Synthesis of Polydimethylsiloxane-Modified Polyurethane and the Structure and Properties of Its Antifouling Coatings

Abstract
:1. Introduction
2. Materials and Methods
2.1. Raw Materials
2.2. Synthesis of Polyurethane
2.3. Preparation of Coating Samples
2.4. Characterization
2.4.1. Structural Analysis
2.4.2. Mechanical Properties
2.4.3. Surface Properties
2.4.4 Antifouling Property
3. Results and Discussions
3.1. Influence of Polydimethylsiloxane (PDMS)
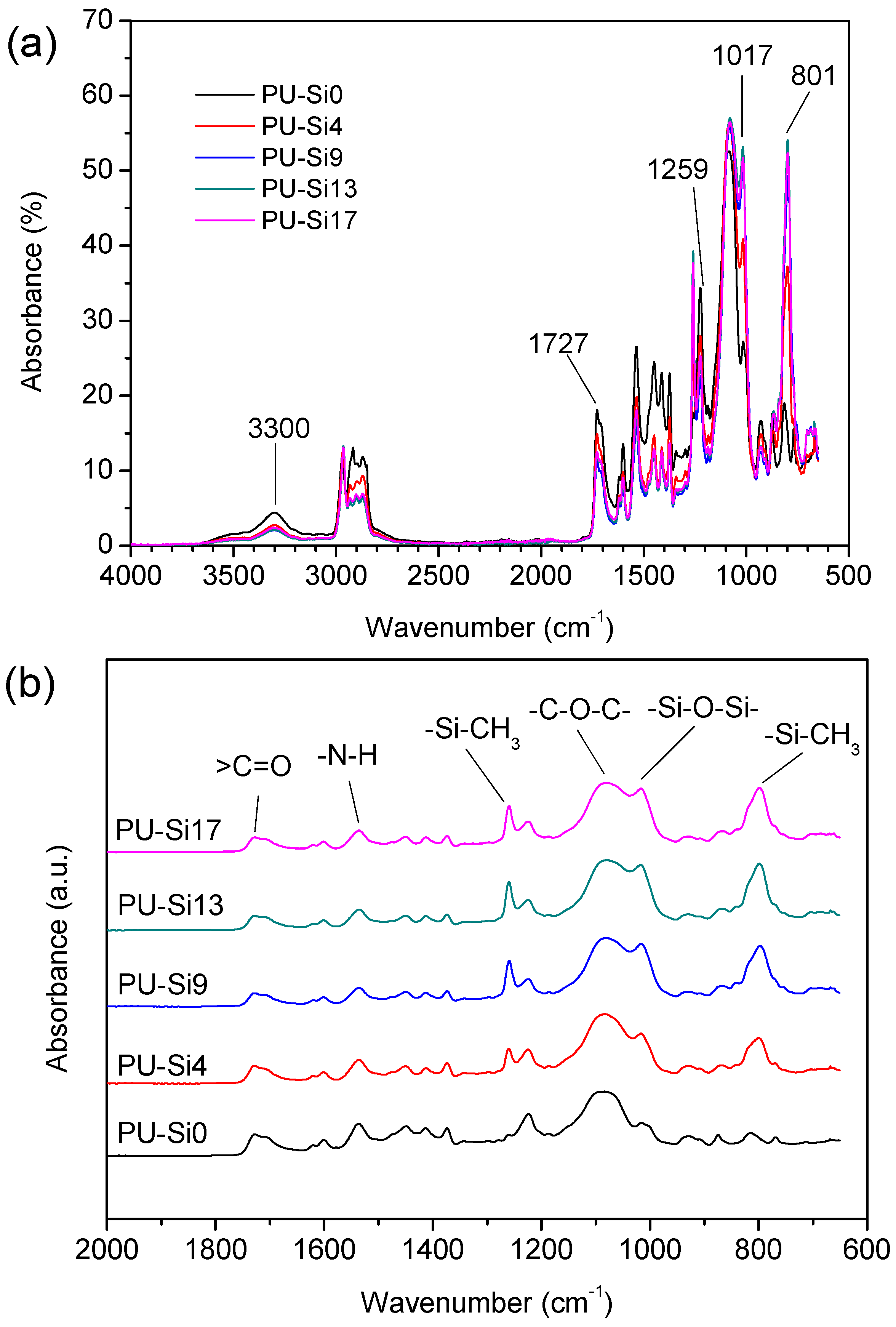
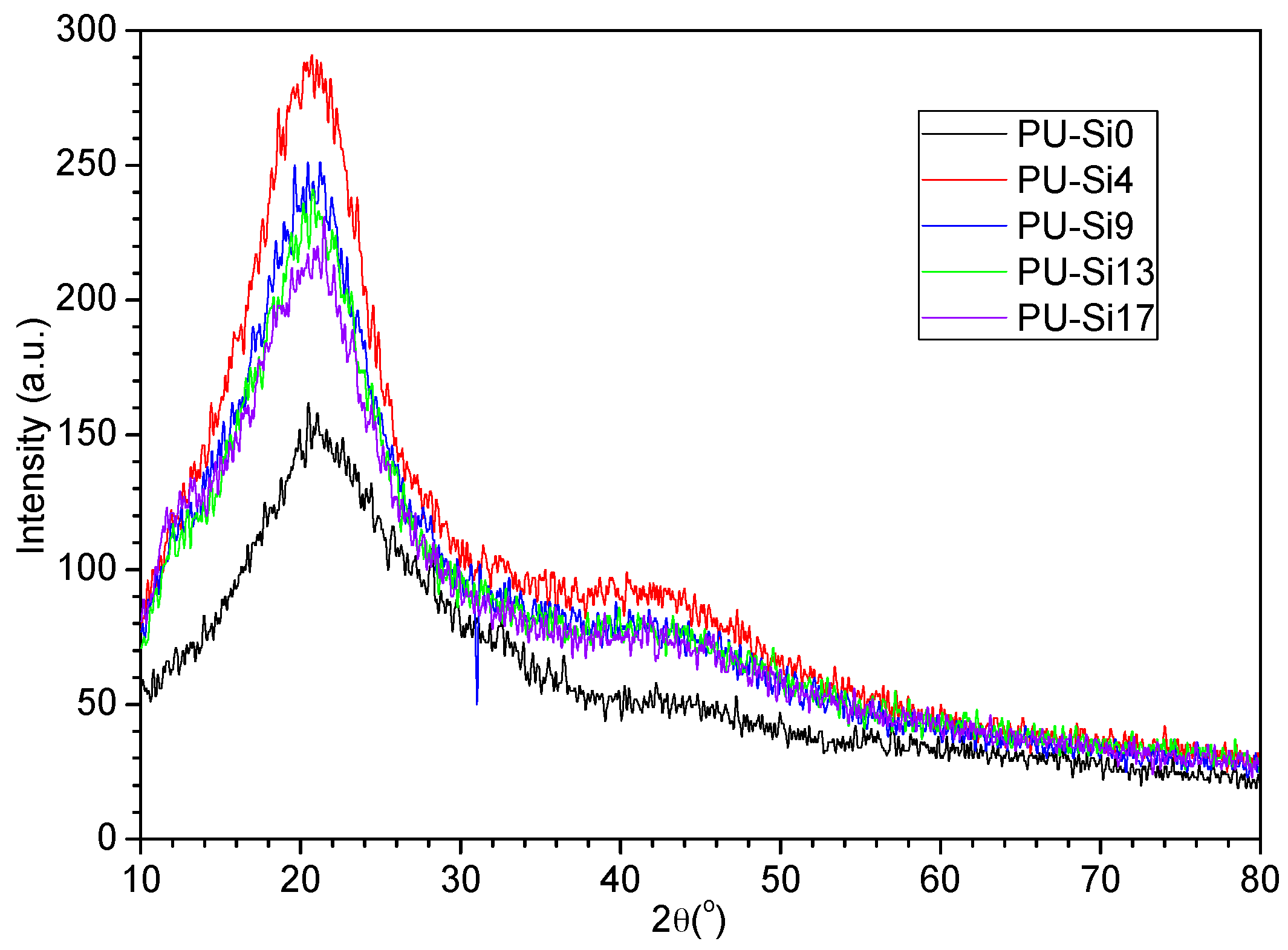
3.1.1. Molecular Structure
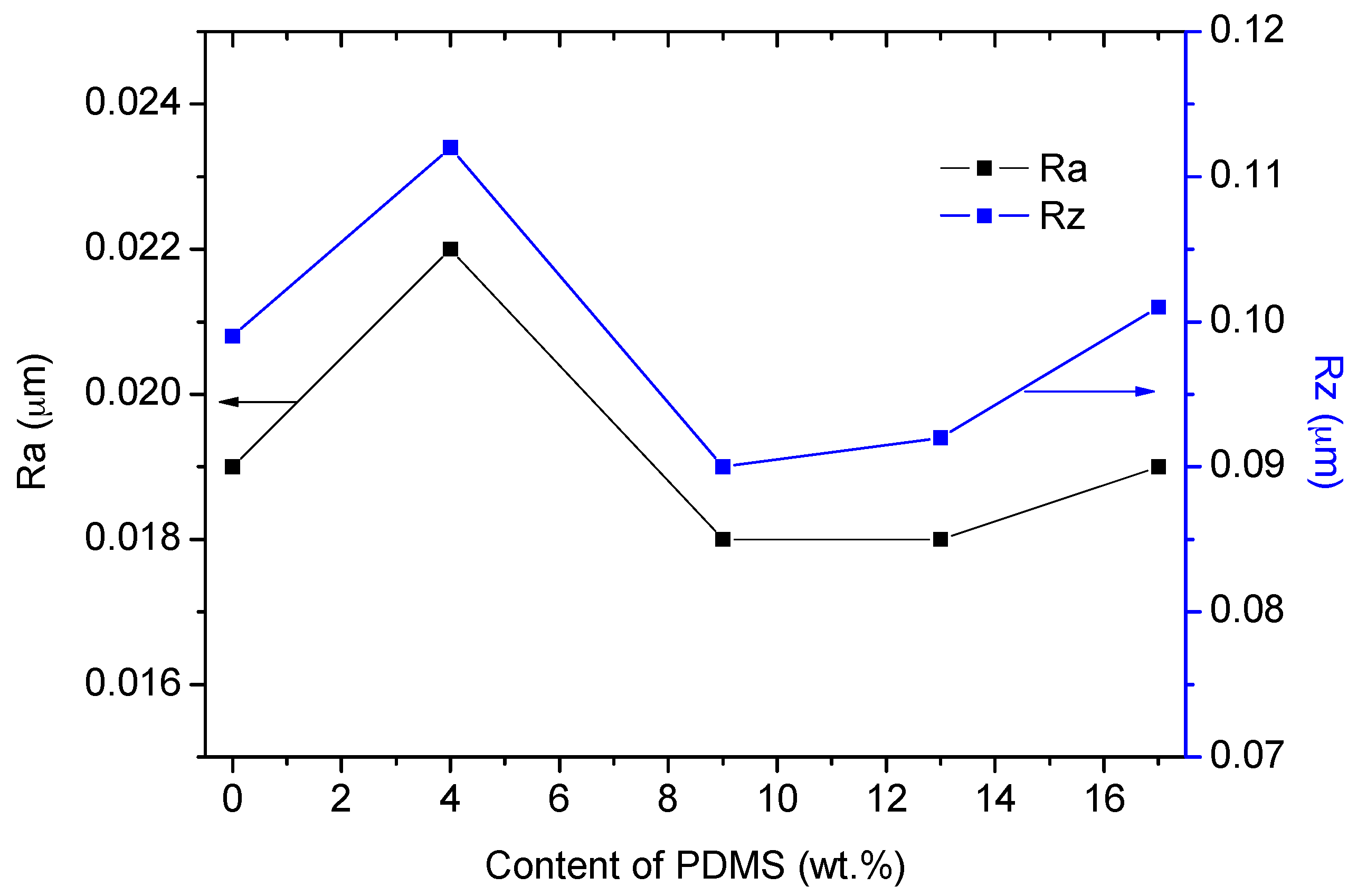
3.1.2. Surface Topography
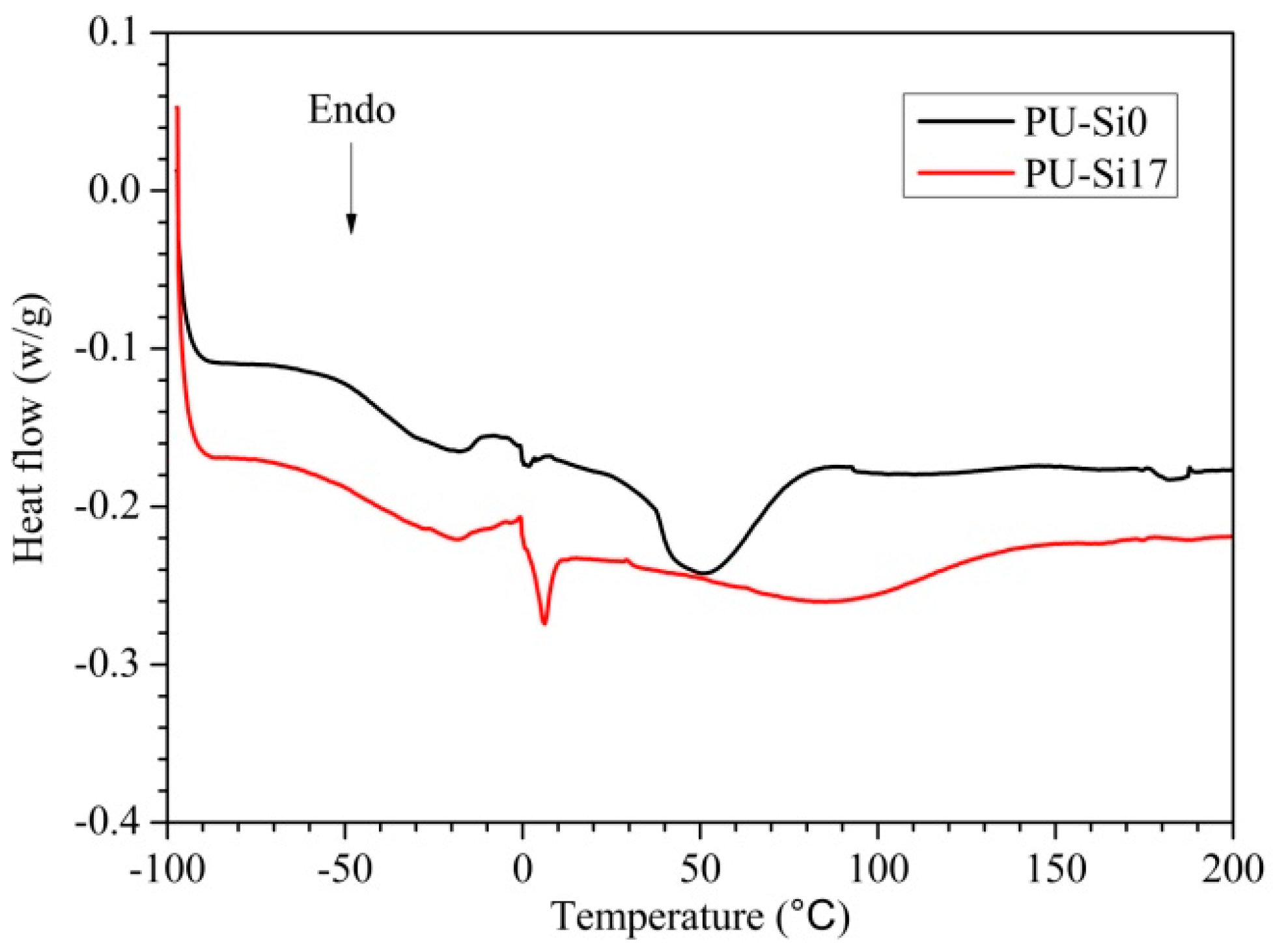
3.1.3. Mechanical Properties
3.1.4. Interface Characterization
3.2. Influence of Seawater Immersion
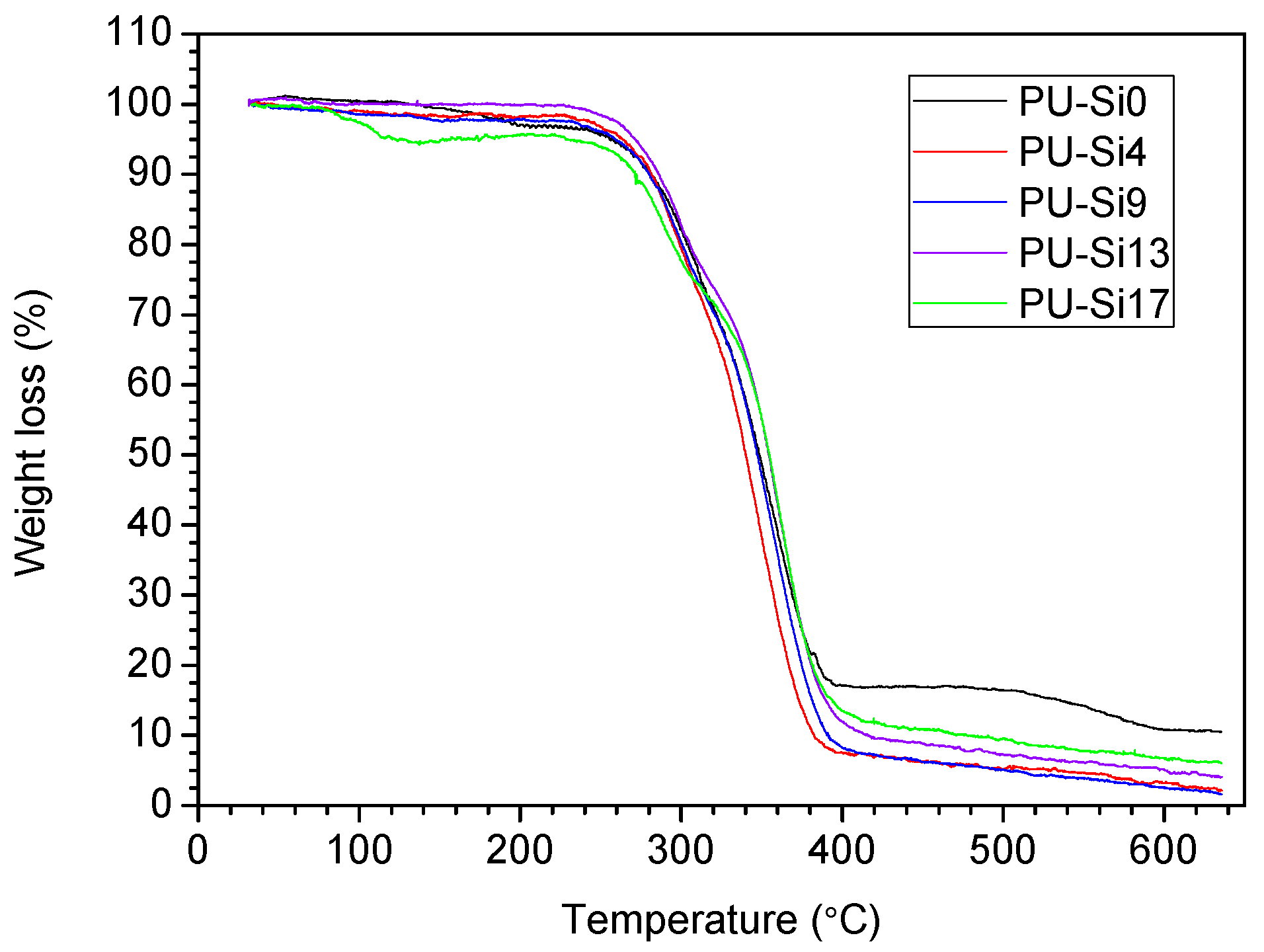
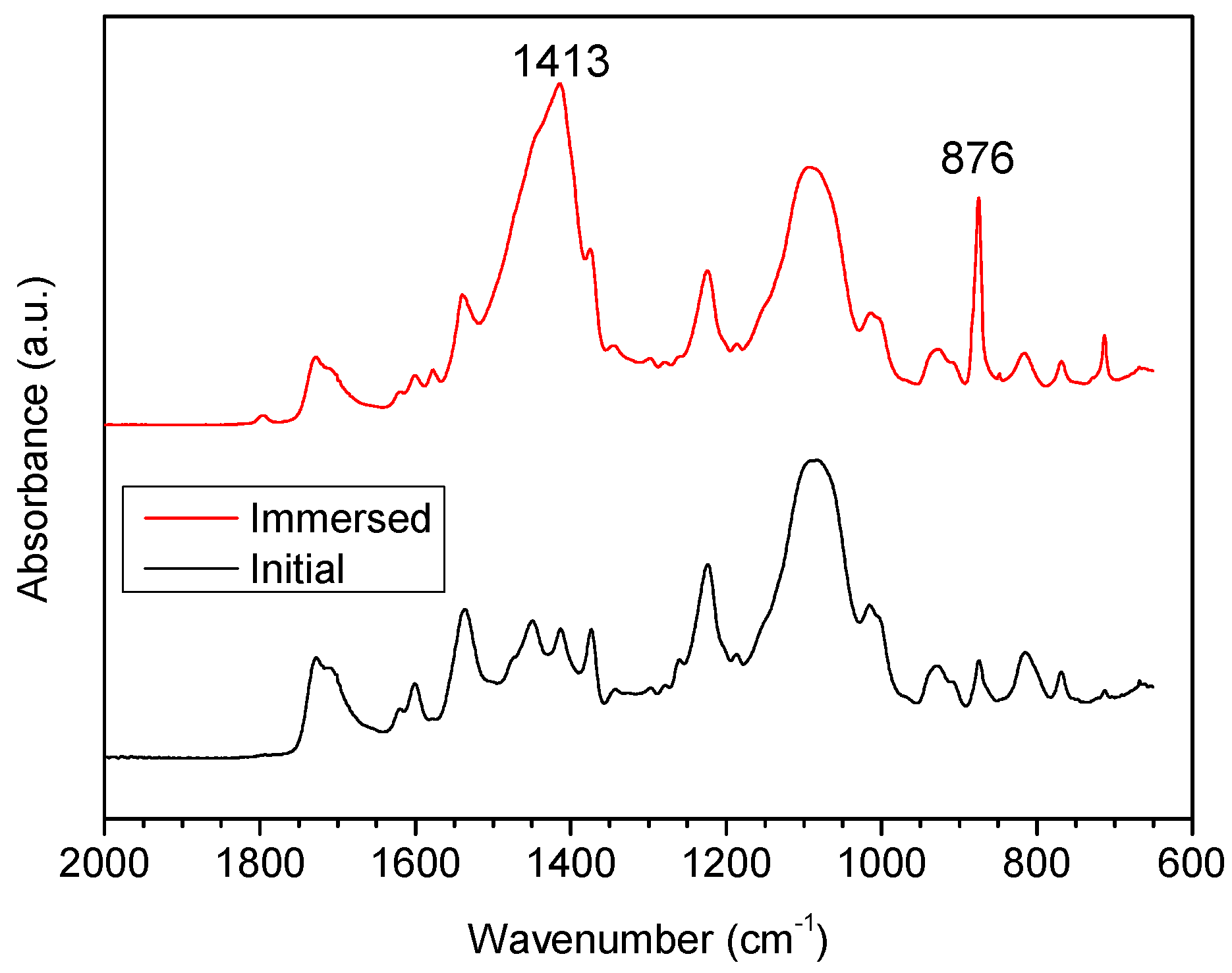
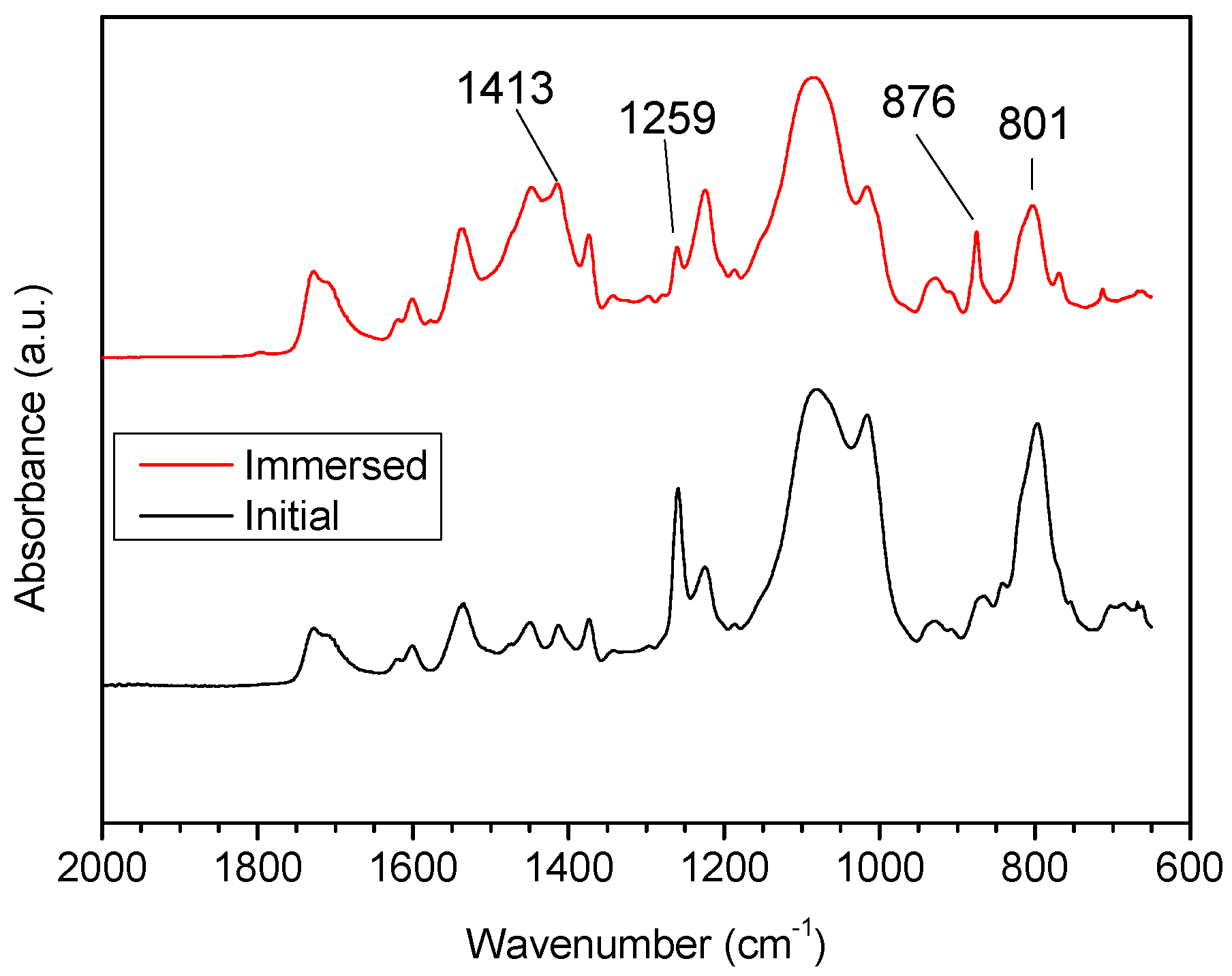
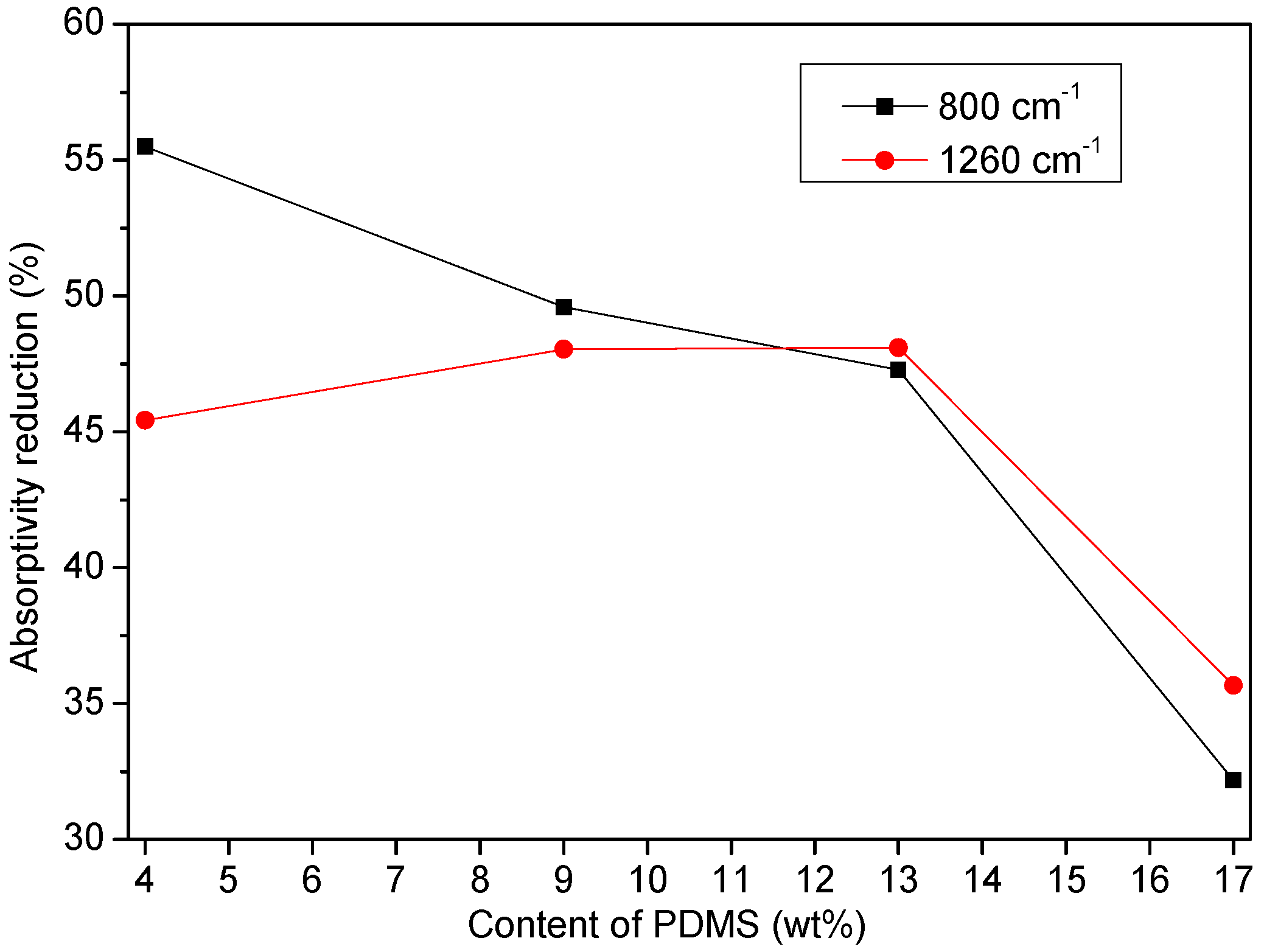
3.2.1. Fourier Transform Infrared (FTIR) Analysis
3.2.2. Atomic Force Microscopy (AFM) Analysis
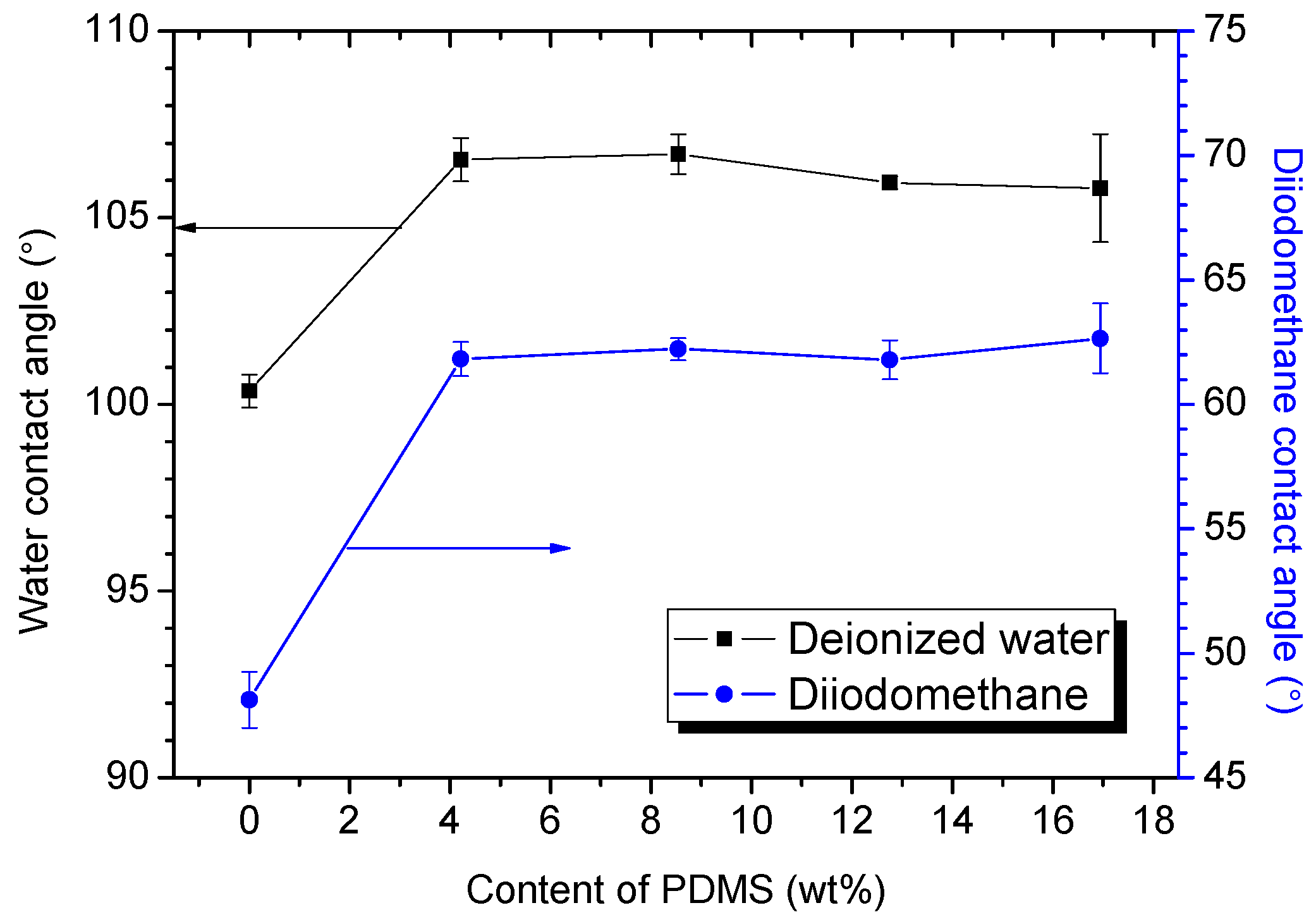
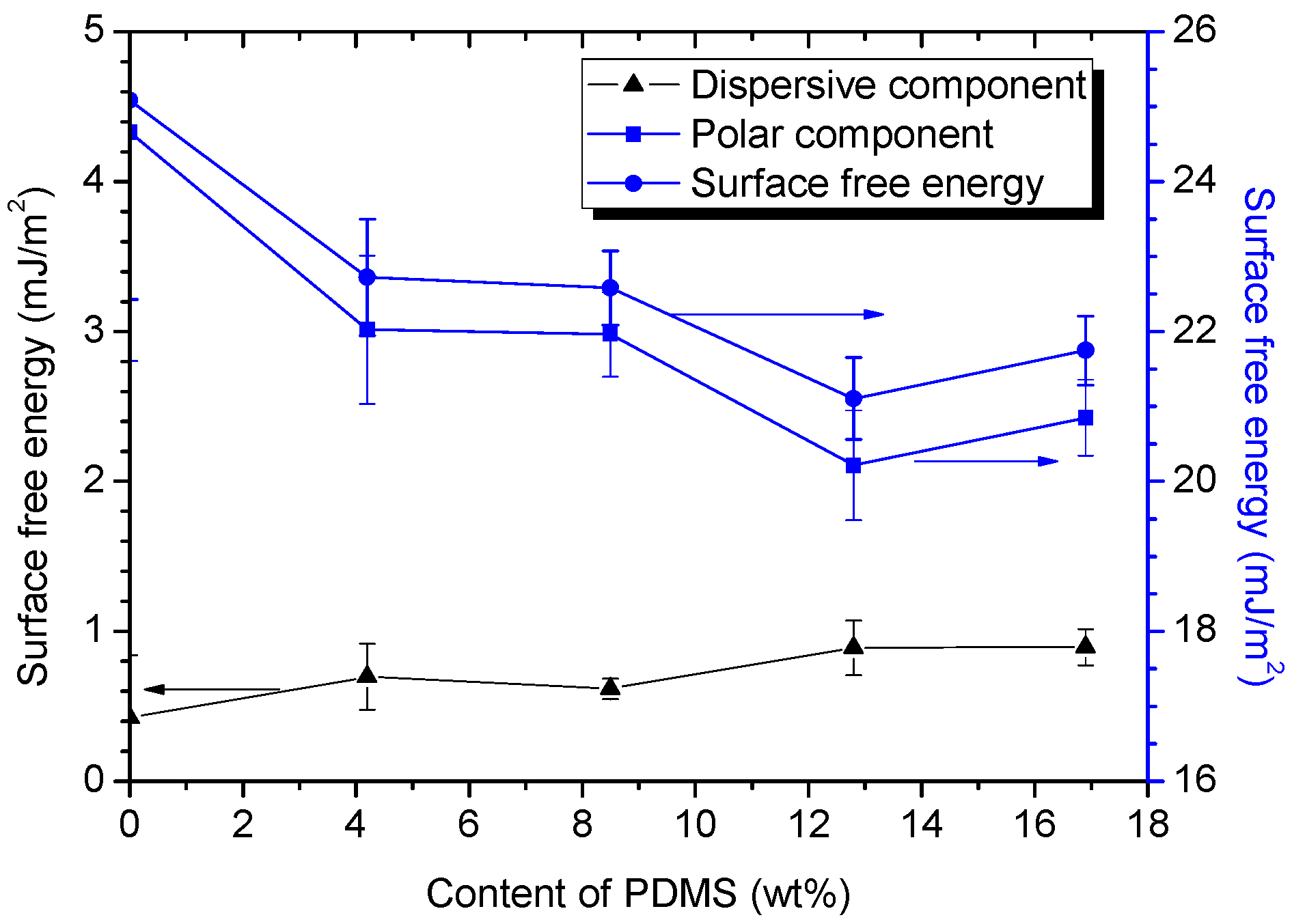
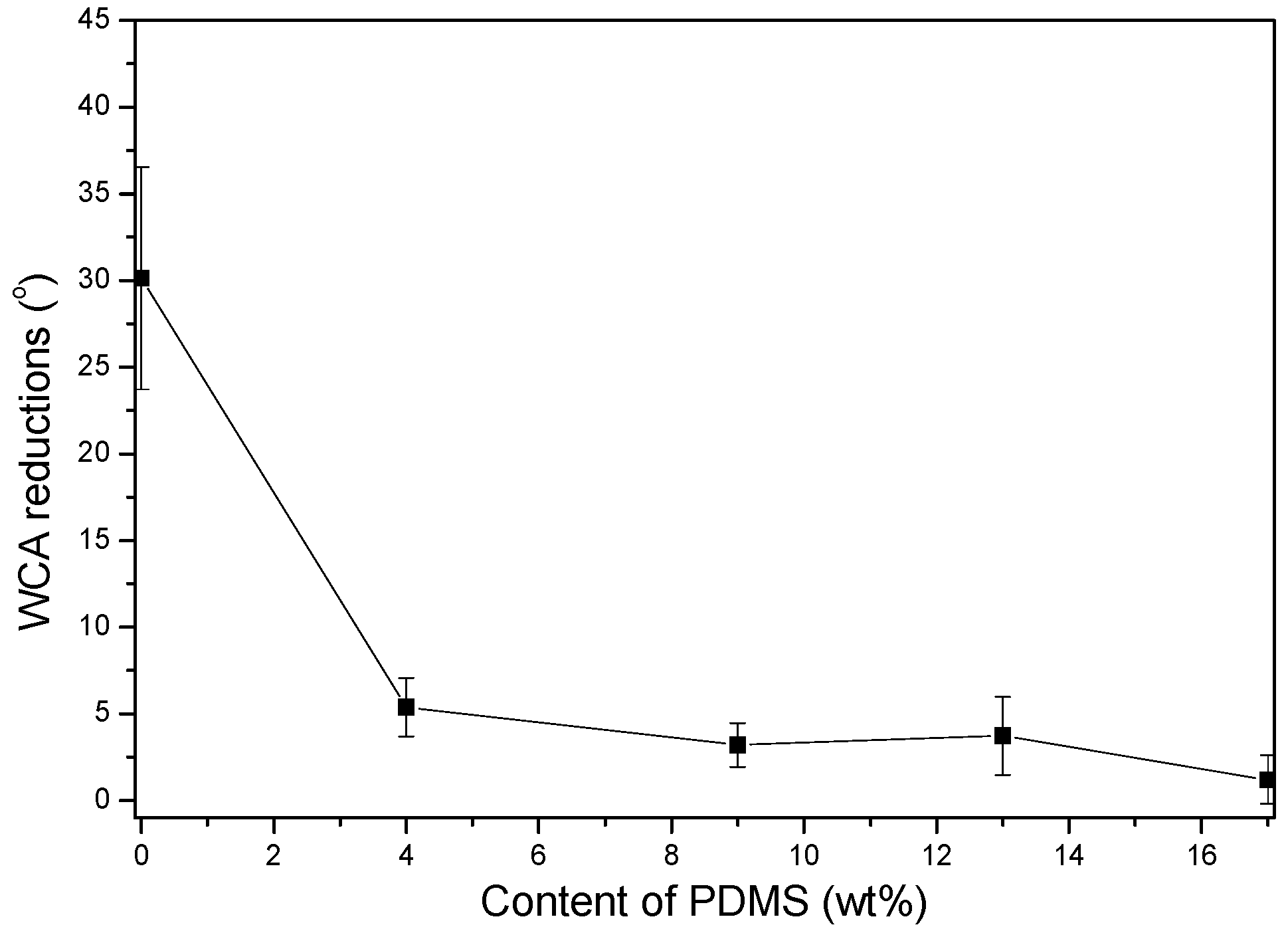
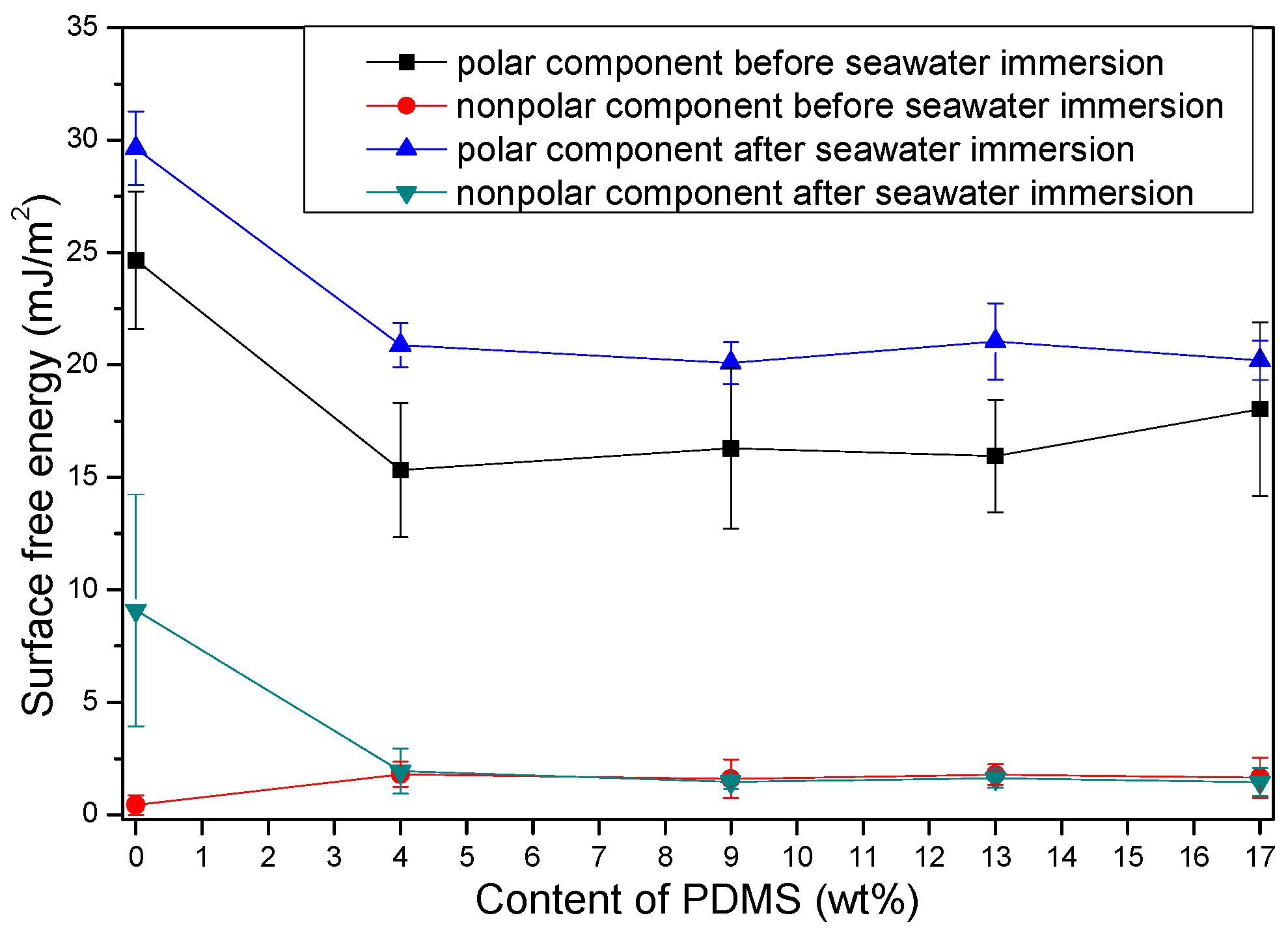
3.2.3. Water Contact Angle (WCA) and Surface Free Energy
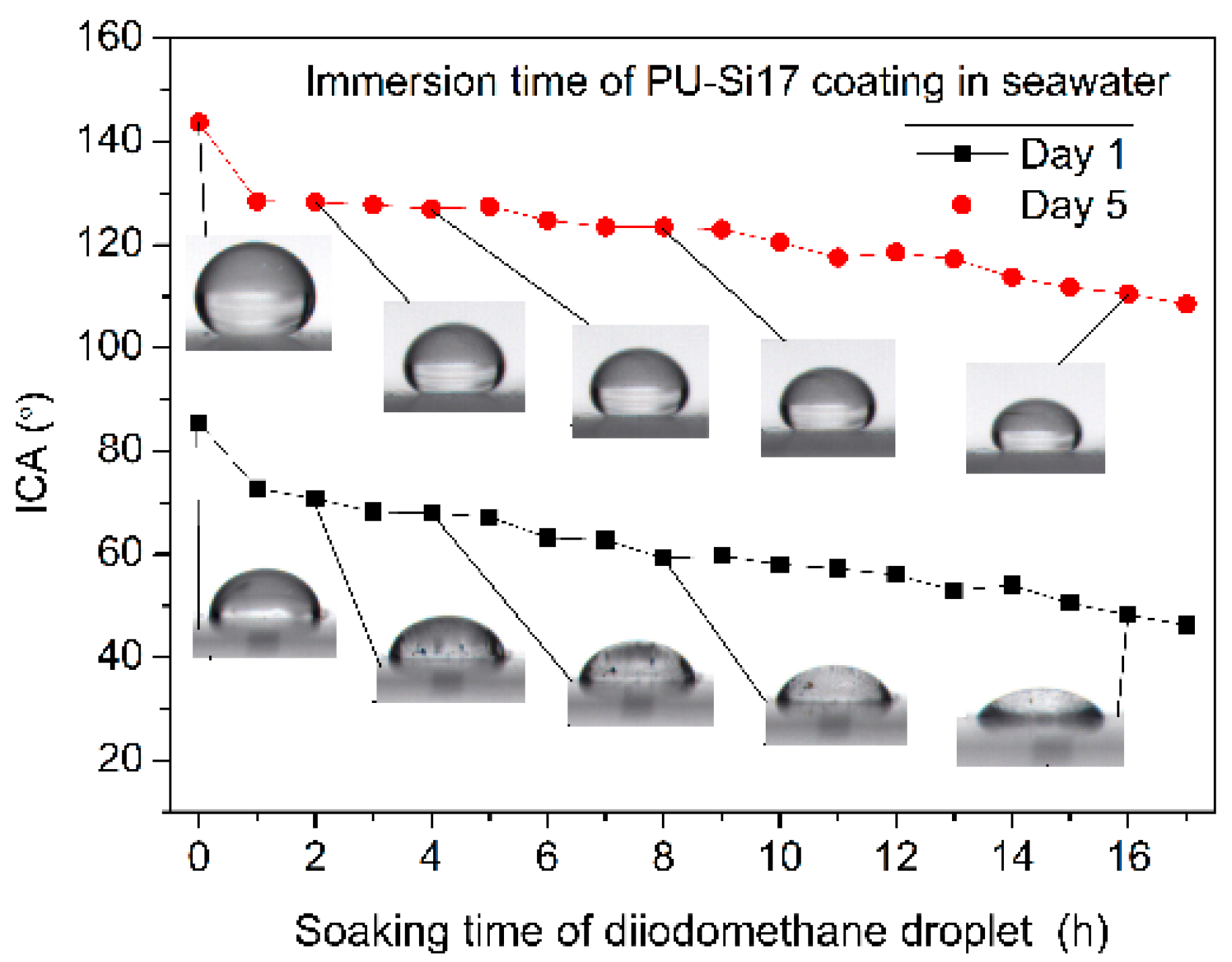
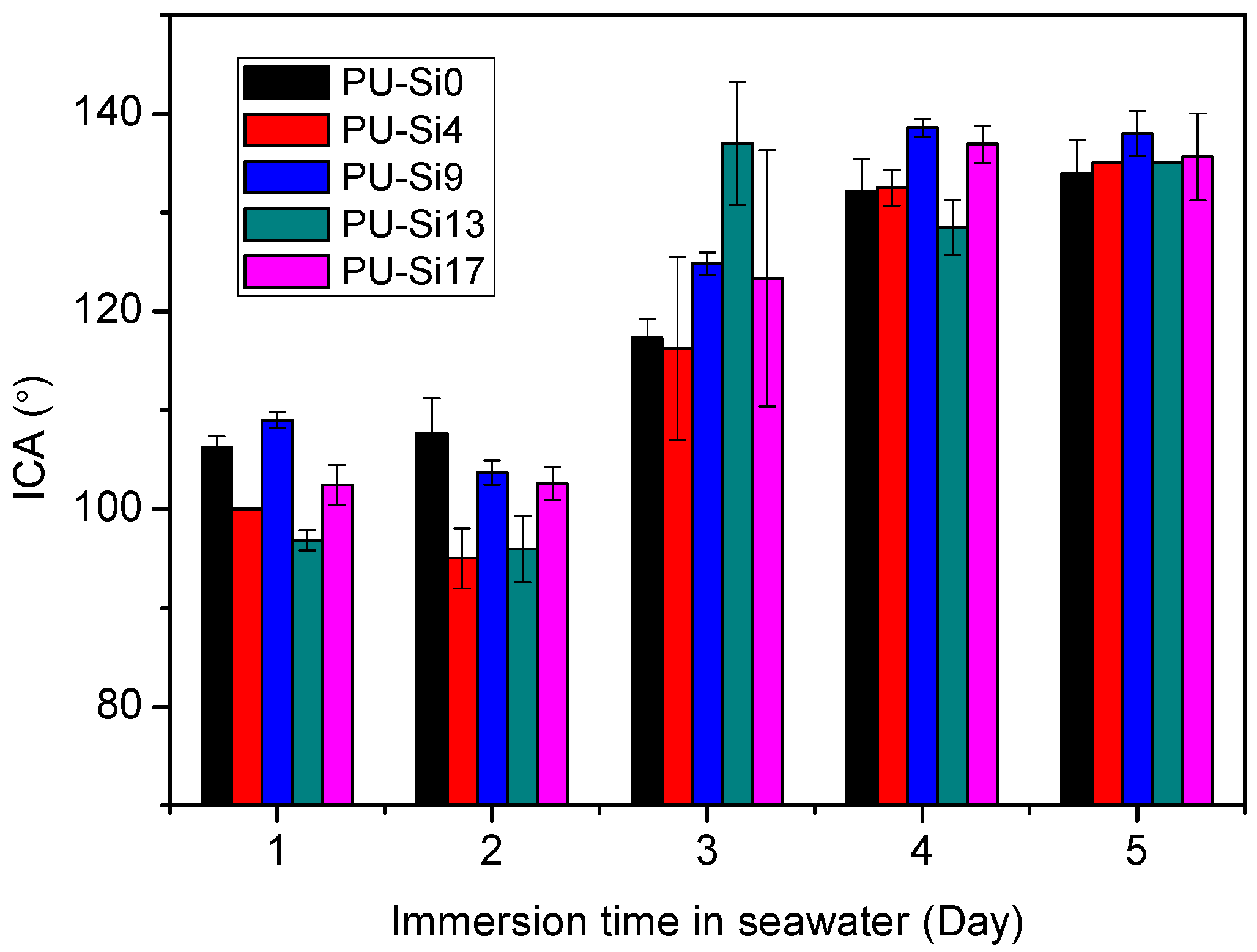
3.2.4. Interface Contact Angles (ICA) In-Situ Measurements during Immersion in Seawater
3.3. Antifouling Properties of the PU-Sis Samples
4. Conclusions
- (1)
- Clear microphase separation was observed in polyurethane, but decreased or even disappeared after immersion in seawater. Incorporating PDMS weakened H-bonding between the soft and hard phases, and increased phase separation. The size of the soft and hard domains also increased. Phase separation was not observed on the surface of samples containing PDMS, because the surface layer was enriched in PDMS.
- (2)
- The flexible PDMS segments decreased the Tg of polyurethane. This improved the flexibility and elasticity of polyurethane, so fouling organisms could be more readily removed.
- (3)
- PDMS chains migrated toward the film surface, forming a uniform hydrophobic layer with a low surface free energy. This surface enrichment of PDMS increased with increasing PDMS content. Because of this enrichment, PU-Si4, PU-Si9, PU-Si13 and PU-Si17 exhibited low surface energies, and were stable toward immersion in seawater. During seawater immersion, polar groups have an upward tendency.
- (4)
- Immersed panels tests in seawater demonstrated the antifouling activity of the PDMS-modified polyurethane. However, it was not the case that the higher the PDMS content, the better the antifouling performance of the PDMS-modified polyurethane. The best modified polyurethane is with 8.5 wt % PDMS. It is a promising material for fouling-release coatings.
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Galhenage, T.P.; Webster, D.C.; Moreira, A.M.S.; Burgett, R.J.; Stafslien, S.J.; Vanderwal, L.; Finlay, J.A.; Franco, S.C.; Clare, A.S. Poly(ethylene) glycol-modified, amphiphilic, siloxane–polyurethane coatings and their performance as fouling-release surfaces. J. Coat. Technol. Res. 2017, 14, 307–322. [Google Scholar] [CrossRef]
- Cavas, L.; Yildiz, P.G.; Mimigianni, P.; Sapalidis, A.; Nitodas, S. Reinforcement effects of multiwall carbon nanotubes and graphene oxide on PDMS marine coatings. J. Coat. Technol. Res. 2018, 15, 105–120. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Prabowo, R.E.; Ohshiro, Y.; Shimono, T.; Jones, D.; Kawai, H.; Otani, M.; Oshino, A.; Inagawa, S.; Akaya, T. The introduction to Japan of the Titan barnacle, Mega balanus coccopoma (Darwin 1854) (Cirripedia: Balanomorpha) and the role of shipping in its translocation. Biofouling 2009, 25, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, G.A.; Forrest, B.M. A preliminary assessment of biofouling and non-indigenous marine species associated with commercial slow-moving vessels arriving in New Zealand. Biofouling 2010, 26, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Selim, M.S.; Shenashen, M.A.; El-Safty, S.A.; Higazy, S.A.; Selim, M.M.; Isago, H.; Elmarakbi, A. Recent progress in marine foul-release polymeric nanocomposite coatings. Prog. Mater. Sci. 2017, 87, 1–32. [Google Scholar] [CrossRef]
- Kavanagh, C.J.; Quinn, R.D.; Swain, G.W. Observation of barnacle detachment from silicones using high-speed video. J. Adhes. 2005, 81, 43–868. [Google Scholar] [CrossRef]
- Chambers, L.D.; Stokes, K.R.; Walsh, F.C.; Wood, R.J.K. Modern approaches to marine antifouling coatings. Surf. Coat. Technol. 2006, 201, 3642–3652. [Google Scholar] [CrossRef]
- Almeida, E.; Diamantino, T.C.; Sousa, O.D. Marine paints: The particular case of antifouling paints. Prog. Org. Coat. 2007, 59, 2–20. [Google Scholar] [CrossRef]
- Jiang, M.; Zhao, X.L.; Ding, X.B.; Zheng, Z.H.; Peng, Y.X. A novel approach to fluorinated polyurethane by macromonomer copolymerization. Eur. Polym. J. 2005, 41, 1798–1803. [Google Scholar] [CrossRef]
- Ameduri, B.; Boutevin, B.; Kostov, G. Fluoroelastomers: Synthesis, properties and applications. Prog. Polym. Sci. 2001, 26, 105–187. [Google Scholar] [CrossRef]
- Eduok, U.; Faye, O.; Szpunar, J. Recent developments and applications of protective silicone coatings: A review of PDMS functional materials. Prog. Org. Coat. 2017, 111, 124–163. [Google Scholar] [CrossRef]
- Fatima, J.; Tuula, T.P. Incorporation of polydimethylsiloxane into polyurethanes and characterization of copolymers. Eur. Polym. J. 2011, 47, 1694–1708. [Google Scholar]
- Brady, R.F. A fracture mechanical analysis of fouling release from nontoxic antifouling coatings. Prog. Org. Coat. 2001, 43, 188–192. [Google Scholar] [CrossRef]
- Gui, T.J. Preparation and Characterization of the Organic Silicone/Fluorine Antifouling Coatings with Low Surface Energy. Ph.D. Thesis, Ocean University of China, Qingdao, China, 2008. [Google Scholar]
- Baier, R.E.; Shafrin, E.G.; Zisman, W.A. Adhesion: mechanisms that assist or impede it. Science 1968, 162, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Thünemann, A.F.; Kublickas, R.H. Low surface energy polysiloxane complexes. J. Mater. Chem. 2001, 11, 381–384. [Google Scholar] [CrossRef]
- Yilgör, I.; Yilgör, E.; Wilkes, G.L. Critical parameters in designing segmented polyurethanes and their effect on morphology and properties: A comprehensive review. Polymer 2015, 58, A1–A36. [Google Scholar] [CrossRef]
- Ge, Z.; Luo, Y.J. Synthesis and characterization of siloxane-modified two-component waterborne polyurethane. Prog. Org. Coat. 2013, 76, 1522–1526. [Google Scholar] [CrossRef]
- Pergal, M.V.; Dzunuzovic, J.V.; Poreba, R.; Ostojic, S.; Radulovic, A.; Spirkova, M. Microstructure and properties of poly(urethane-siloxane)s based on hyperbranched polyester of the fourth pseudo generation. Prog. Org. Coat. 2013, 76, 743–756. [Google Scholar] [CrossRef]
- Jia, B.D.; Xing, Y.Z.; Jing, C. A new core-shell type fluorinated acrylic and siliconated polyurethane hybrid emulsion. J. Coat. Technol. Res. 2007, 4, 283–288. [Google Scholar]
- Majumdar, P.; Stafslien, S.; Daniels, J.; Webster, D.C. High throughput combinatorial characterization of thermosetting siloxane–urethane coatings having spontaneously formed microtopographical surfaces. J. Coat. Technol. Res. 2007, 4, 131–138. [Google Scholar] [CrossRef]
- Sommer, S.; Ekin, A.; Webster, D.C. A preliminary study on the properties and fouling-release performance of siloxane-polyurethane coatings prepared from poly(dimethylsiloxane) (PDMS) macromers. Biofouling 2010, 26, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Pieper, R.J.; Ekin, A.; Webster, D.C.; Cassé, F.; Callow, J.A.; Callow, M.E. Combinatorial approach to study the effect of acrylic polyol composition on the properties of crosslinked siloxane-polyurethane fouling-release coatings. J. Coat. Technol. Res. 2007, 4, 453–461. [Google Scholar] [CrossRef]
- Zhang, Y.; Qi, Y.H.; Zhang, Z.P. Synthesis of PPG-TDI-BDO polyurethane and the influence of hard segment content on its structure and antifouling properties. Prog. Org. Coat. 2016, 97, 115–121. [Google Scholar] [CrossRef]
- Owens, D.K.; Wendt, R.D. Liquid spreading on rough metal surfaces. J. Mater. Sci. 1980, 15, 431–437. [Google Scholar]
- Choi, T.; Weksler, J.; Padsalgikar, A.; Runt, J. Influence of soft segment composition on phase separated microstructure of polydimethylsiloxane-based segmented polyurethane copolymers. Polymer 2009, 50, 2320–2327. [Google Scholar] [CrossRef]
- Choi, T.; Weksler, J.; Padsalgikar, A.; Runt, J. Microstructural organization of polydimethylsiloxane soft segment polyurethanes derived from a single macrodiol. Polymer 2010, 51, 4375–4382. [Google Scholar] [CrossRef]
- Rahman, M.M.; Kim, H.D. Characterization of waterborne polyurethane adhesives containing different soft segments. J. Adhes. Sci. Technol. 2007, 21, 81–96. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomer | HSH220 | HSH330 | PDMS2200 | BDO | TDI100 |
|---|---|---|---|---|---|
| Functional group | −OH | −OH | −OH | −OH | −N=C=O |
| Functionality | 2 | 3 | 2 | 2 | 2 |
| Molecular weight | 2000 | 3000 | 2000 | 90.121 | 174.15 |
| Amount of Monomer (wt %) | PU-Si0 | PU-Si4 | PU-Si9 | PU-Si13 | PU-Si17 |
|---|---|---|---|---|---|
| HSH220 | 67.7 | 64.1 | 59.9 | 56.1 | 52.5 |
| HSH330 | 7.6 | 7.1 | 6.7 | 6.3 | 5.8 |
| PDMS2200 | 0.0 | 4.2 | 8.5 | 12.8 | 16.9 |
| TDI | 20.3 | 20.2 | 20.5 | 20.4 | 20.4 |
| BDO | 4.4 | 4.4 | 4.4 | 4.4 | 4.4 |
| Mechanical Property | PU-Si0 | PU-Si4 | PU-Si9 | PU-Si13 | PU-Si17 |
|---|---|---|---|---|---|
| Tensile strength (MPa) | 0.51 | 1.09 | 1.05 | 1.37 | 2.09 |
| Elongation at break (%) | 2070 | 955 | 2442 | 1258 | 1315 |
| Elastic modulus (MPa) | 0.69 | 0.79 | 0.40 | 1.10 | 1.13 |
| Specimen | PU-Si0 | PU-Si4 | PU-Si9 | PU-Si13 | PU-Si17 | Silicone Coating |
|---|---|---|---|---|---|---|
| Coverage (%) | 26.7 | 8.2 | 1.6 | 2.9 | 22.1 | 100 |
| 4.15 | 3.68 | 2.67 | 4.41 | 4.72 | 2.86 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-P.; Song, X.-F.; Cui, L.-Y.; Qi, Y.-H. Synthesis of Polydimethylsiloxane-Modified Polyurethane and the Structure and Properties of Its Antifouling Coatings. Coatings 2018, 8, 157. https://doi.org/10.3390/coatings8050157
Zhang Z-P, Song X-F, Cui L-Y, Qi Y-H. Synthesis of Polydimethylsiloxane-Modified Polyurethane and the Structure and Properties of Its Antifouling Coatings. Coatings. 2018; 8(5):157. https://doi.org/10.3390/coatings8050157
Chicago/Turabian StyleZhang, Zhan-Ping, Xiao-Fei Song, Li-Ying Cui, and Yu-Hong Qi. 2018. "Synthesis of Polydimethylsiloxane-Modified Polyurethane and the Structure and Properties of Its Antifouling Coatings" Coatings 8, no. 5: 157. https://doi.org/10.3390/coatings8050157
APA StyleZhang, Z.-P., Song, X.-F., Cui, L.-Y., & Qi, Y.-H. (2018). Synthesis of Polydimethylsiloxane-Modified Polyurethane and the Structure and Properties of Its Antifouling Coatings. Coatings, 8(5), 157. https://doi.org/10.3390/coatings8050157