Corrosion Potential Modulation on Lead Anodes Using Water Oxidation Catalyst Coatings


Abstract
1. Introduction
2. Materials and Methods
3. Results
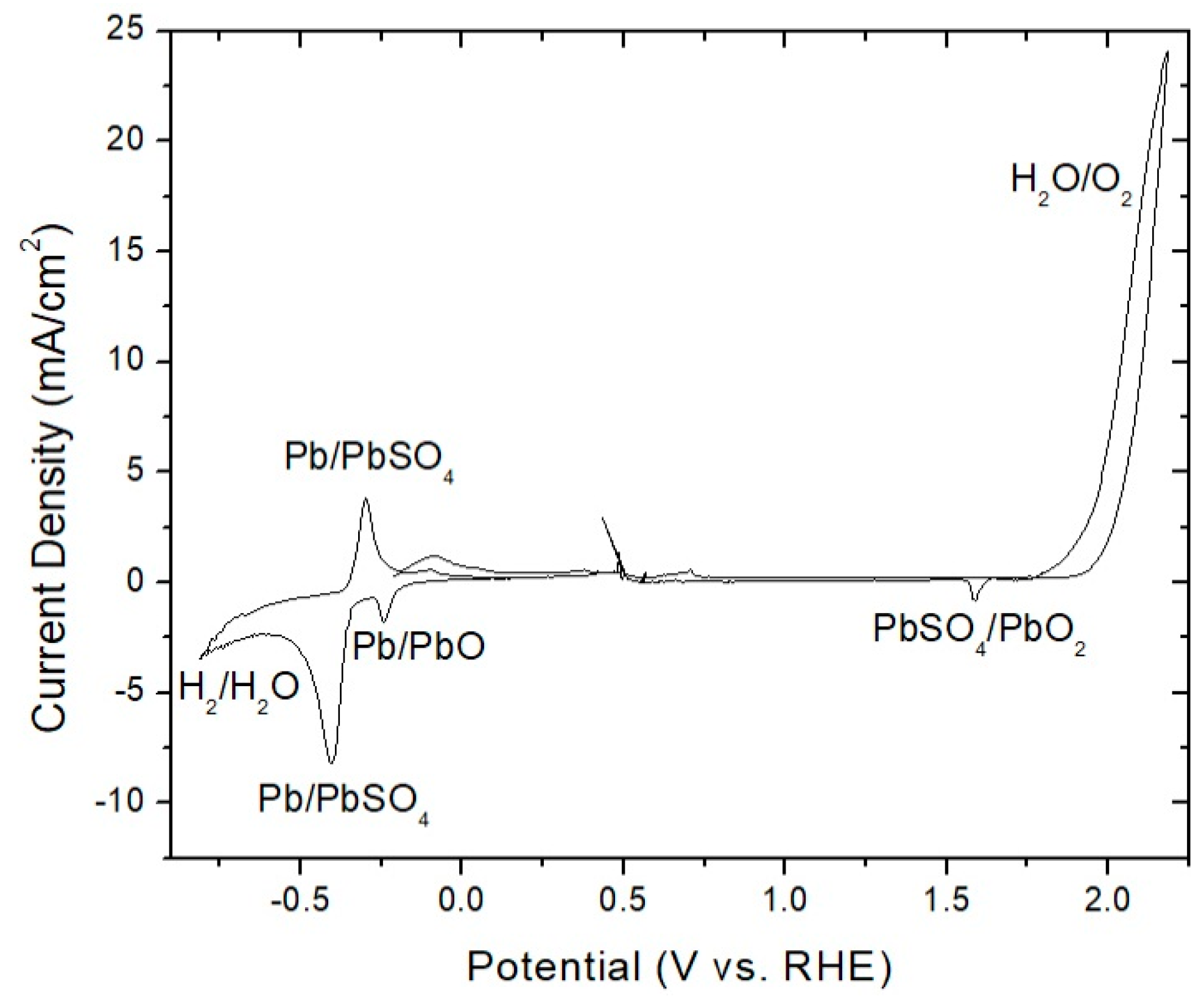
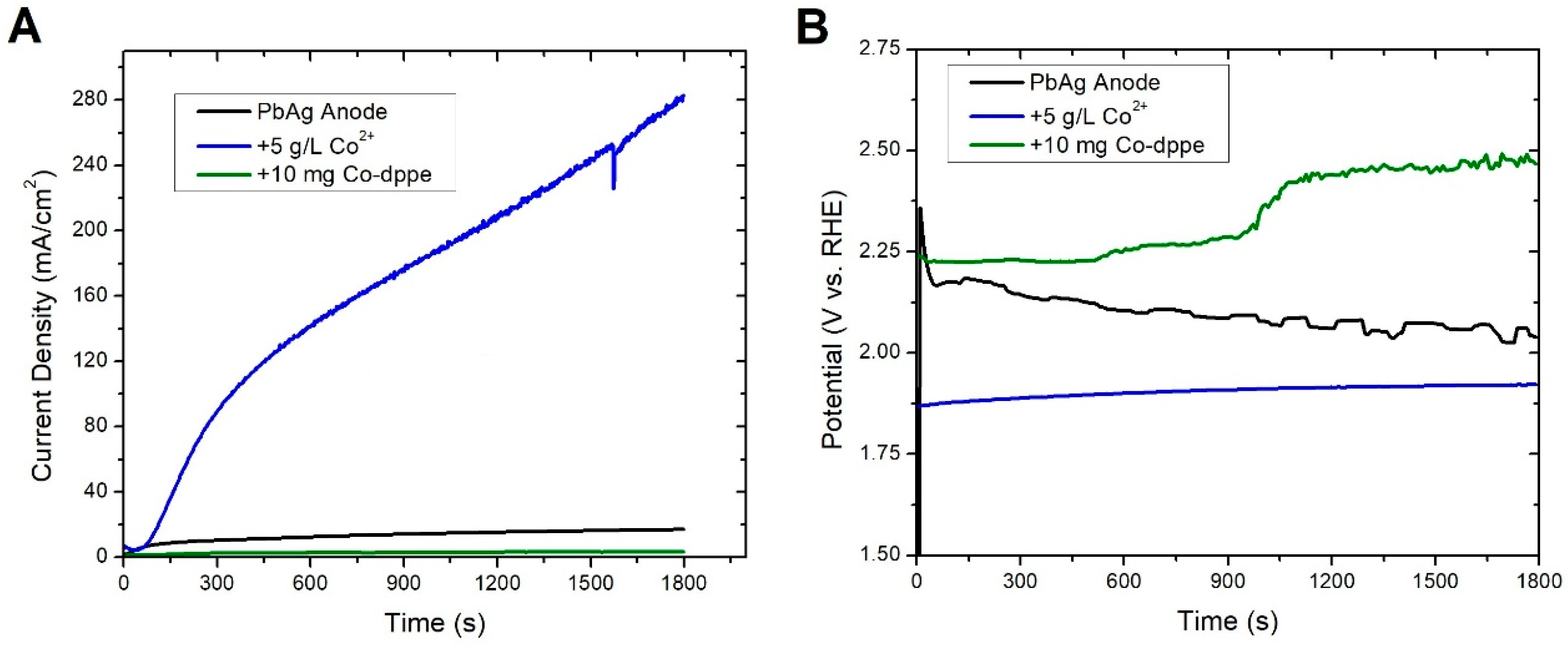
3.1. Pb Anodes
- Pre-electrolysis: The electrode surface upon immersion into 1.6 M H2SO4, prior to electrolysis. We observed that this state persists for days, with an Ecorr on an unmodified PbAg surface of −0.25 V vs. RHE.
- Highly polarized: This state is accessed by measuring OCP immediately after sufficient electrode polarization for the formation of oxygen bubbles. It possesses a short lifetime (<1 h) and exhibits the same Ecorr between trials independent of Co2+/Mn2+/Co-dppe presence or concentration. These attributes suggest that it is indicative of a charged silver species, as proposed previously, with an Ecorr of approximately 1.69 V vs. RHE.
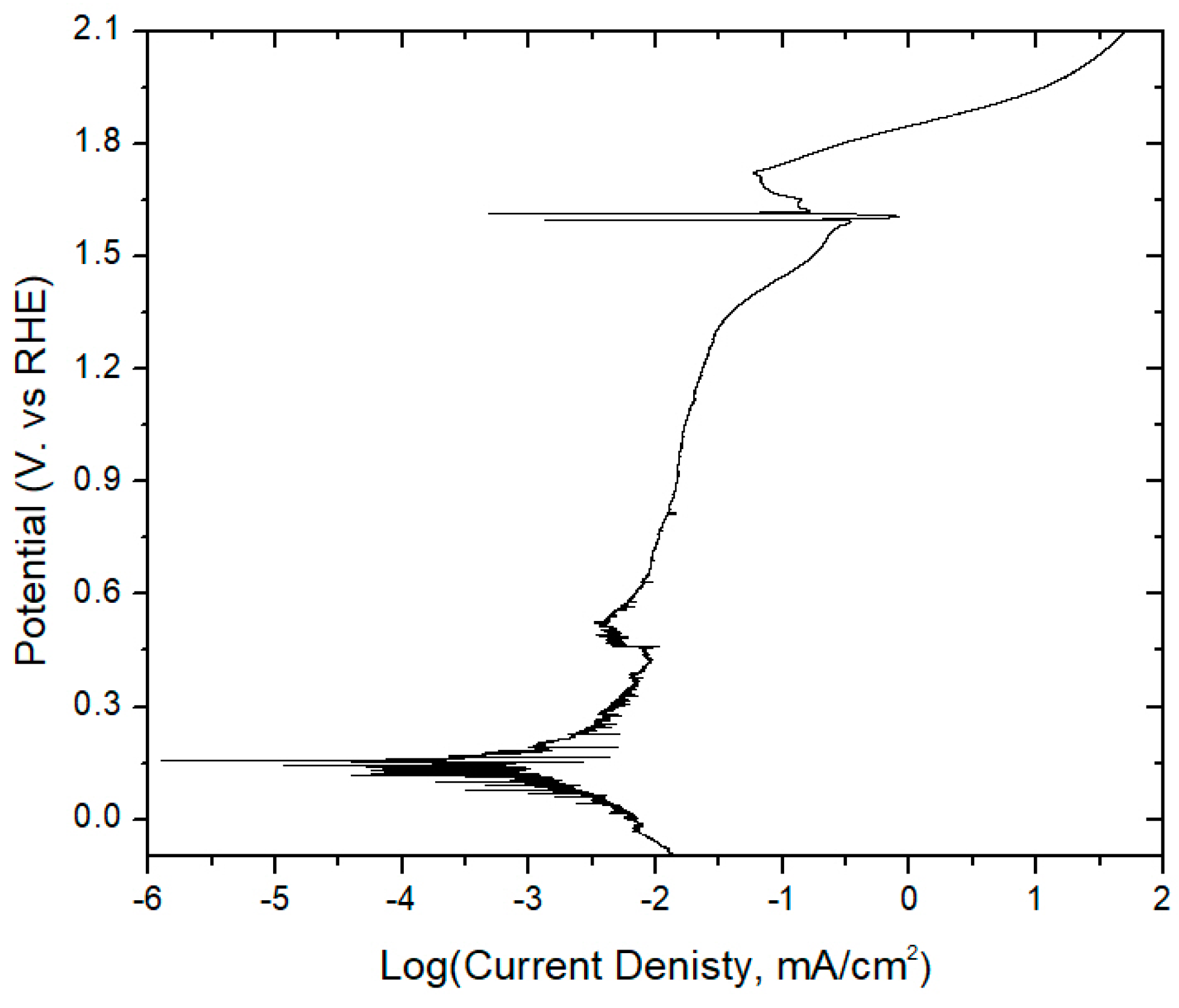
- Polarized dynamic equilibrium: This equilibrium state is defined as the OCP having the lowest measured current reached during potentiodynamic polarization (0.14 V vs. RHE for unmodified PbAg; see Table S1 and Figure S1) immediately after electrolysis and varies dependent on the electrode surface coating (Co2+/Mn2+/Co-dppe).
- Resting equilibrium: This state corresponds to the second OCP reached during potentiodynamic polarization; the same value is also reached if the electrode is left sitting in the H2SO4 electrolyte for a day after electrolysis (0.01 V vs. RHE for unmodified PbAg). Similar to the polarized equilibrium potential, this OCP varies dependent on the electrode surface coating or formation of a dense mixed oxide layer.
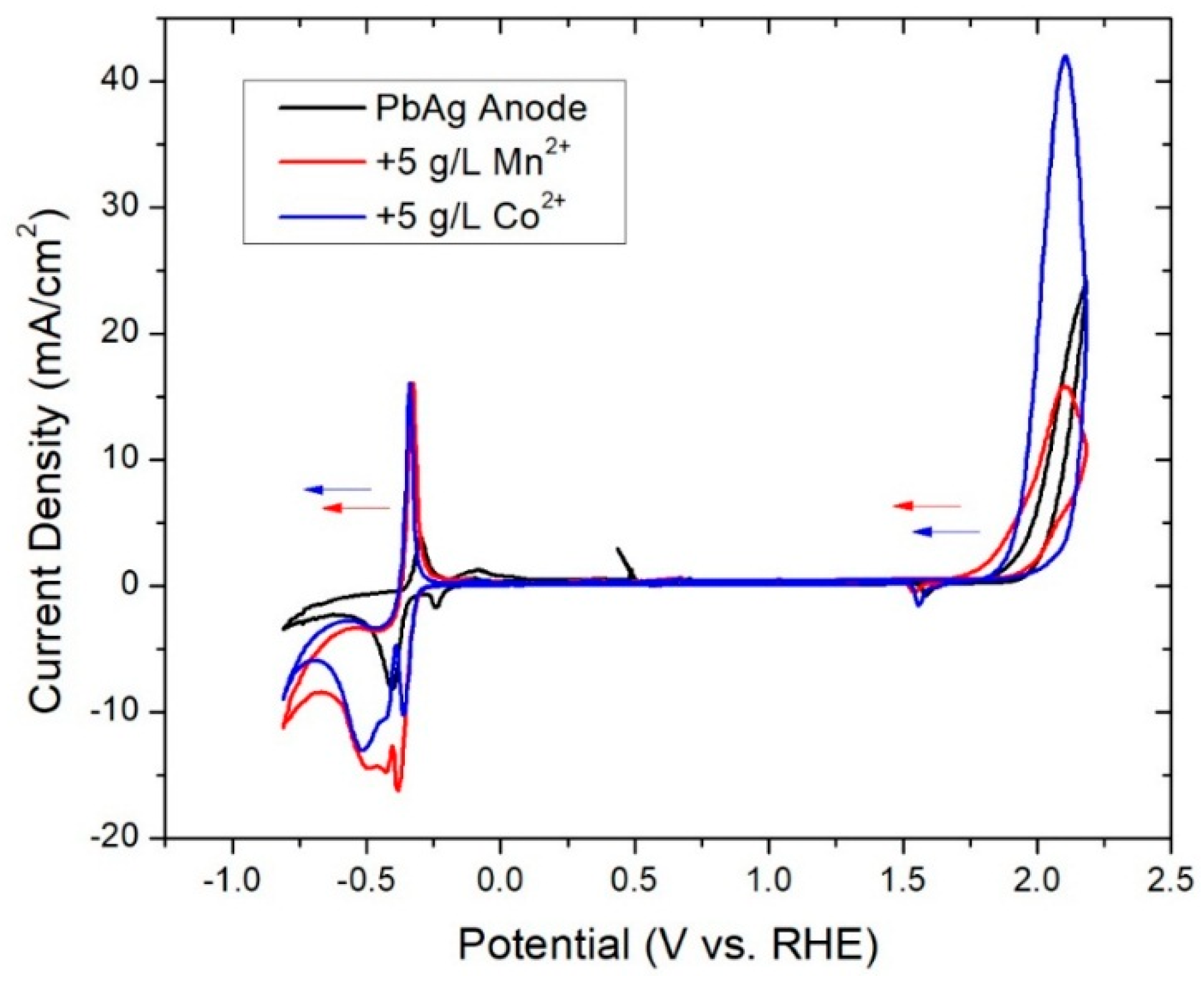
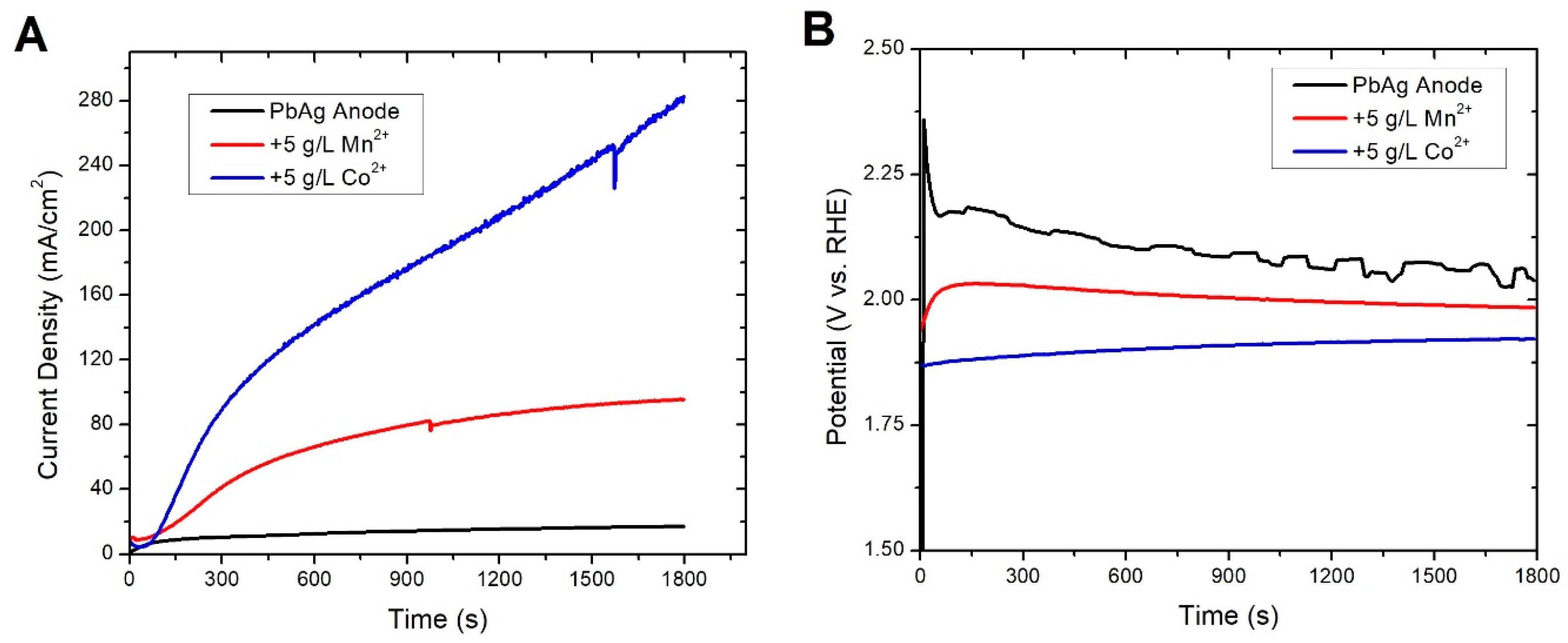
3.2. In-Situ Coatings
3.3. Ex-Situ Coating
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hunter, B.M.; Gray, H.B.; Muller, A.M. Earth-Abundant Heterogeneous Water Oxidation Catalysts. Chem. Rev. 2016, 116, 14120–14136. [Google Scholar] [CrossRef] [PubMed]
- Clancy, M.; Bettles, C.J.; Stuart, A.; Birbilis, N. The influence of alloying elements on the electrochemistry of lead anodes for electrowinning of metals: A review. Hydrometallurgy 2013, 131–132, 144–157. [Google Scholar] [CrossRef]
- Sheehan, S.W.; Cave, E.R.; Kuhl, K.P.; Flanders, N.; Smeigh, A.L.; Co, D.T. Commercializing Solar Fuels within Today’s Markets. Chem 2017, 3, 3–7. [Google Scholar] [CrossRef]
- Li, Y.; Gong, M.; Liang, Y.; Feng, J.; Kim, J.E.; Wang, H.; Hong, G.; Zhang, B.; Dai, H. Advanced zinc-air batteries based on high-performance hybrid electrocatalysts. Nat. Commun. 2013, 4, 1805. [Google Scholar] [CrossRef] [PubMed]
- Winand, R. Electrodeposition of metals and alloys—New results and perspectives. Electrochim. Acta 1994, 39, 1091–1105. [Google Scholar] [CrossRef]
- Tunnicliffe, M.; Mohammadi, F.; Alfantazi, A. Polarization Behavior of Lead-Silver Anodes in Zinc Electrowinning Electrolytes. J. Electrochem. Soc. 2012, 159, C170–C180. [Google Scholar] [CrossRef]
- Zhang, W.; Houlachi, G. Electrochemical studies of the performance of different Pb–Ag anodes during and after zinc electrowinning. Hydrometallurgy 2010, 104, 129–135. [Google Scholar] [CrossRef]
- McGinnity, J.J.; Nicol, M.J. The role of silver in enhancing the electrochemical activity of lead and lead-silver alloy anodes. Hydrometallurgy 2014, 144–145, 133–139. [Google Scholar] [CrossRef]
- Mohammadi, M.; Alfantazi, A. The performance of Pb-MnO2 and Pb-Ag anodes in 2 Mn(II)-containing sulphuric acid electrolyte solutions. Hydrometallurgy 2015, 153, 134–144. [Google Scholar] [CrossRef]
- Cachet, C.; Le Pape-Rerolle, C.; Wiart, R. Influence of Co2+ and Mn2+ ions on the kinetics of lead anodes for zinc electrowinning. J. Appl. Electrochem. 1999, 29, 813–820. [Google Scholar] [CrossRef]
- Bloomfield, A.J.; Sheehan, S.W.; Collom, S.L.; Crabtree, R.H.; Anastas, P.T. A heterogeneous water oxidation catalyst from dicobalt octacarbonyl and 1, 2-bis(diphenylphosphino) ethane. New J. Chem. 2014, 38, 1540–1545. [Google Scholar] [CrossRef]
- Bloomfield, A.J.; Sheehan, S.W.; Collom, S.L.; Anastas, P.T. Performance Enhancement for Electrolytic Systems through the Application of a Cobalt-based Heterogeneous Water Oxidation Catalyst. ACS Sustain. Chem. Eng. 2015, 3, 1234–1240. [Google Scholar] [CrossRef]
- Ivanov, I.; Stefanov, Y.; Noncheva, Z.; Petrova, M.; Dobrev, T.; Mirkova, L.; Vermeersch, R.; Demaerel, J.P. Insoluble anodes used in hydrometallurgy: Part I. Corrosion resistance of lead and lead alloy anodes. Hydrometallurgy 2000, 57, 109–124. [Google Scholar] [CrossRef]
- Carr, J.P.; Hampson, N.A. The Lead Dioxide Electrode. Chem. Rev. 1972, 72, 679–703. [Google Scholar] [CrossRef]
- Takehara, Z.-I. Dissolution and precipitation reactions of lead sulfate in positive and negative electrodes in lead acid battery. J. Power Sources 2000, 85, 29–37. [Google Scholar] [CrossRef]
- Delahy, P.; Pourbaix, M.; Van Rysselberghe, P. Potential-pH Diagram of Lead and its Applications to the Study of Lead Corrosion and to the Lead Storage Battery. J. Electrochem. Soc. 1951, 98, 57–64. [Google Scholar] [CrossRef]
- Koshel, N.D.; Gerasika, N.S.; Kostyrya, M.V. Nonstationary processes that occur on nonpolarizable lead surface in sulfuric acid. Surf. Eng. Appl. Electrochem. 2016, 52, 23–31. [Google Scholar] [CrossRef]
- Vanysek, P. Electrochemical Series. In Handbook of Chemistry and Physics, 3rd ed.; Haynes, W.M., Ed.; CRC Press: Boca Riton, FL, USA, 2012. [Google Scholar]
- Lai, Y.Q.; Li, Y.; Jiang, L.X.; Lv, X.J.; Li, J.; Liu, Y.X. Electrochemical performance of a Pb/Pb-MnO2 composite anode in sulfuric acid solution containing Mn2+. Hydrometallurgy 2012, 115–116, 64–70. [Google Scholar] [CrossRef]
- Newnham, R.H. Corrosion rates of lead based anodes for zinc electrowinning at high current densities. J. Appl. Electrochem. 1992, 22, 116–124. [Google Scholar] [CrossRef]
- Nikoloski, A.N.; Nicol, M.J. Addition of Cobalt to Lead Anodes used for Oxygen Evolution—A Literature Review. Miner. Process. Extr. Metall. Rev. 2009, 31, 30–57. [Google Scholar] [CrossRef]
- Zhang, W.; Ghali, E.; Houlachi, G. Performance of Pb-Ag anodes during polarisation and decay periods in zinc electrowinning. Can. Metall. Q. 2013, 52, 123–131. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, L.X.; Lv, X.J.; Lai, Y.Q.; Zhang, H.L.; Li, J.; Liu, Y.X. Oxygen evolution and corrosion behaviors of co-deposited Pb/Pb-MnO2 composite anode for electrowinning of nonferrous metals. Hydrometallurgy 2011, 109, 252–257. [Google Scholar] [CrossRef]
- Mondschein, J.S.; Callejas, J.F.; Read, C.G.; Chen, J.Y.C.; Holder, C.F.; Badding, C.K.; Schaak, R.E. Crystalline Cobalt Oxide Films for Sustained Electrocatalytic Oxygen Evolution under Strongly Acidic Conditions. Chem. Mater. 2017, 29, 950–957. [Google Scholar] [CrossRef]
- Meruva, R.K.; Meyerhoff, M.E. Mixed Potential Response Mechanism of Cobalt Electrodes toward Inorganic Phosphate. Anal. Chem. 1996, 68, 2022–2026. [Google Scholar] [CrossRef] [PubMed]
- Olesen, B.H.; Avci, R.; Lewandowski, Z. Manganese dioxide as a potential cathodic reactant in corrosion of stainless steels. Corros. Sci. 2000, 42, 211–227. [Google Scholar] [CrossRef]
- Fukushima, S. Studies on Electrolytic Refining of Zinc. VII Effects of Manganese on Corrosion of Lead Anode. Sci. Rep. Res. Inst. Tohoku Univ. Ser. A 1960, 12, 456–468. [Google Scholar]
- Kulandaisamy, S.; Rethinaraj, J.P.; Shockalingam, S.C.; Visvanathan, S.; Venkateswaran, K.V.; Ramachandran, P.; Nandakumar, V. Performance of catalytic activated anodes in the electrowinning of metals. J. Appl. Electrochem. 1997, 27, 579–583. [Google Scholar] [CrossRef]
- Chen, C.; Bloomfield, A.J.; Sheehan, S.W. Selective Electrochemical Oxidation of Lactic Acid Using Iridium-Based Catalysts. Ind. Eng. Chem. Res. 2017, 56, 3560–3567. [Google Scholar] [CrossRef]
- Goberna-Ferrón, S.; Soriano-López, J.; Galán-Mascarós, J.R. Activity and Stability of the Tetramanganese Polyanion [Mn4(H2O)2(PW9O34)2]10− during Electrocatalytic Water Oxidation. Inorganics 2015, 3, 332–340. [Google Scholar] [CrossRef]
- Li, J.; Güttinger, R.; Moré, R.; Song, F.; Wan, W.; Patzke, G.R. Frontiers of water oxidation: The quest for true catalysts. Chem. Soc. Rev. 2017, 46, 6124–6147. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Frei, H. Nanostructured cobalt and manganese oxide clusters as efficient water oxidation catalysts. Energy Environ. Sci. 2010, 3, 1018–1027. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution Additive | Pre-Electrolysis Ecorr (V) | Highly Polarized Ecorr (V) | Polarized Eq. Ecorr (V) | Resting Eq. Ecorr (V) |
|---|---|---|---|---|
| None | −0.26 | 1.69 | 0.14 | 0.01 |
| 5 g·L−1 Co2+ | −0.24 | 1.69 | 1.60 | 0.04 |
| 5 g·L−1 Mn2+ | −0.21 | 1.69 | 0.99 | 0.12 |
| Solution Additive | Pb/PbSO4 E1/2 (V) | Pb/PbO EPb/PbO (V) | PbSO4/PbO2 EPbSO4/PbO2 (V) | OER Onset EOER (V) |
|---|---|---|---|---|
| None | −0.31 | −0.24 | 1.59 | 1.92 |
| 5 g·L−1 Co2+ | −0.50 | −0.36 | 1.56 | 1.89 |
| 5 g·L−1 Mn2+ | −0.46 | −0.32 | 1.57 | 1.82 |
| Solution Additive | Pre-Electrolysis Ecorr (V) | Highly Polarized Ecorr (V) | Polarized Eq. Ecorr (V) | Resting Eq. Ecorr (V) |
|---|---|---|---|---|
| None | −0.26 | 1.69 | 0.14 | 0.01 |
| Co-dppe | −0.11 | 1.69 | 0.01 | −0.06 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotyk, J.F.K.; Chen, C.; Sheehan, S.W. Corrosion Potential Modulation on Lead Anodes Using Water Oxidation Catalyst Coatings. Coatings 2018, 8, 246. https://doi.org/10.3390/coatings8070246
Kotyk JFK, Chen C, Sheehan SW. Corrosion Potential Modulation on Lead Anodes Using Water Oxidation Catalyst Coatings. Coatings. 2018; 8(7):246. https://doi.org/10.3390/coatings8070246
Chicago/Turabian StyleKotyk, Juliet F. Khosrowabadi, Chi Chen, and Stafford W. Sheehan. 2018. "Corrosion Potential Modulation on Lead Anodes Using Water Oxidation Catalyst Coatings" Coatings 8, no. 7: 246. https://doi.org/10.3390/coatings8070246
APA StyleKotyk, J. F. K., Chen, C., & Sheehan, S. W. (2018). Corrosion Potential Modulation on Lead Anodes Using Water Oxidation Catalyst Coatings. Coatings, 8(7), 246. https://doi.org/10.3390/coatings8070246