Two-Dimensional SiP, SiAs, GeP and GeAs as Promising Candidates for Photocatalytic Applications



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Method
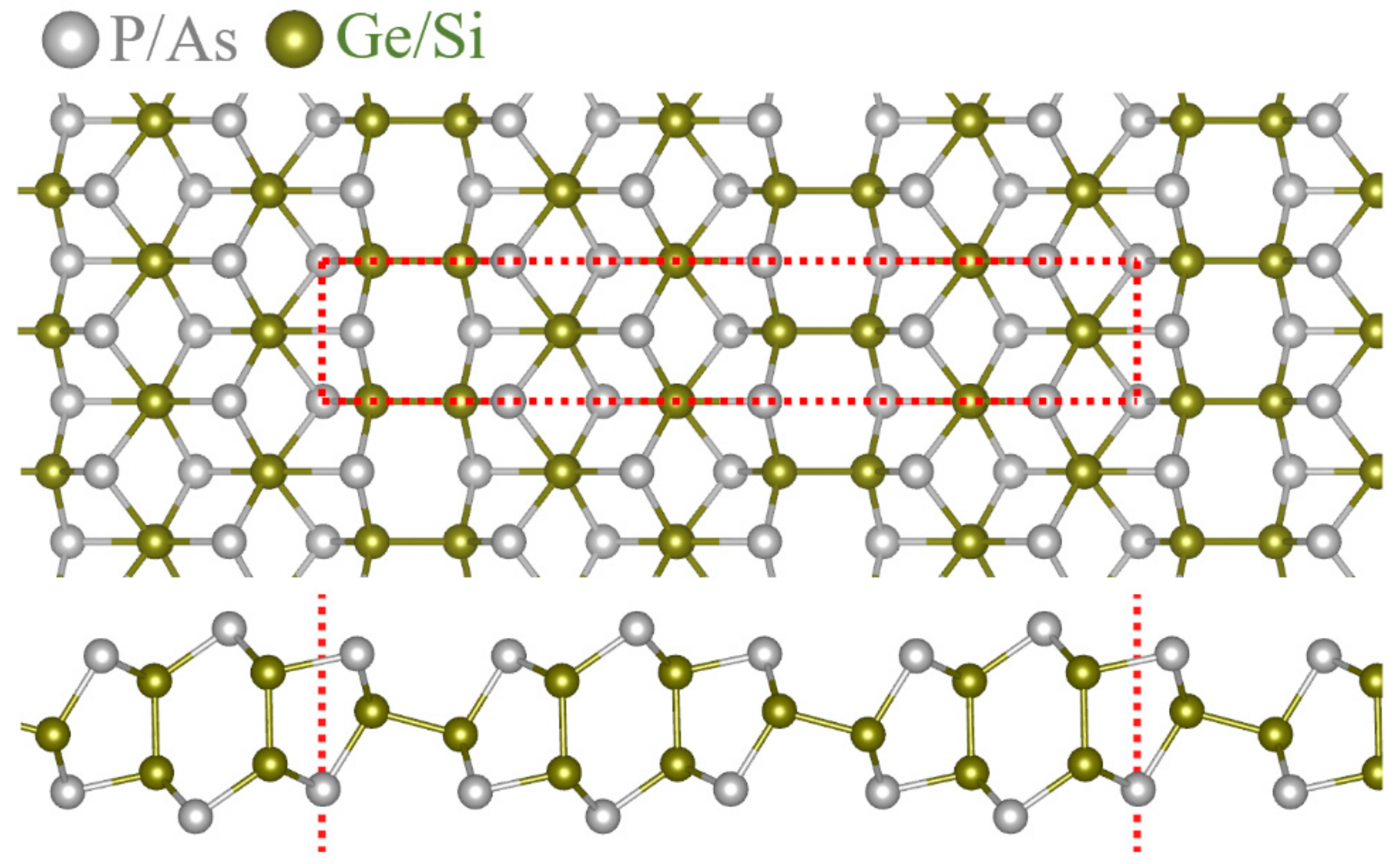
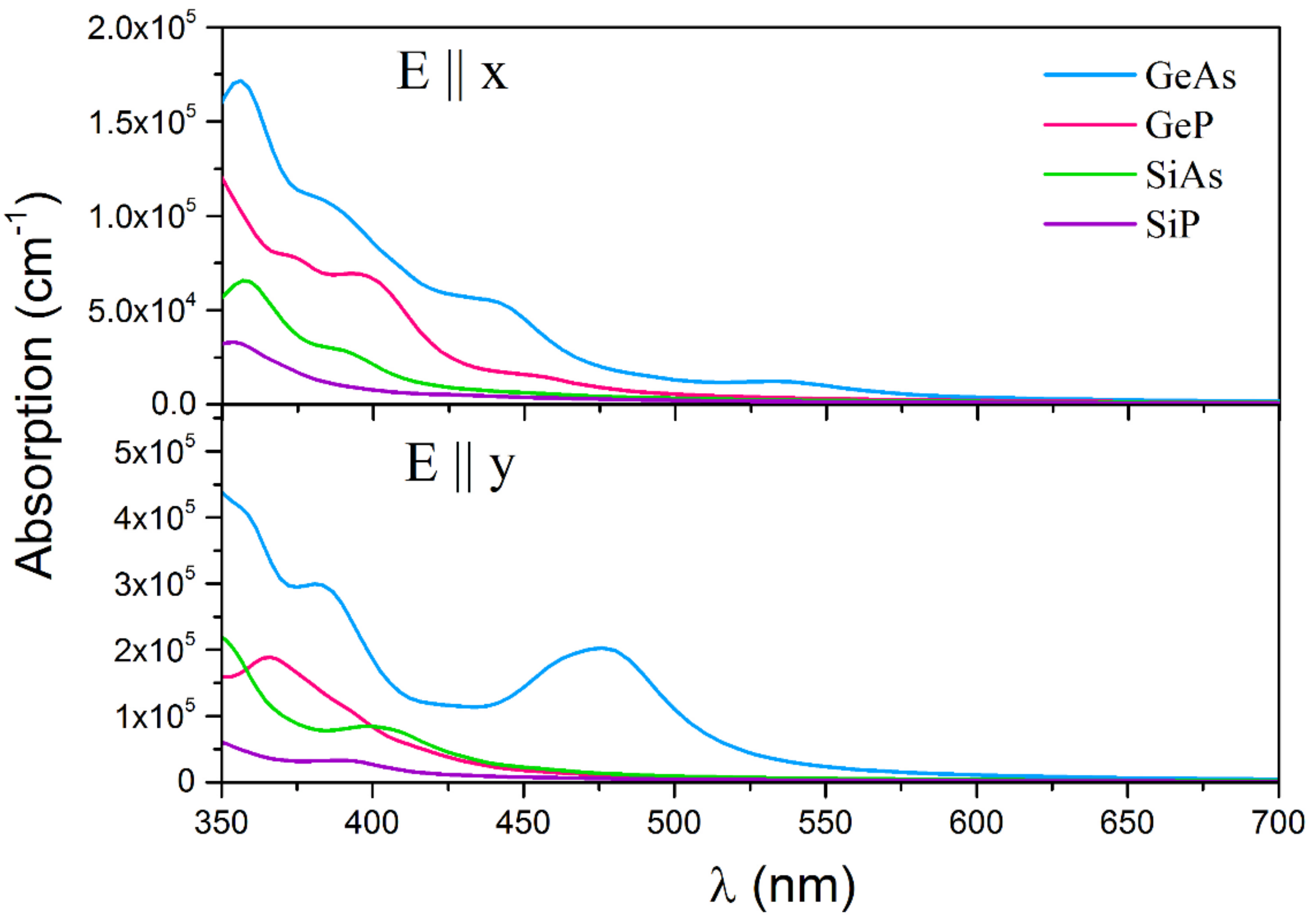
3. Results and Discussions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Novoselov, K.; Geim, A.K.; Morozov, S.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Watanabe, K.; Tsuda, O.; Taniguchi, T. Deep ultraviolet light-emitting hexagonal boron nitride synthesized at atmospheric pressure. Science 2007, 317, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Ci, L.; Lu, H.; Sorokin, P.B.; Jin, C.; Ni, J.; Kvashnin, A.G.; Kvashnin, D.G.; Lou, J.; Yakobson, B.I.; et al. Large scale growth and characterization of atomic hexagonal boron nitride layers. Nano Lett. 2010, 10, 3209–3215. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Y. Strain-induced self-doping in silicene and germanene from first-principles. Solid State Commun. 2013, 155, 6–11. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef]
- Watts, M.C.; Picco, L.; Russell-Pavier, F.S.; Cullen, P.L.; Miller, T.S.; Bartuś, S.P.; Payton, O.D.; Skipper, N.T.; Tileli, V.; Howard, C.A. Production of phosphorene nanoribbons. Nature 2019, 568, 216–220. [Google Scholar] [CrossRef]
- Gusmão, R.; Sofer, Z.; Pumera, M. Functional protection of exfoliated black phosphorus by noncovalent modification with anthraquinone. ACS Nano 2018, 12, 5666–5673. [Google Scholar] [CrossRef]
- Sturala, J.; Sofer, Z.; Pumera, M. Chemistry of layered pnictogens: Phosphorus, arsenic, antimony, and bismuth. Angew. Chem. Int. Ed. 2019, 131, 7551–7557. [Google Scholar] [CrossRef]
- Ares, P.; Palacios, J.J.; Abellán, G.; Gómez-Herrero, J.; Zamora, F. Recent progress on antimonene: A new bidimensional material. Adv. Mater. 2018, 30, 1703771. [Google Scholar] [CrossRef]
- Jariwala, D.; Sangwan, V.K.; Lauhon, L.; Marks, T.J.; Hersam, M.C. Emerging device applications for semiconducting two-dimensional transition metal dichalcogenides. ACS Nano 2014, 8, 1102–1120. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.; Kenneth, D.; Shi-Zhang, Q. Advent of 2D rhenium disulfide (ReS2): Fundamentals to applications. Adv. Funct. Mater. 2017, 27, 1606129. [Google Scholar] [CrossRef]
- Shengxue, Y.; Yanhan, Y.; Minghui, W.; Chunguang, H.; Wanfu, S.; Yongji, G.; Li, H.; Chengbao, J.; Yongzhe, Z. Highly in-plane optical and electrical anisotropy of 2D germanium arsenide. Adv. Funct. Mater. 2018, 28, 1707379. [Google Scholar] [CrossRef]
- Liu, E.; Fu, Y.; Wang, Y.; Feng, Y.; Liu, H.; Wan, X.; Zhou, W.; Wang, B.; Shao, L.; Ho, C.H.; et al. Integrated digital inverters based on two-dimensional anisotropic ReS2 field-effect transistors. Nat. Commun. 2015, 6, 6991. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Hu, M.; Li, D.; He, Y.; Zhao, R.; Jiang, X.; Song, S.; Wang, L.; Fan, C. A nano and micro-integrated protein chip based on quantum dot probes and a microfluidic network. Nano Res. 2008, 1, 490–496. [Google Scholar] [CrossRef]
- Yang, H.; Jussila, H.; Autere, A.; Komsa, H.P.; Ye, G.; Chen, X.; Hasan, T.; Sun, Z. Optical waveplates based on birefringence of anisotropic two-dimensional layered materials. ACS Photonics 2017, 4, 3023–3030. [Google Scholar] [CrossRef]
- Beck, C.G. Crystallography of SiP and SiAs single crystals and of SiP precipitates in Si. J. Appl. Phys. 1966, 37, 4683. [Google Scholar] [CrossRef]
- Donohue, P.C.; Young, H.S. Synthesis, structure, and superconductivity of new high pressure phases in the systems GeP and GeAs. J. Solid State Chem. 1970, 1, 143–149. [Google Scholar] [CrossRef]
- Ashton, M.; Sinnott, S.B.; Hennig, R.G. Computational discovery and characterization of polymorphic two-dimensional IV–V materials. Appl. Phys. Lett. 2016, 109, 192103. [Google Scholar] [CrossRef]
- Liang, L.; Weike, W.; Penglai, G.; Xiangde, Z.; Bei, D.; Xingqiang, S.; Guoying, G.; Huiqiao, L.; Tianyou, Z. 2D GeP: An unexploited low-symmetry semiconductor with strong in-plane anisotropy. Adv. Mater. 2018, 30, 1706771. [Google Scholar] [CrossRef]
- Cheng, A.Q.; He, Z.; Zhao, J.; Zeng, H.; Chen, R.S. Monolayered silicon and germanium monopnictide semiconductors: Excellent stability, high absorbance, and strain engineering of electronic properties. ACS Appl. Mater. Interfaces 2018, 10, 5133–5139. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, B.; Rabczuk, T. Anisotropic mechanical properties and strain tuneable band-gap in single-layer SiP, SiAs, GeP and GeAs. Phys. E Low Dimens. Syst. Nanostruct. 2018, 103, 273–278. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Cao, C.; He, Y. Optical anisotropy and strain tunable optical, electronic and structural properties in monolayer GeP: A computational study. Phys. E Low Dimens. Syst. Nanostruct. 2019, 113, 172–180. [Google Scholar] [CrossRef]
- Wang, X.; Deng, G.; Yu, W.; Wang, B.; Li, X.; Liu, Y.; Zhao, Y.; Wang, Q.; Zhang, L.; Cai, X. A class of two-dimensional SiAs monolayers with novel electronic and optical properties from ab initio investigations. Eur. Phys. J. Plus 2019, 134, 287. [Google Scholar] [CrossRef]
- Shi, L.B.; Cao, S.; Yang, M. Strain behavior and carrier mobility for novel two-dimensional semiconductor of GeP: First principles calculations. Phys. E Low Dimens. Syst. Nanostruct. 2019, 107, 124–130. [Google Scholar] [CrossRef]
- Shojaei, F.; Hahn, J.R.; Kang, H.S. Electronic structure and photocatalytic band offset of few-layer GeP2. J. Mater. Chem. A 2017, 5, 22146–22155. [Google Scholar] [CrossRef]
- Mortazavi, H. Could art therapy reduce the death anxiety of patients with advanced cancer? An interesting question that deserves to be investigated. Indian J. Palliat. Care 2018, 24, 387–388. [Google Scholar] [CrossRef]
- Shojaei, F.; Kang, H.S. Electronic structures and Li-diffusion properties of group IV-V layered materials: Hexagonal germanium phosphide and germanium arsenide. J. Phys. Chem. C 2016, 120, 23842–23850. [Google Scholar] [CrossRef]
- Tabatabaeichehr, M.; Mortazavi, H.; Abadi, M.H.; Moayed, L. Sexual desire and related factors in middle-aged and elderly married women: A cross-sectional study in Iran. Open Access Maced. J. Med Sci. 2018, 6, 1906–1911. [Google Scholar] [CrossRef]
- Tagani, M.B. Electrical and mechanical properties of a fully hydrogenated two-dimensional polyaniline sheet. Comput. Mater. Sci. 2018, 153, 126–133. [Google Scholar] [CrossRef]
- Tagani, M.B.; Vishkayi, S.I. Polyaniline (C3N) nanoribbons: Magnetic metal, semiconductor, and half-metal. J. Appl. Phys. 2018, 124, 84304. [Google Scholar] [CrossRef]
- Vishkayi, S.I.; Tagani, M.B. Current-voltage characteristics of borophene and borophane sheets. Phys. Chem. Chem. Phys. 2017, 19, 21461–21466. [Google Scholar] [CrossRef]
- Bafekry, A.; Shayesteh, S.F.; Peeters, F.M. C3N monolayer: Exploring the emerging of novel electronic and magnetic properties with adatom adsorption, functionalizations, electric field, charging, and strain. J. Phys. Chem. C 2019, 123, 12485–12499. [Google Scholar] [CrossRef]
- Peng, Y.; Li, J.; Tang, X.; Yang, W.; Chen, X.; Fan, C.; Wang, K. Molecular dynamics study on the tribological properties of phosphorene/polyethylene composites. Coatings 2019, 9, 342. [Google Scholar] [CrossRef]
- Liu, T.; Qin, H.; Yang, D.; Zhang, G. First principles study of gas molecules adsorption on monolayered β-SnSe. Coatings 2019, 9, 390. [Google Scholar] [CrossRef]
- Bafekry, A.; Ghergherehchi, M.; Shayesteh, S.F.; Peeters, F. Adsorption of molecules on C3N nanosheet: A first-principle calculations. Chem. Phys. 2019, 526, 110442. [Google Scholar] [CrossRef]
- Shi, L.B.; Yang, M.; Cao, S.; You, Q.; Niu, Y.Y.; Wang, Y.Z. Elastic behavior and intrinsic carrier mobility for monolayer SnS and SnSe: First-principles calculations. Appl. Surf. Sci. 2019, 492, 435–448. [Google Scholar] [CrossRef]
- Makaremi, M.; Grixti, S.; Butler, K.T.; Ozin, G.A.; Singh, C.V. Band engineering of carbon nitride monolayers by N-Type, P-Type, and isoelectronic doping for photocatalytic applications. ACS Appl. Mater. Interfaces 2018, 10, 11143–11151. [Google Scholar] [CrossRef]
- Bafekry, A.; Ghergherehchi, M.; Shayesteh, S.F. Tuning the electronic and magnetic properties of antimonene nanosheets via point defects and external fields: First-principles calculations. Phys. Chem. Chem. Phys. 2019, 21, 10552–10566. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Shahrokhi, M. Quasi-particle energies and optical excitations of ZnS monolayer honeycomb structure. Appl. Surf. Sci. 2016, 390, 377–384. [Google Scholar] [CrossRef]
- Tachibana, Y.; Vayssieres, L.; Durrant, J.R. Artificial photosynthesis for solar water-splitting. Nat. Photonics 2012, 6, 511–518. [Google Scholar] [CrossRef]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Kreher, K. Fundamentals of semiconductors—physics and materials properties. Z. Phys. Chem. 1997, 198, 275. [Google Scholar] [CrossRef]
- Seitz, L.C.; Chen, Z.; Forman, A.J.; Pinaud, B.A.; Benck, J.D.; Jaramillo, T.F. Modeling practical performance limits of photoelectrochemical water splitting based on the current state of materials research. ChemSusChem 2014, 7, 1372–1385. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Zhou, L.; Shinde, A.; Suram, S.K.; Montoya, J.H.; Winston, D.; Gregoire, J.M.; Persson, K.A. Electrochemical stability of metastable materials. Chem. Mater. 2017, 29, 10159–10167. [Google Scholar] [CrossRef]
- Jain, A.; Wang, Z.; Nørskov, J.K. Stable two-dimensional materials for oxygen reduction and oxygen evolution reactions. ACS Energy Lett. 2019, 4, 1410–1411. [Google Scholar] [CrossRef]
- Gubaev, K.; Podryabinkin, E.V.; Hart, G.L.; Shapeev, A.V. Accelerating high-throughput searches for new alloys with active learning of interatomic potentials. Comput. Mater. Sci. 2019, 156, 148–156. [Google Scholar] [CrossRef]
- Kostiuchenko, T.; Körmann, F.; Neugebauer, J.; Shapeev, A. Impact of lattice relaxations on phase transitions in a high-entropy alloy studied by machine-learning potentials. Npj Comput. Mater. 2019, 5, 55. [Google Scholar] [CrossRef]
- Podryabinkin, E.V.; Tikhonov, E.V.; Shapeev, A.V.; Oganov, A.R. Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning. Phys. Rev. B 2019, 99, 064114. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortazavi, B.; Shahrokhi, M.; Cuniberti, G.; Zhuang, X. Two-Dimensional SiP, SiAs, GeP and GeAs as Promising Candidates for Photocatalytic Applications. Coatings 2019, 9, 522. https://doi.org/10.3390/coatings9080522
Mortazavi B, Shahrokhi M, Cuniberti G, Zhuang X. Two-Dimensional SiP, SiAs, GeP and GeAs as Promising Candidates for Photocatalytic Applications. Coatings. 2019; 9(8):522. https://doi.org/10.3390/coatings9080522
Chicago/Turabian StyleMortazavi, Bohayra, Masoud Shahrokhi, Gianaurelio Cuniberti, and Xiaoying Zhuang. 2019. "Two-Dimensional SiP, SiAs, GeP and GeAs as Promising Candidates for Photocatalytic Applications" Coatings 9, no. 8: 522. https://doi.org/10.3390/coatings9080522
APA StyleMortazavi, B., Shahrokhi, M., Cuniberti, G., & Zhuang, X. (2019). Two-Dimensional SiP, SiAs, GeP and GeAs as Promising Candidates for Photocatalytic Applications. Coatings, 9(8), 522. https://doi.org/10.3390/coatings9080522