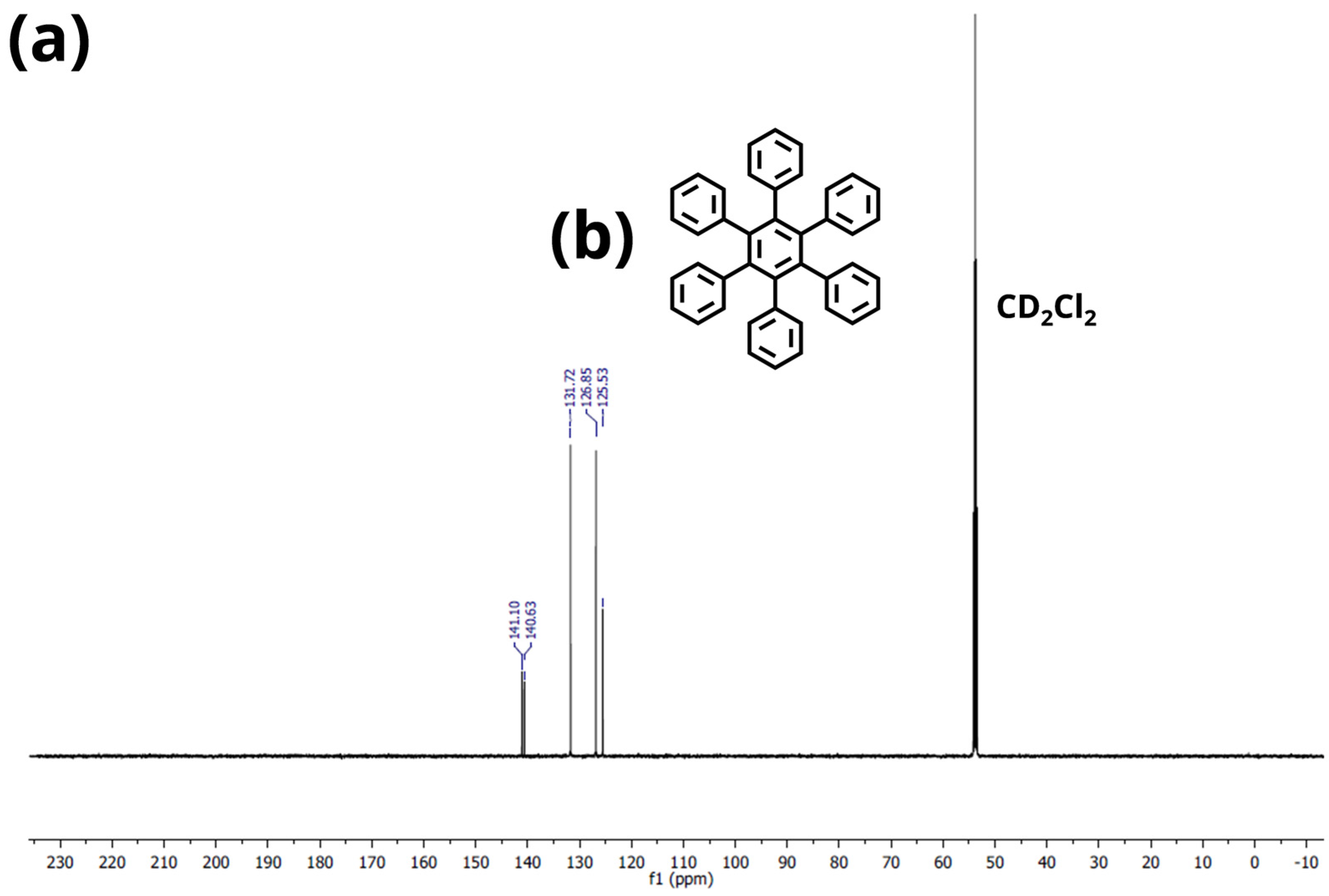
Appendix A.2. NMR Spectroscopy


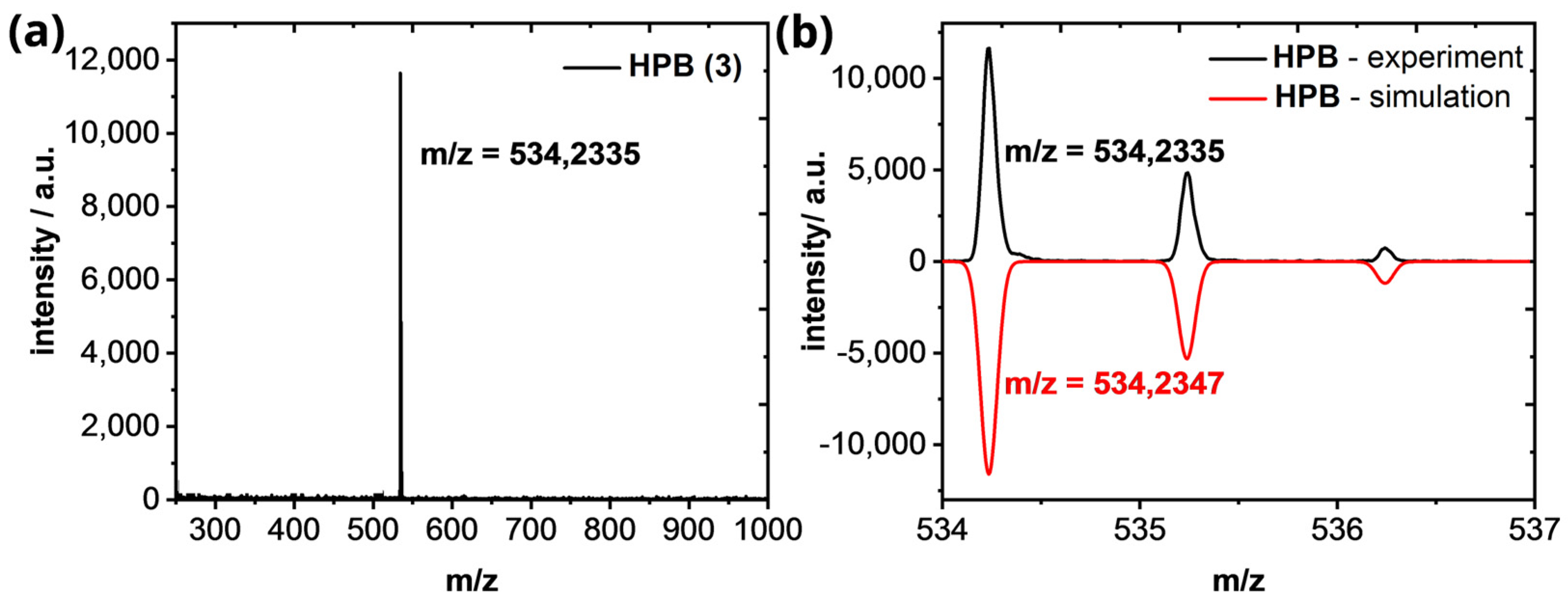
Appendix A.3. High-Resolution Mass Spectrometry of HPB (3) and HBC (1)


Appendix B. XRD Measurement

Appendix C. SEM Imaging

References
- Jäger, H.; Cherif, C.; Kirsten, M.; Behnisch, T.; Wolz, D.S.; Böhm, R.; Gude, M. Influence of processing parameters on the properties of carbon fibres—An overview. Mater. Werkst. 2016, 47, 1044–1057. [Google Scholar] [CrossRef]
- Böhm, R.; Thieme, M.; Wohlfahrt, D.; Wolz, D.; Richter, B.; Jäger, H. Reinforcement Systems for Carbon Concrete Composites Based on Low-Cost Carbon Fibers. Fibers 2018, 6, 56. [Google Scholar] [CrossRef]
- Jäger, H.; Frohs, W. Industrial Carbon and Graphite Materials; Wiley: Hoboken, NJ, USA, 2021; Volume 1. [Google Scholar]
- Chae, H.G.; Choi, Y.H.; Minus, M.L.; Kumar, S. Carbon nanotube reinforced small diameter polyacrylonitrile based carbon fiber. Compos. Sci. Technol. 2009, 69, 406–413. [Google Scholar] [CrossRef]
- Chien, A.-T.; Liu, H.C.; Newcomb, B.A.; Xiang, C.; Tour, J.M.; Kumar, S. Polyacrylonitrile fibers containing graphene oxide nanoribbons. ACS Appl. Mater. Interfaces 2015, 7, 5281–5288. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.G.; Minus, M.L.; Rasheed, A.; Kumar, S. Stabilization and carbonization of gel spun polyacrylonitrile/single wall carbon nanotube composite fibers. Polymer 2007, 13, 3781–3789. [Google Scholar] [CrossRef]
- O’Connell, M.J. Carbon Nanotubes: Properties and Applications; Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Chang, H.; Luo, J.; Gulgunje, P.V.; Kumar, S. Structural and Functional Fibers. Annu. Rev. Mater. Res. 2017, 47, 331–359. [Google Scholar] [CrossRef]
- Lyons, K.M. Tensile Testing and Stabilisation/Carbonisation Studies of Polyacrylonitrile/Carbon Nanotube Composite Fibers. Master’s Thesis, School of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, GA, USA, 2012. [Google Scholar]
- Morgan, P. Carbon Fibers and Their Composites; Taylor & Francis: Boca Raton, FL, USA, 2005. [Google Scholar]
- Hill, J.P.; Jin, W. Self-Assembled Hexa-peri-Hexabenzocoronene Graphitic Nanotube. Science 2004, 304, 1481–1483. [Google Scholar] [CrossRef]
- Kirsten, M.; Freudenberg, C.; Cherif, C. Carbonfasern, der Werkstoff des 21. Jahrhunderts. Beton Stahlbetonbau 2015, 110, 8–15. [Google Scholar] [CrossRef]
- Stintz, M. Reine Technologien: Lehrveranstaltung; Institute of Process Engineering and Environmental Technology, TU Dresden: Dresden, Germany, 2017. [Google Scholar]
- Arbab, S.; Mirbaha, H.; Zeinolebadi, A.; Nourpanah, P. Indicators for evaluation of progress in thermal stabilization reactions of polyacrylonitrile fibers. J. Appl. Polym. Sci. 2014, 131, 40343. [Google Scholar] [CrossRef]
- Hou, Y.; Sun, T.; Wang, H.; Wu, D. A new method for the kinetic study of cyclization reaction during stabilization of polyacrylonitrile fibers. J. Mater. Sci. 2008, 43, 4910–4914. [Google Scholar] [CrossRef]
- Tsai, J.-S.; Hsu, H.-N. Determination of the aromatization index for oxidized polyacrylonitrile fibre by the differential scanning calorimetry method. J. Mater. Sci. Lett. 1992, 11, 1403–1405. [Google Scholar] [CrossRef]
- ISO 11566; Carbon Fibre—Determination of the Tensile Properties of Single-Filament Specimens. Composites and reinforcement fibres, Vernier; ISO-International Organization for Standardization: Geneva, Switzerland, 1996.
- DIN EN ISO 5079; Fasern—Bestimmung der Höchstzugkraft und Höchstzugkraftdehnung an Spinnfasern. Normenausschuss Materialprüfung: Physikalisch-technologische Prüfverfahren für Textilien; DIN-Deutsches Institut für Normung: Berlin, Germany, 1996.
- Nunna, S.; Creighton, C.; Fox, B.L.; Naebe, M.; Maghe, M.; Tobin, M.J.; Bambery, K.; Vongsvivut, J.; Hameed, N. The effect of thermally induced chemical transformations on the structure and properties of carbon fibre precursors. J. Mater. Chem. A 2017, 5, 7372–7382. [Google Scholar] [CrossRef]
- Nunna, S.; Maghe, M.; Fakhrhoseini, S.; Polisetti, B.; Naebe, M. A Pathway to Reduce Energy Consumption in the Thermal Stabilization Process of Carbon Fiber Production. Energies 2018, 11, 1145. [Google Scholar] [CrossRef]
- Nunna, S.; Naebe, M.; Hameed, N.; Creighton, C.; Naghashian, S.; Jennings, M.J.; Atkiss, S.; Setty, M.; Fox, B.L. Investigation of progress of reactions and evolution of radial heterogeneity in the initial stage of thermal stabilization of PAN precursor fibres. Polym. Degrad. Stab. 2016, 125, 105–114. [Google Scholar] [CrossRef]
- Zhu, Y.; Wilding, M.A. Estimation, using infrared spectroscopy, of the cyclization of poly(acrylonitrile) during the stabilization stage of carbon fibre production. J. Mater. Sci. 1996, 31, 3831–3837. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, B.; Sun, L.; Zhang, H. Study on the thermal oxidative stabilization reactions and the formed structures in polyacrylonitrile during thermal treatment. Polym. Degrad. Stab. 2017, 140, 104–113. [Google Scholar] [CrossRef]
- Kim, M.A.; Jang, D.; Tejima, S.; Cruz-Silva, R.; Joh, H.I.; Kim, H.C.; Lee, S.; Endo, M. Strengthened PAN-based carbon fibers obtained by slow heating rate carbonization. Sci. Rep. 2016, 6, 22988. [Google Scholar] [CrossRef]
- Chae, H.G.; Sreekumar, T.V.; Uchida, T.; Kumar, S. A comparison of reinforcement efficiency of various types of carbon nanotubes in polyacrylonitrile fiber. Polymer 2005, 46, 10925–10935. [Google Scholar] [CrossRef]
- Fitzer, E.; Müller, D.J. Zur Bildung von gewinkelten Leiterpolymeren in Polyacrylnitril-Fasern. Die Makrornolekulare Chernie 1971, 144, 117–133. [Google Scholar] [CrossRef]
- Heine, M. Optimierung der Reaktionsbedingungen von thermoplastischen Polymer-Fasern zur Kohlenstoffaser-Herstellung am Beispiel von Polyacrylnitril. Ph.D. Thesis, Faculty of Chemistry, University of Karlsruhe, Karlsruhe, Germany, 1988. [Google Scholar]
- Meinl, J.; Kirsten, M.; Cherif, C.; Michaelis, A. Influence of PAN-Fiber Stretching during Thermal Treatment on the Stabilization Reactions. AJAC 2016, 7, 282–293. [Google Scholar] [CrossRef]
- Thünemann, A.F.; Ruland, W. Lamellar Mesophases in Polyacrylonitrile: A Synchrotron Small-Angle X-ray Scattering Study. Macromolecules 2000, 33, 2626–2631. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Zhou, T.; Liu, X.; Yuan, Q.; Zhang, A. New understanding on the reaction pathways of the polyacrylonitrile copolymer fiber pre-oxidation: Online tracking by two-dimensional correlation FTIR spectroscopy. RSC Adv. 2016, 6, 4397–4409. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, P.; Zhang, L.; Ma, Z.; Liang, J.; Fong, H. Thermo-chemical reactions occurring during the oxidative stabilization of electrospun polyacrylonitrile precursor nanofibers and the resulting structural conversions. Carbon 2009, 47, 1087–1095. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Lee, S.; Park, S.; Jo, S.M.; Lee, H.-S.; Joh, H.-I. Continuous and rapid stabilization of polyacrylonitrile fiber bundles assisted by atmospheric pressure plasma for fabricating large-tow carbon fibers. Carbon 2015, 94, 412–416. [Google Scholar] [CrossRef]
- Park, S.; Kil, H.-S.; Choi, D.; Song, S.-K.; Lee, S. Rapid stabilization of polyacrylonitrile fibers achieved by plasma-assisted thermal treatment on electron-beam irradiated fibers. J. Ind. Eng. Chem. 2019, 69, 449–454. [Google Scholar] [CrossRef]
- Habeeb, S.A.; Rajabi, L.; Dabirian, F. Production of polyacrylonitrile/boehmite nanofibrous composite tubular structures by opposite-charge electrospinning with enhanced properties from a low-concentration polymer solution. Polym. Compos. 2019, 41, 1649–1661. [Google Scholar] [CrossRef]
- Feng, X.; Pisula, W.; Takase, M.; Dou, X.; Enkelmann, V.; Wagner, M.; Ding, N.; Müllen, K. Synthesis, Helical Organization, and Fibrous Formation of C3 Symmetric Methoxy-Substituted Discotic Hexa- peri -hexabenzocoronene. Chem. Mater. 2008, 20, 2872–2874. [Google Scholar] [CrossRef]
- Yoshida, K.; Morimoto, I.; Mitsudo, K.; Tanaka, H. RhCl3/amine-catalyzed [2+2+2] cyclization of alkynes. Tetrahedron 2008, 64, 5800–5807. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
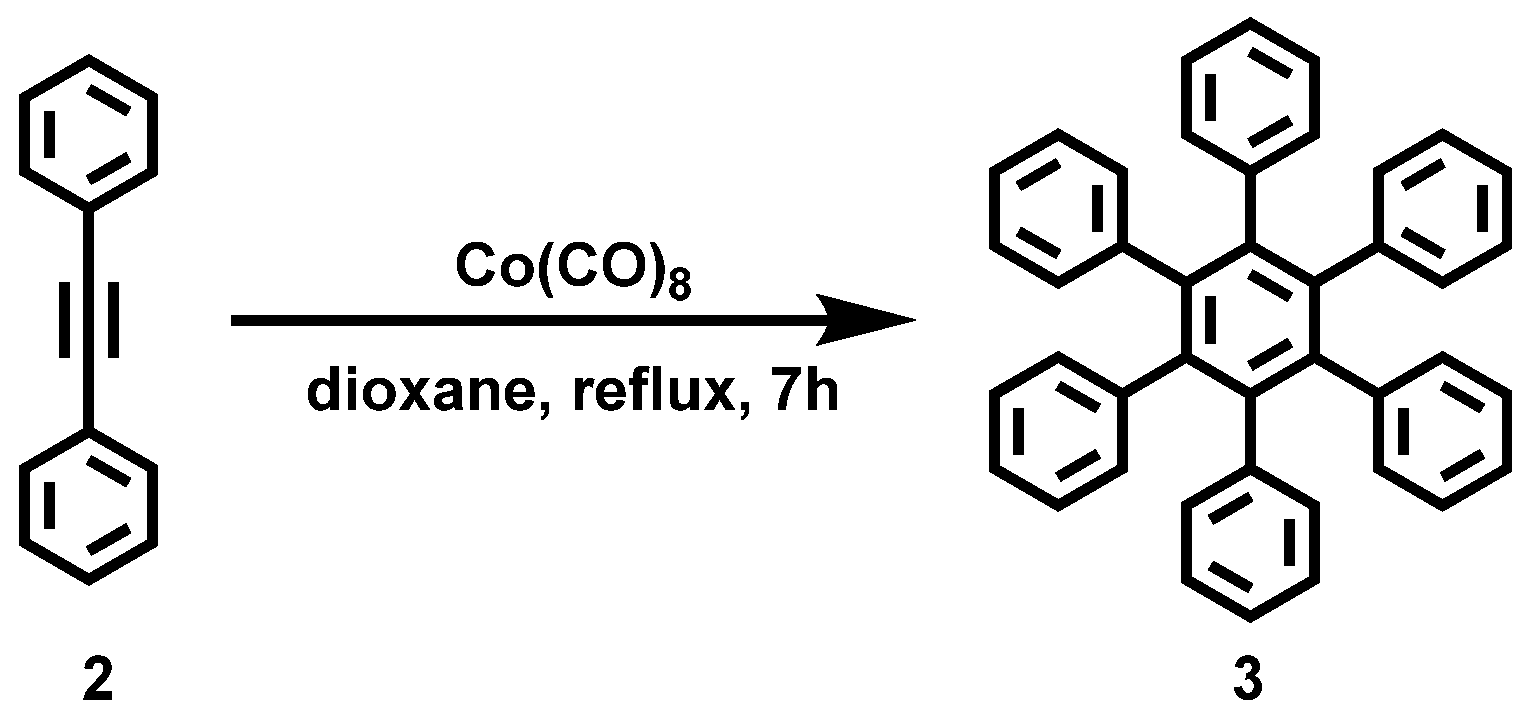{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PAN-Based Fiber | HBC/PAN-Based Fiber | |
|---|---|---|
| Concentration | Homogeneous PAN 18 wt.% | PAN 18 wt.% + HBC 0.1 wt.% |
| Dope temperature | 70 ℃ | |
| Stretching ratio | 1:4 | |
| Coagulation bath temp. | 15 °C | |
| Linear density | 188.2 tex | 210.3 tex |
| Trio 1 > DuoHZ1 | DuoHZ1 > Trio2 | Trio2 > DuoHZ2 | DuoHZ2 > DuoHZ3 | DuoHZ3 > DuoHZ4 | DuoHZ4 > Trio3 | Total Strain | |
|---|---|---|---|---|---|---|---|
| Strain (%) | −4.16 | 0.60 | 0.99 | 0.3 | 0.32 | 1.41–1.44 | −0.54 |
| Parameters | Low-Temperature Oven | Coupling Element | High-Temperature Oven |
|---|---|---|---|
| Temperature (°C) | 450/550/650/800 | 400 | 900/1050/1200/1400 |
| Tension (cN) | 200–900 | ||
| Dwell Time (min) | ≈4.5 | - | ≈5 |
| SF Basis | SI in % | CI in % | Density in g/cm³ |
|---|---|---|---|
| PAN | 45.9 ± 2.6 | 40.2 | 1.23 ± 0.02 |
| HBC/PAN | 52.7 ± 11.1 | 40.2 | 1.27 ± 0.02 |
| PF | CF | |||
|---|---|---|---|---|
| PAN | HBC/PAN | PAN | HBC/PAN | |
| Orientation factor, f | 0.65 | 0.69 | 0.75 | 0.77 |
| PF Basis | Tensile Strength in MPa | Young’s Modulus in GPa | EAB in % | Density in g/cm³ | |
|---|---|---|---|---|---|
| PF | PAN | 270 ± 20 | 7.1 ± 0.5 | 30.1 ± 6.5 | 1.15 ± 0.02 |
| HBC/PAN | 300 ± 30 | 7.6 ± 1.1 | 33.8 ± 7.0 | 1.16 ± 0.02 | |
| CF | PAN | 1890 ± 346 | 152.5 ± 10.1 | 1.3 ± 0.2 | 1.51 ± 0.02 |
| HBC/PAN | 1933 ± 278 | 191.0 ± 11.3 | 1.1 ± 0.2 | 1.64 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peters, R.; Jang, D.; Wolz, D.S.J.; Lee, S.; Jäger, H.; Richter, M.; Cherif, C.; Vasiutovich, K.; Richter, M.; Feng, X.; et al. Investigation of the Influence of Hexabenzocoronene in Polyacrylonitrile-Based Precursors for Carbon Fibers. Fibers 2023, 11, 14. https://doi.org/10.3390/fib11020014
Peters R, Jang D, Wolz DSJ, Lee S, Jäger H, Richter M, Cherif C, Vasiutovich K, Richter M, Feng X, et al. Investigation of the Influence of Hexabenzocoronene in Polyacrylonitrile-Based Precursors for Carbon Fibers. Fibers. 2023; 11(2):14. https://doi.org/10.3390/fib11020014
Chicago/Turabian StylePeters, Romy, Dawon Jang, Daniel Sebastian Jens Wolz, Sungho Lee, Hubert Jäger, Mirko Richter, Chokri Cherif, Kiryl Vasiutovich, Marcus Richter, Xinliang Feng, and et al. 2023. "Investigation of the Influence of Hexabenzocoronene in Polyacrylonitrile-Based Precursors for Carbon Fibers" Fibers 11, no. 2: 14. https://doi.org/10.3390/fib11020014
APA StylePeters, R., Jang, D., Wolz, D. S. J., Lee, S., Jäger, H., Richter, M., Cherif, C., Vasiutovich, K., Richter, M., Feng, X., Behnisch, T., & Gude, M. (2023). Investigation of the Influence of Hexabenzocoronene in Polyacrylonitrile-Based Precursors for Carbon Fibers. Fibers, 11(2), 14. https://doi.org/10.3390/fib11020014