
Preparation and Grafting Functionalization of Self-Assembled Chitin Nanofiber Film


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
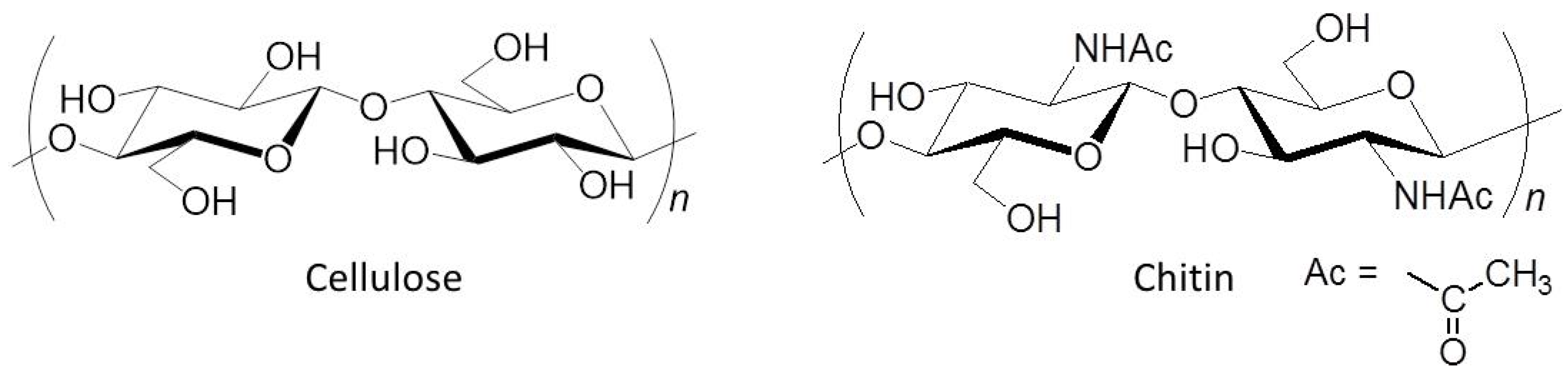
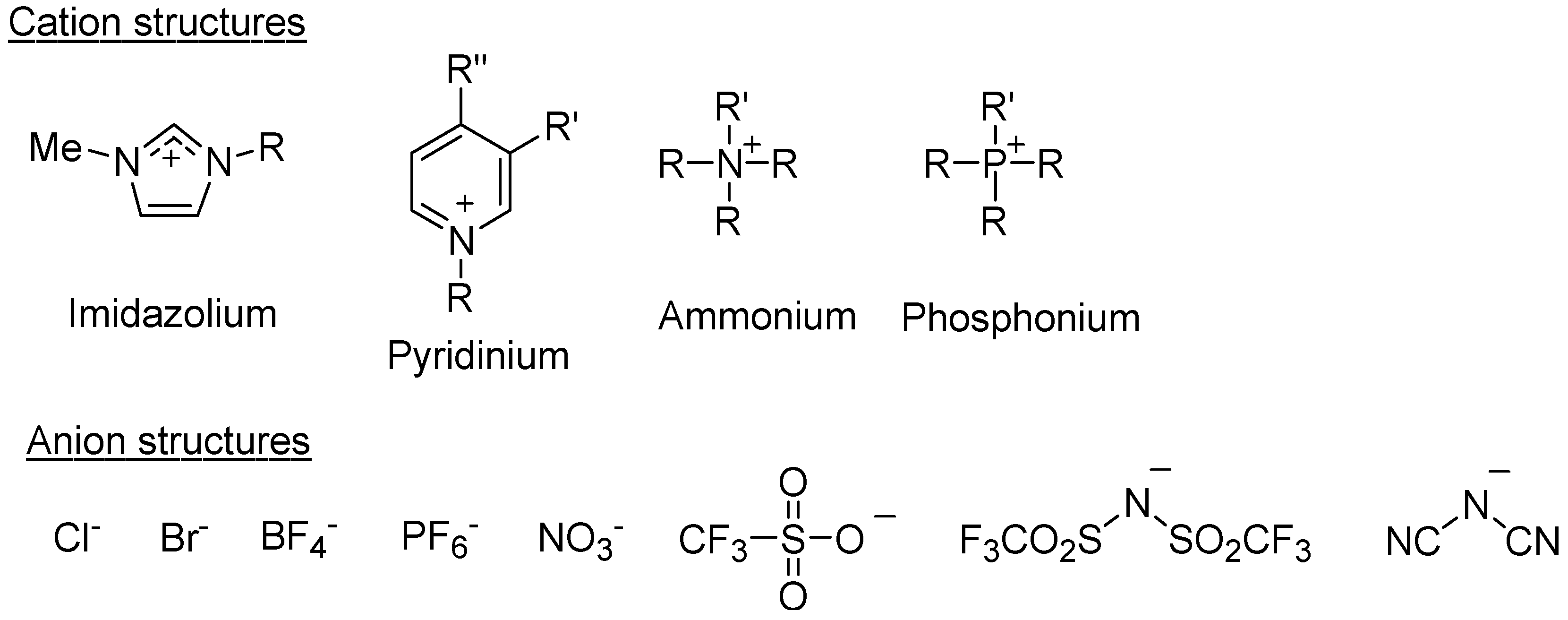
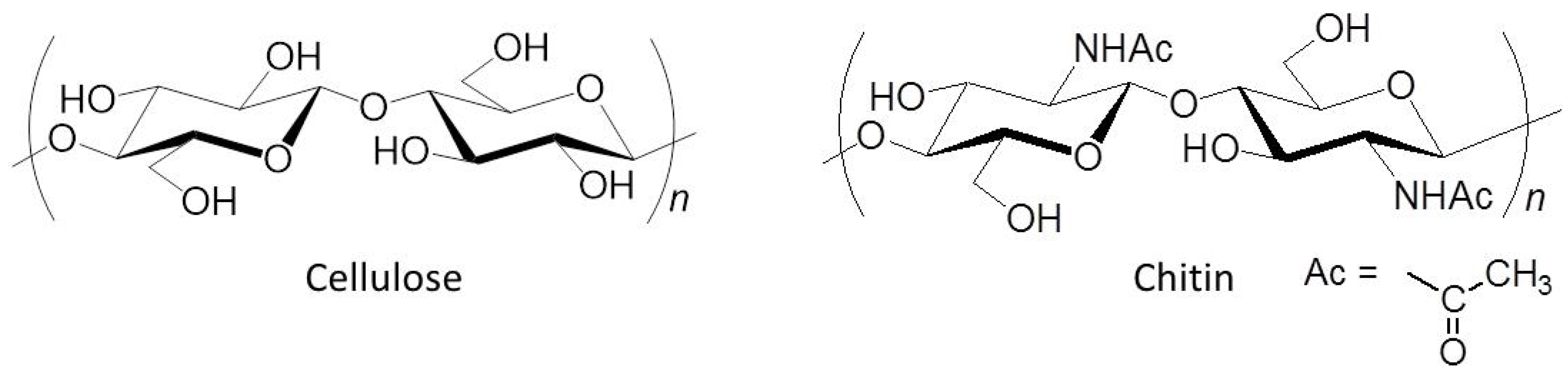
:1. Introduction
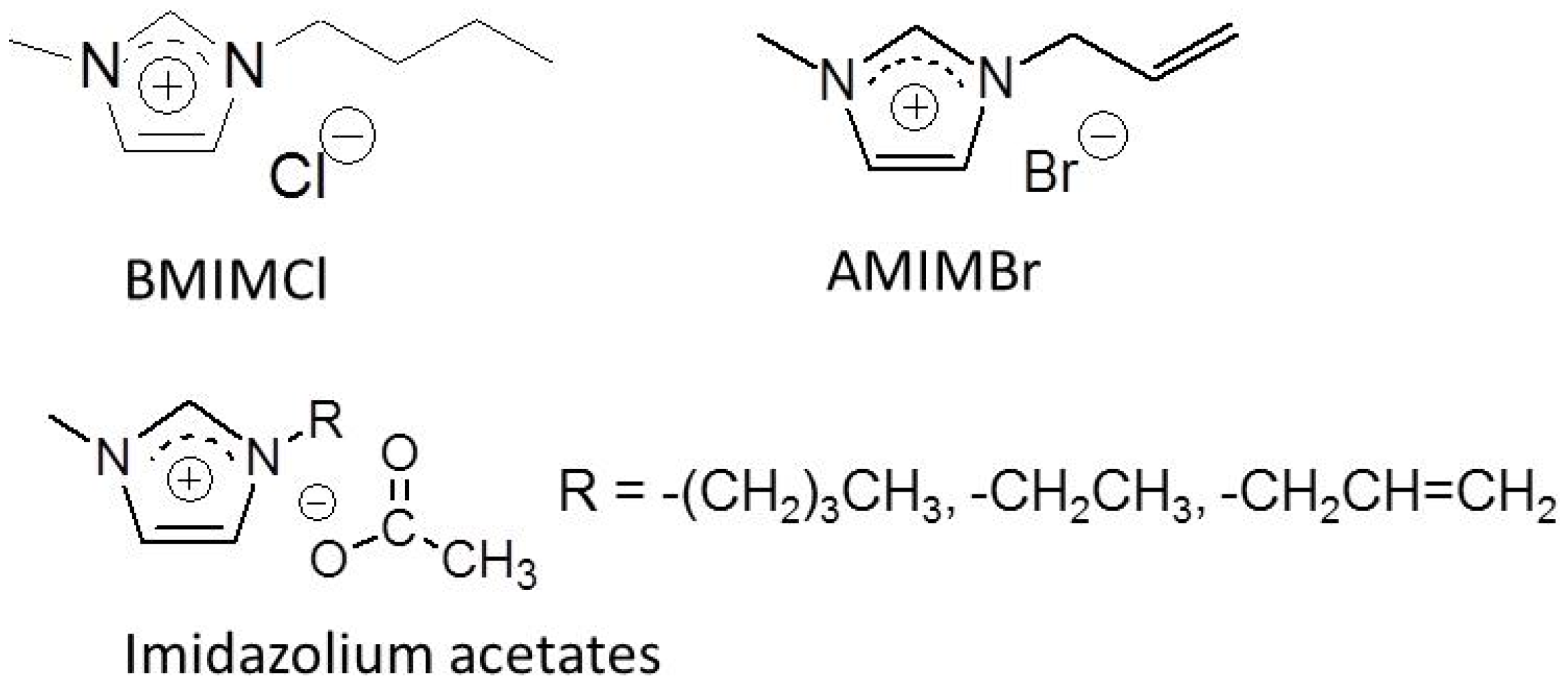
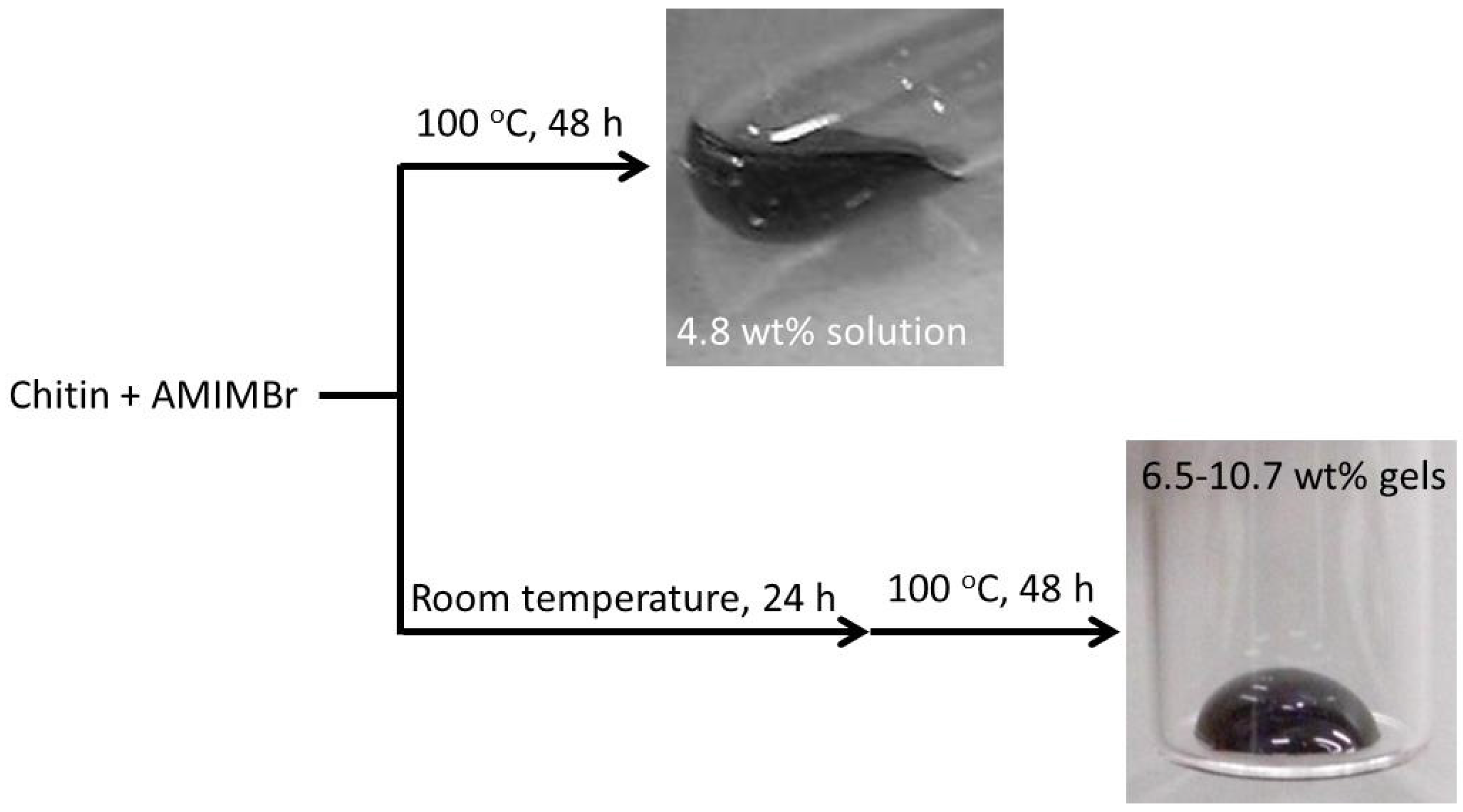
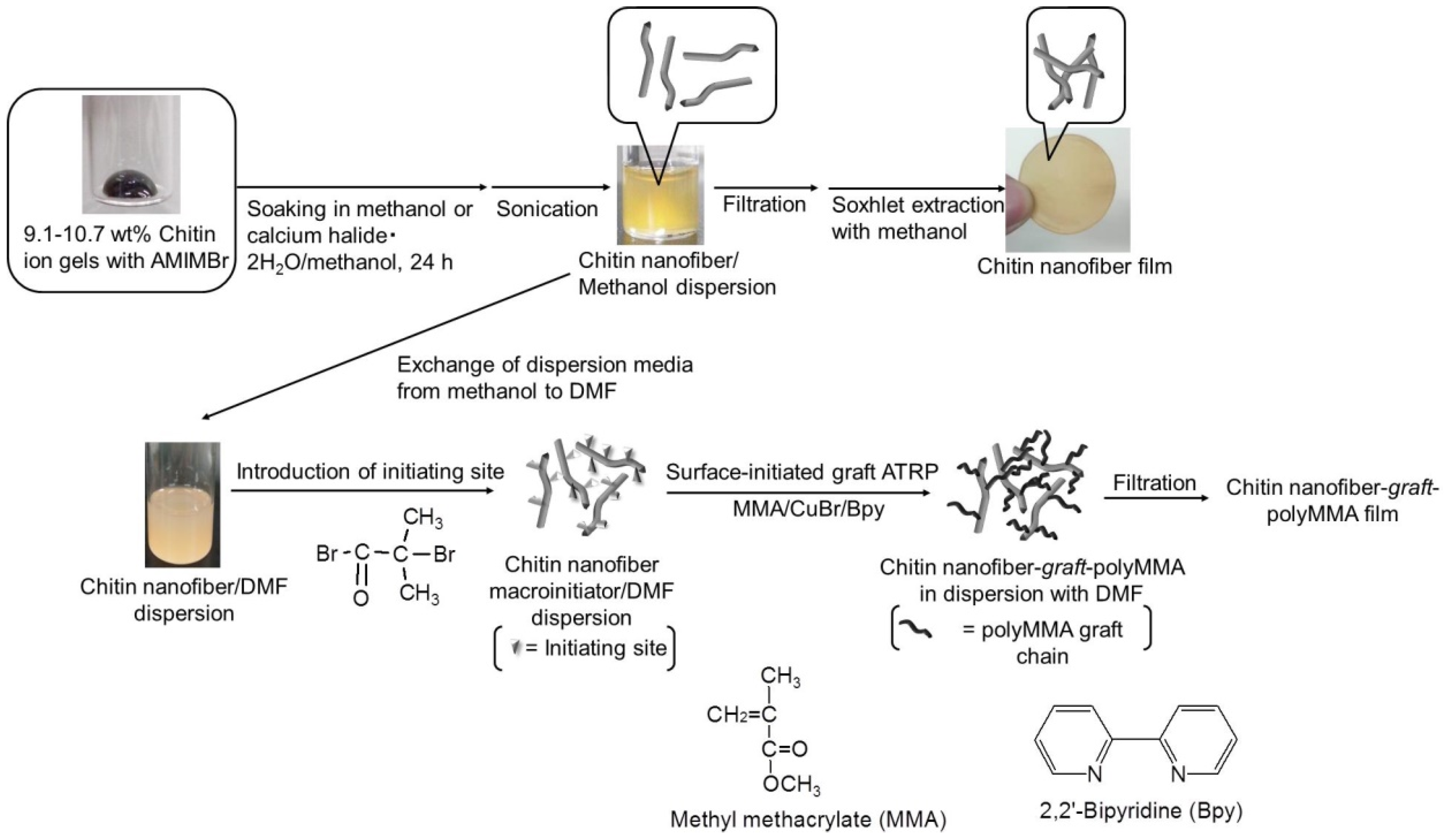
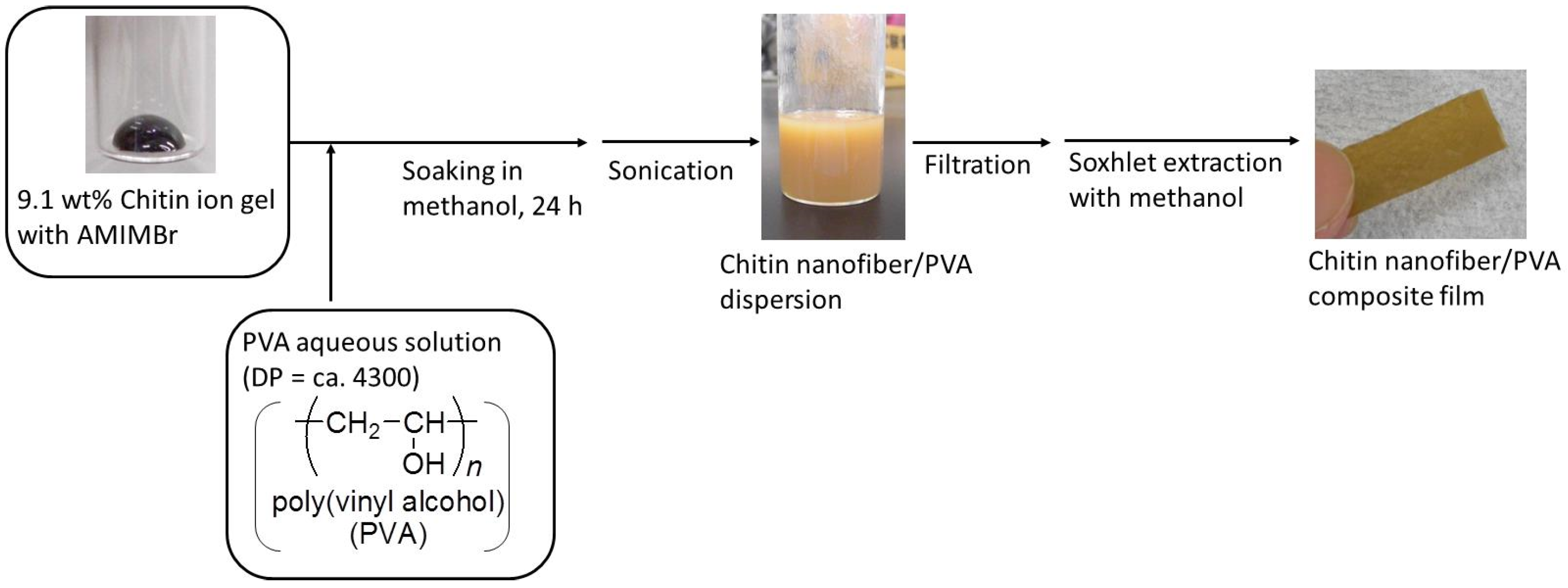
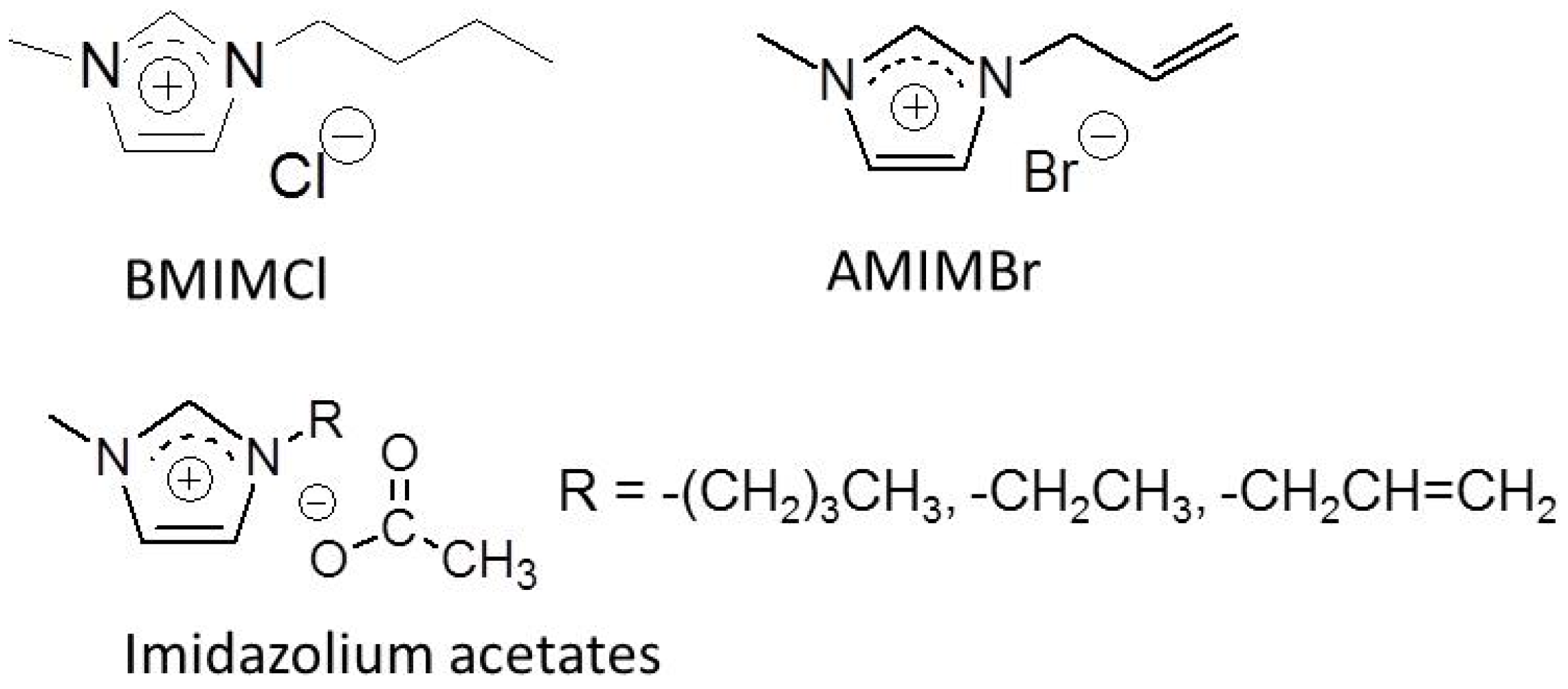
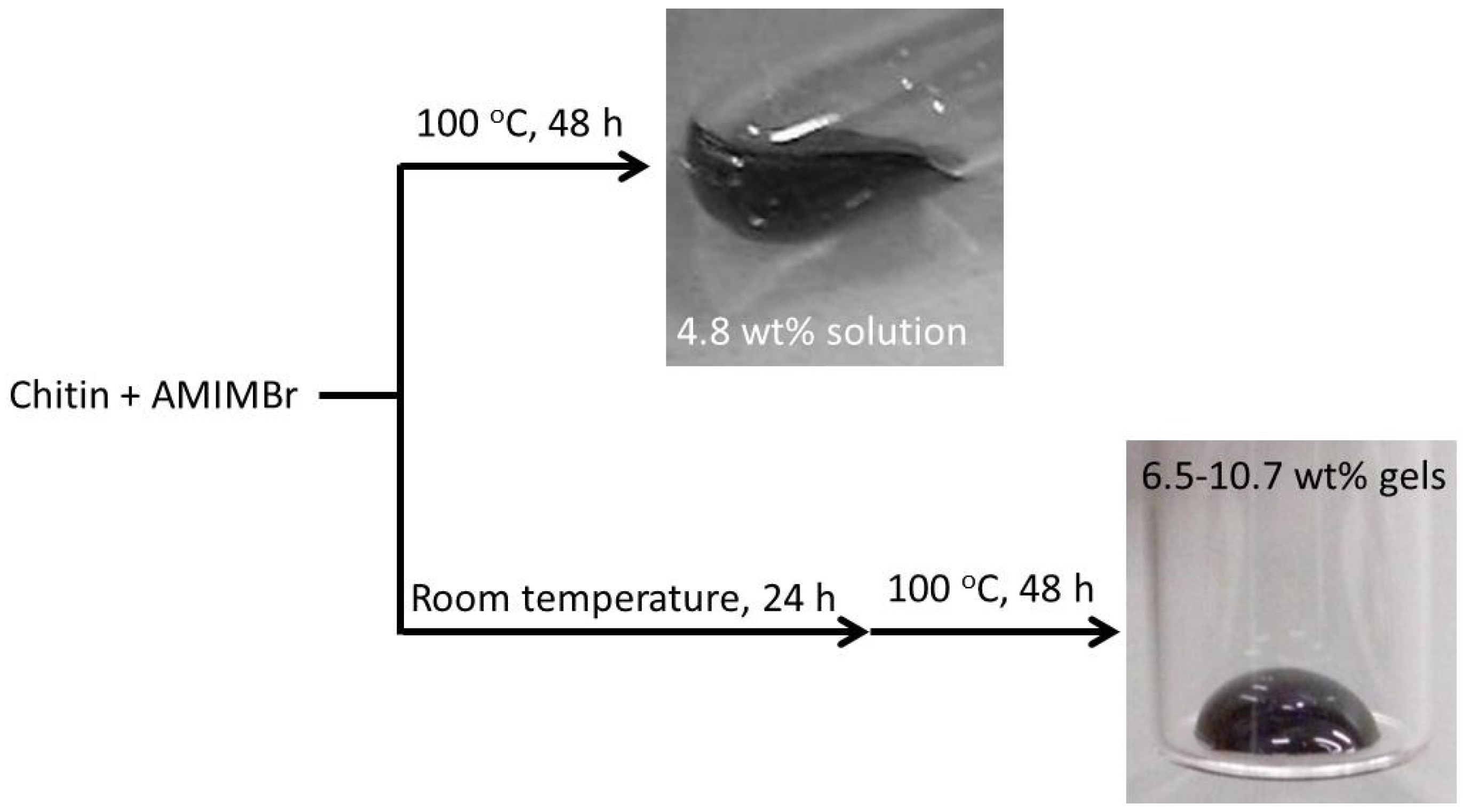
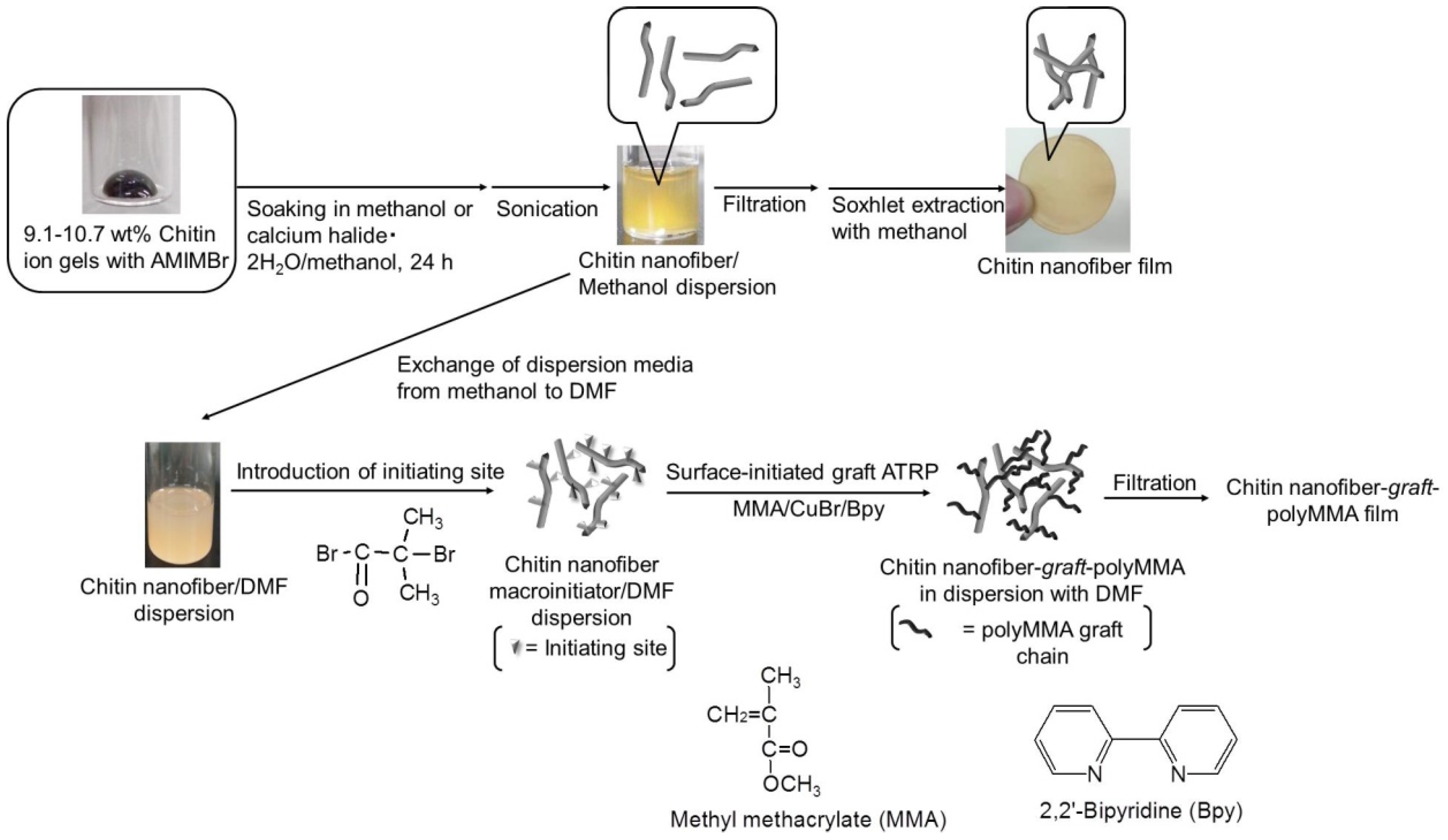
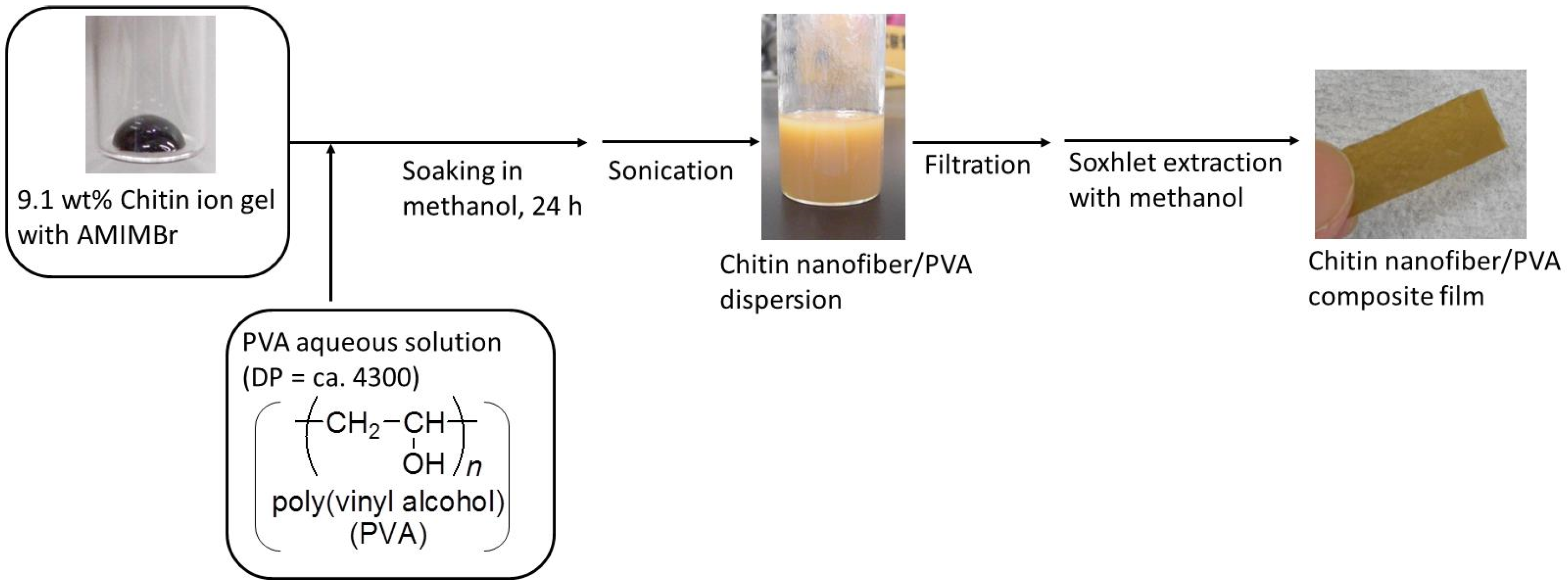
2. Preparation of Self-Assembled Chitin Nanofiber Film from Ion Gel
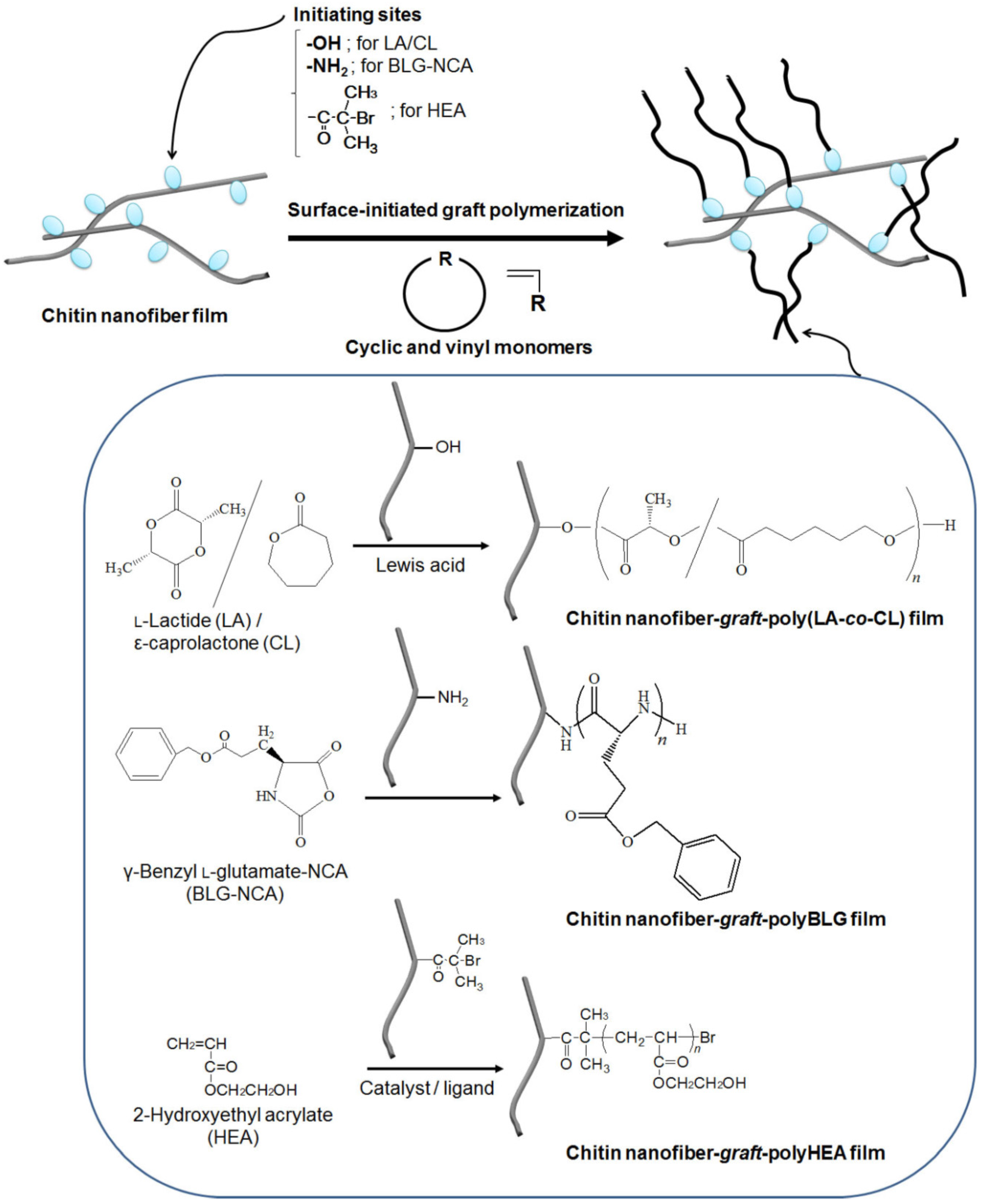
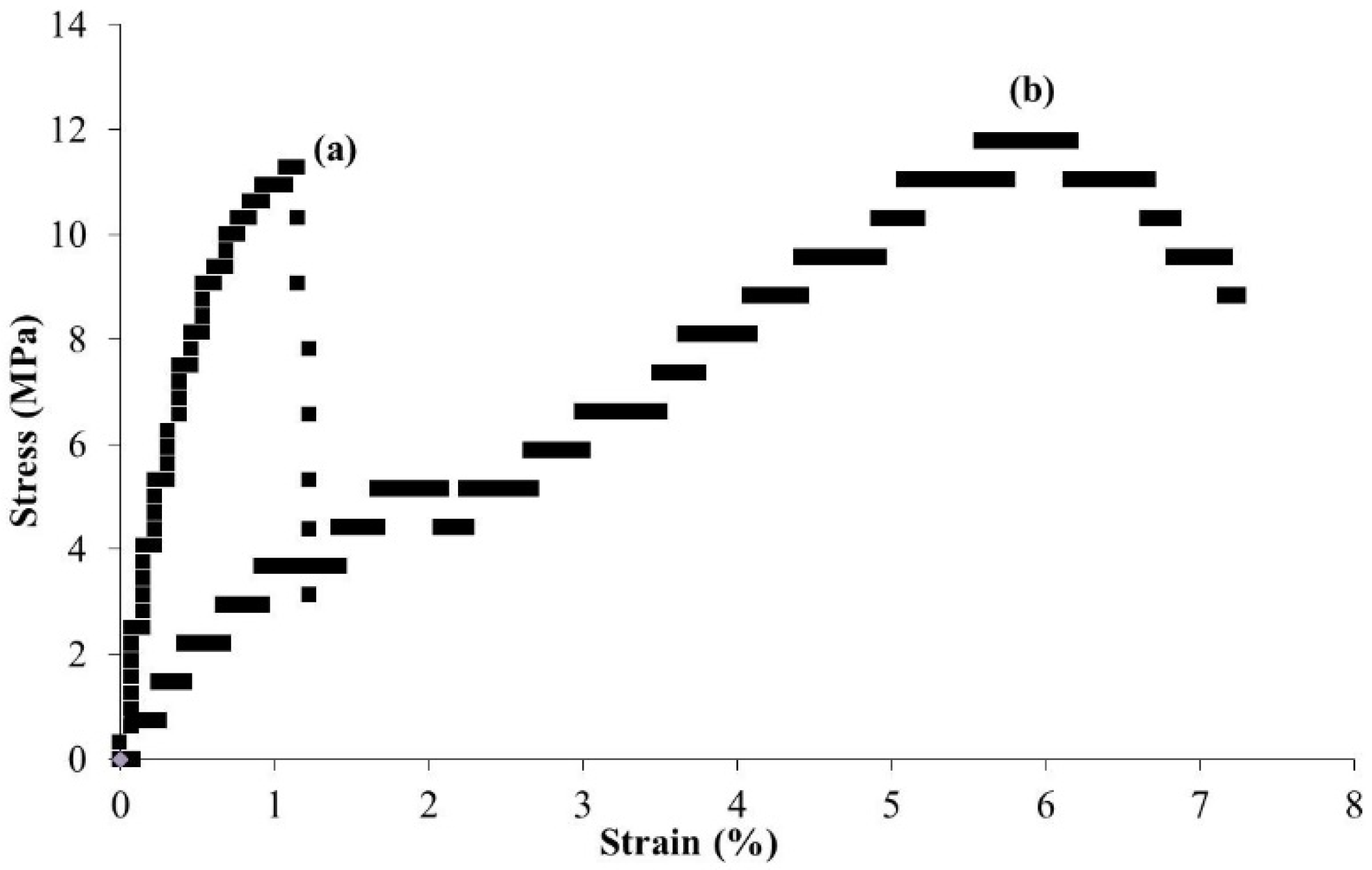
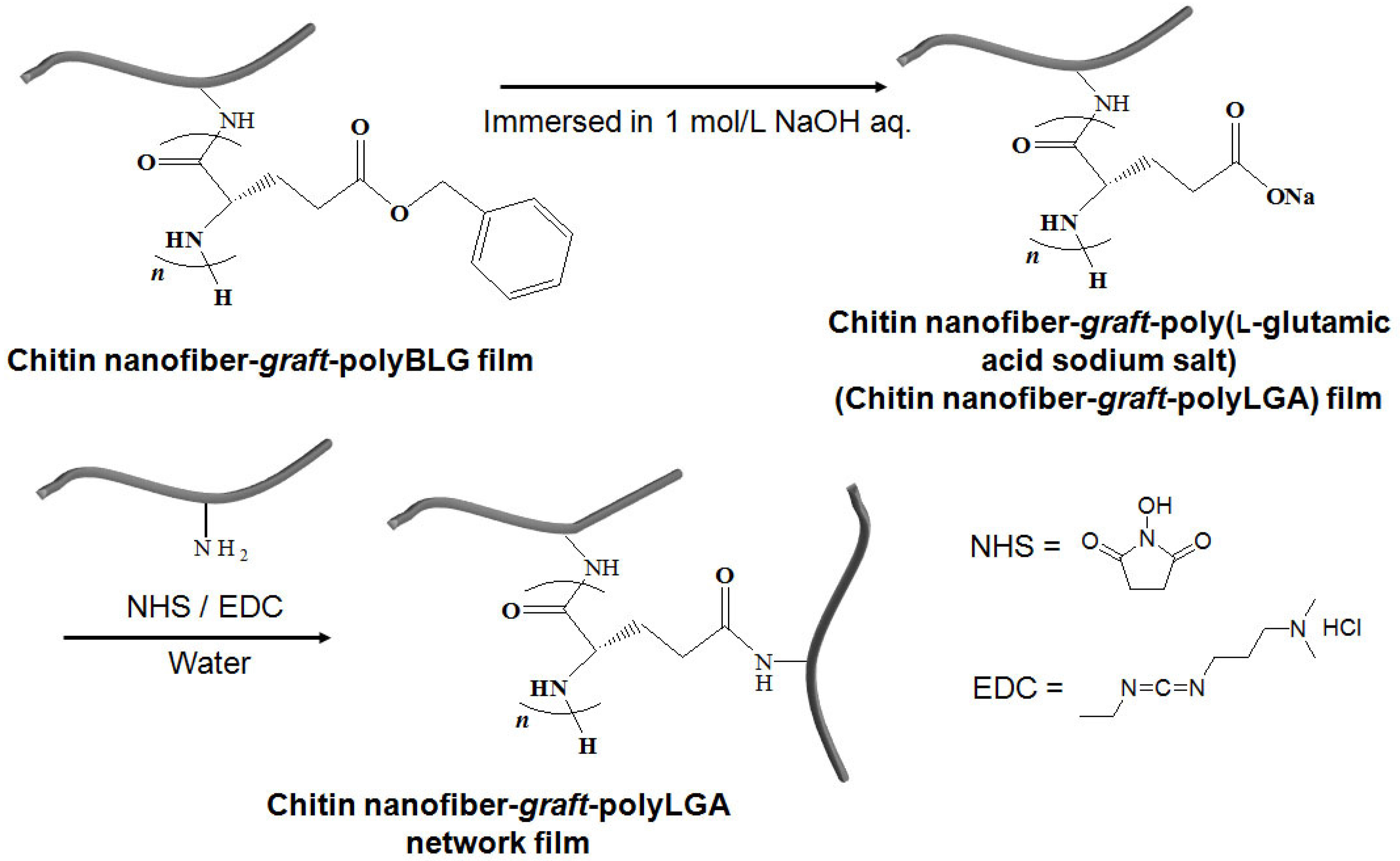
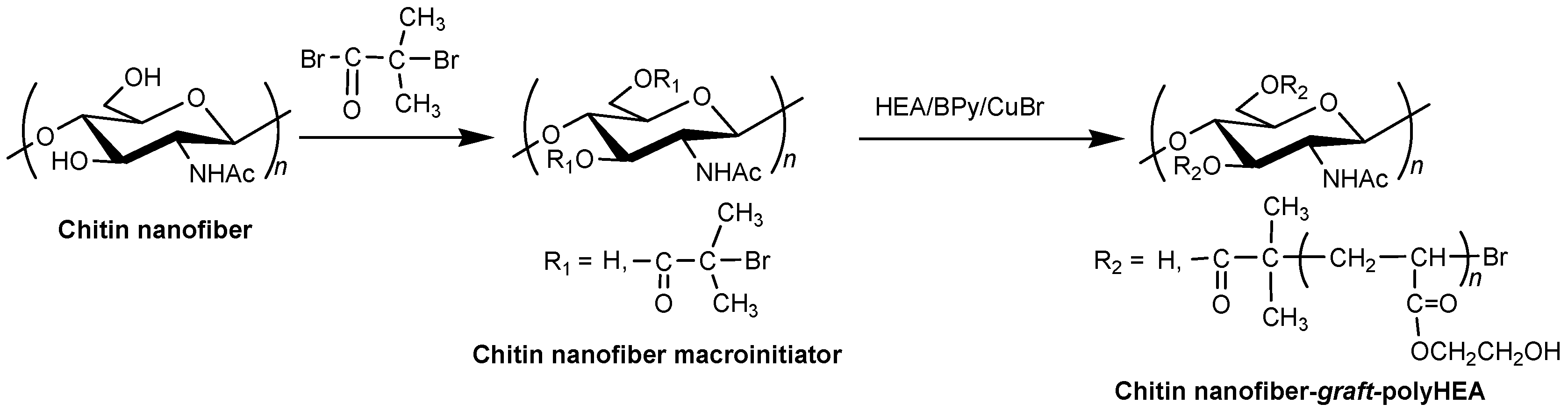
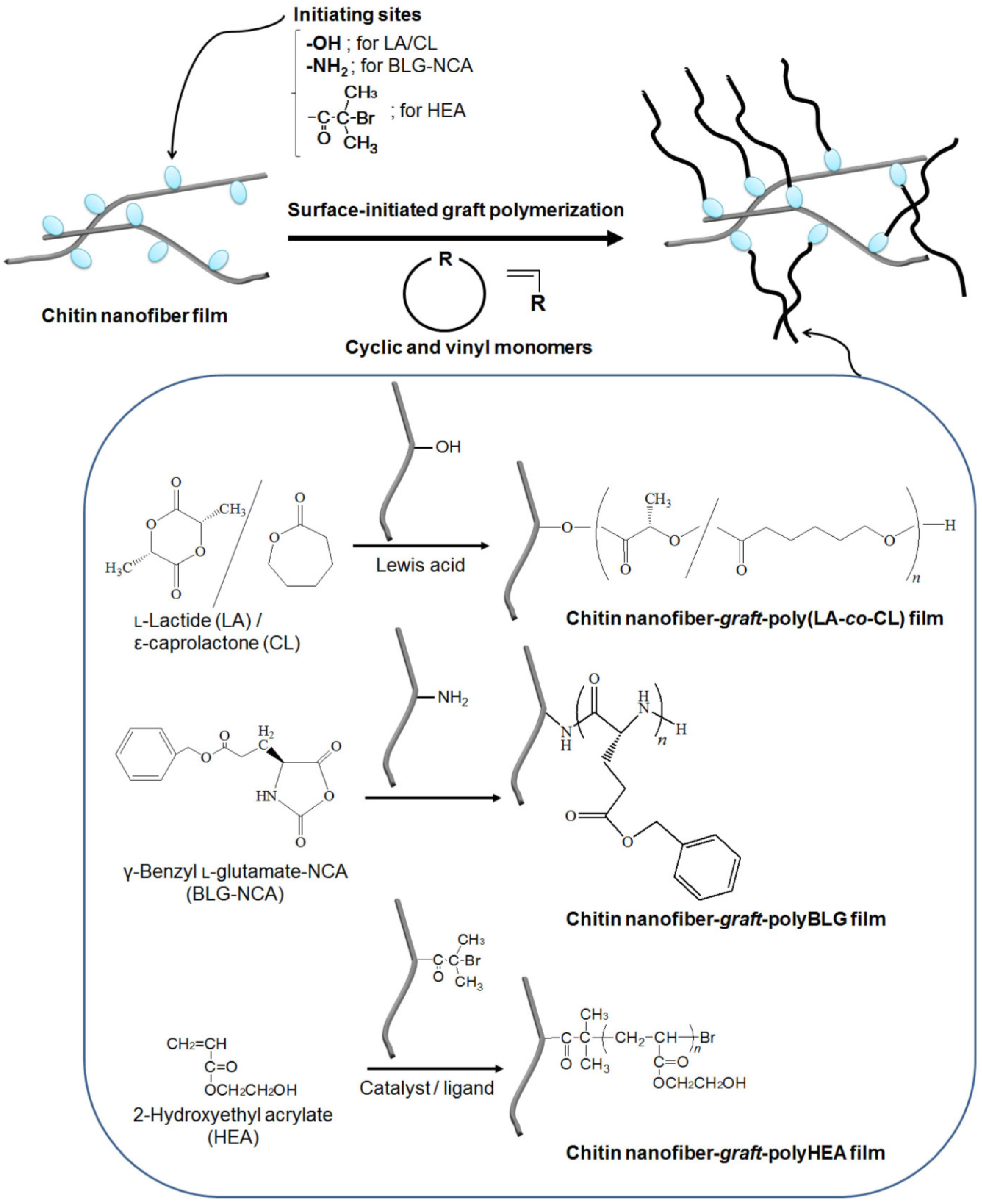
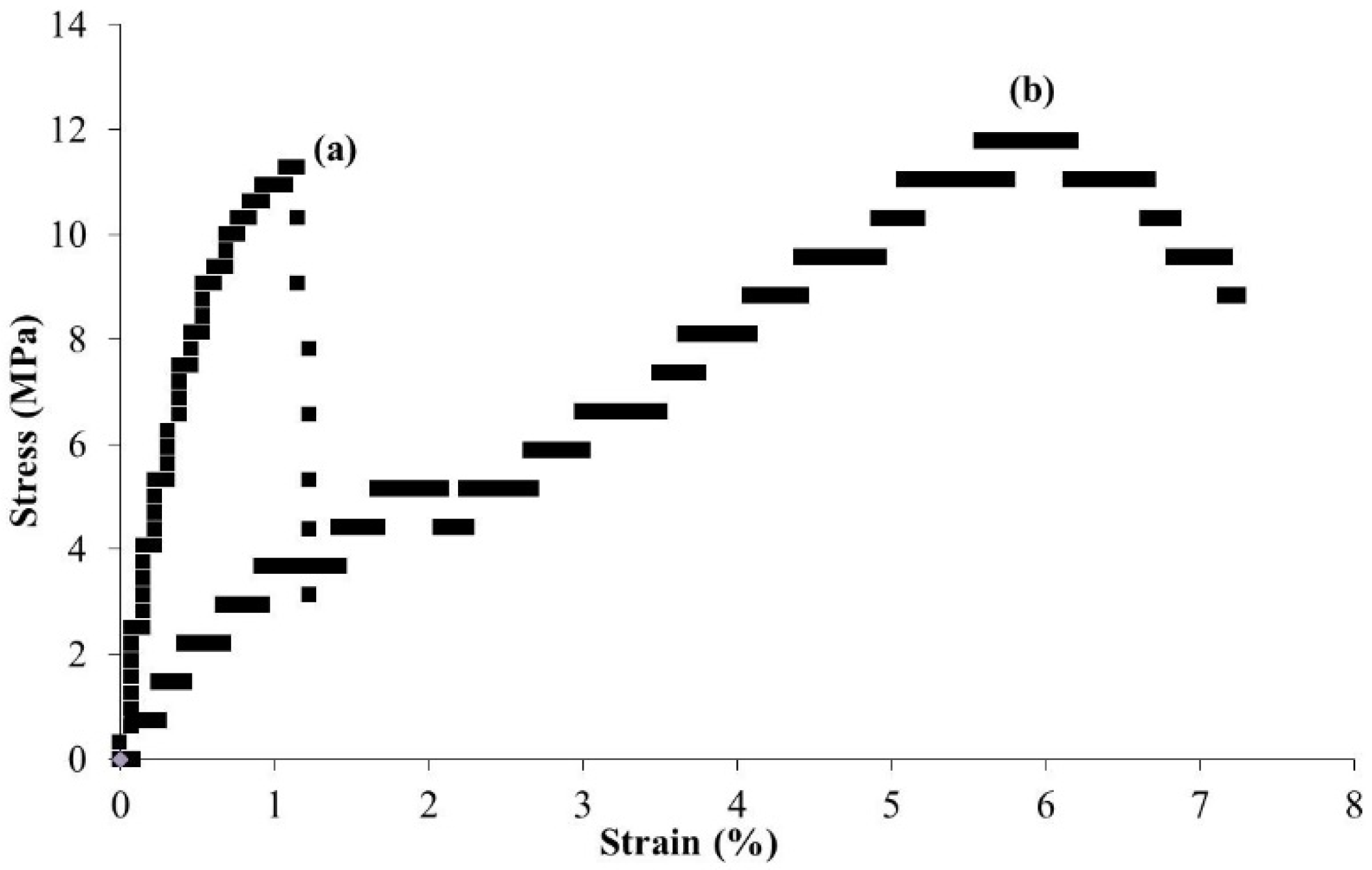
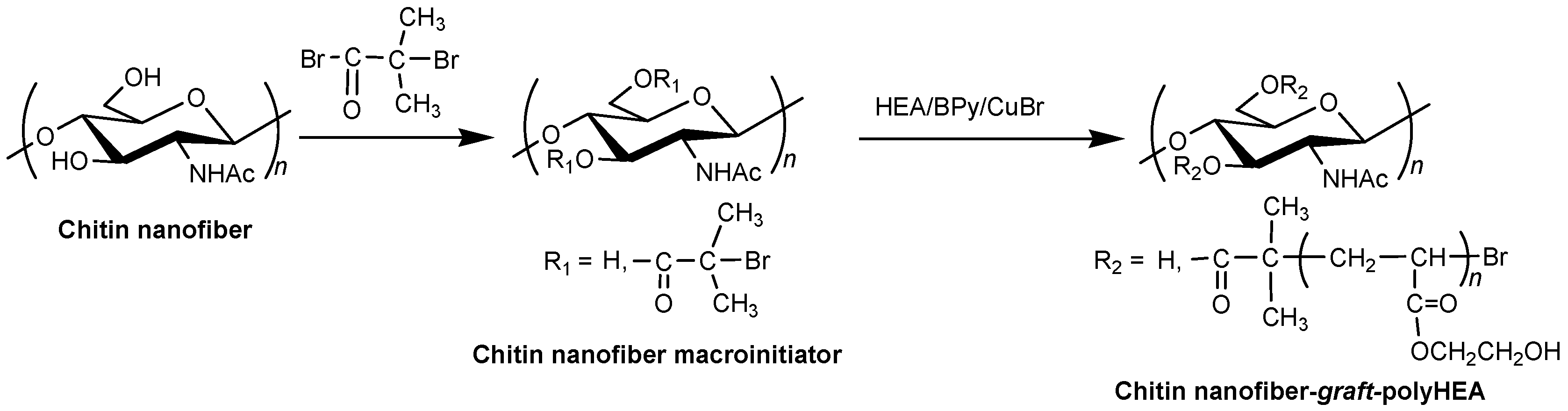
3. Surface-Initiated Graft Polymerization from Self-Assembled Chitin Nanofiber Film
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Schuerch, C. Polysaccharides. In Encyclopedia of Polymer Science and Engineering, 2nd ed.; Mark, H.F., Bilkales, N., Overberger, C.G., Eds.; John Wiley & Sons: New York, NY, USA, 1986; Volume 13, pp. 87–162. [Google Scholar]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 7th ed.; W.H. Freeman: New York, NY, USA, 2012. [Google Scholar]
- Klemm, D.; Heublein, B.; Fink, H.P.; Bohn, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef] [PubMed]
- Kurita, K. Chitin and chitosan: Functional biopolymers from marine crustaceans. Mar. Biotechnol. 2006, 8, 203–226. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Pillai, C.K.S.; Paul, W.; Sharma, C.P. Chitin and chitosan polymers: Chemistry, solubility and fiber formation. Prog. Polym. Sci. 2009, 34, 641–678. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A. Biomedical exploitation of chitin and chitosan via mechano-chemical disassembly, electrospinning, dissolution in imidazolium ionic liquids, and supercritical drying. Mar. Drugs 2011, 9, 1510–1533. [Google Scholar] [CrossRef] [PubMed]
- El Seoud, O.A.; Koschella, A.; Fidale, L.C.; Dorn, S.; Heinze, T. Applications of ionic liquids in carbohydrate chemistry: A window of opportunities. Biomacromolecules 2007, 8, 2629–2647. [Google Scholar] [CrossRef] [PubMed]
- Liebert, T.; Heinze, T. Interaction of ionic liquids with polysaccharides 5. Solvents and reaction media for the modification of cellulose. Bioresources 2008, 3, 576–601. [Google Scholar]
- Feng, L.; Chen, Z.I. Research progress on dissolution and functional modification of cellulose in ionic liquids. J. Mol. Liq. 2008, 142, 1–5. [Google Scholar] [CrossRef]
- Pinkert, A.; Marsh, K.N.; Pang, S.S.; Staiger, M.P. Ionic liquids and their interaction with cellulose. Chem. Rev. 2009, 109, 6712–6728. [Google Scholar] [CrossRef] [PubMed]
- Gericke, M.; Fardim, P.; Heinze, T. Ionic liquids—Promising but challenging solvents for homogeneous derivatization of cellulose. Molecules 2012, 17, 7458–7502. [Google Scholar] [CrossRef] [PubMed]
- Isik, M.; Sardon, H.; Mecerreyes, D. Ionic liquids and cellulose: Dissolution, chemical modification and preparation of new cellulosic materials. Int. J. Mol. Sci. 2014, 15, 11922–11940. [Google Scholar] [CrossRef] [PubMed]
- Welton, T. Room-temperature ionic liquids. Solvents for synthesis and catalysis. Chem. Rev. 1999, 99, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Hallett, J.P.; Welton, T. Room-temperature ionic liquids: Solvents for synthesis and catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef] [PubMed]
- Wasserscheid, P.; Keim, W. Ionic liquids—New “solutions” for transition metal catalysis. Angew. Chem. Int. Ed. 2000, 39, 3772–3789. [Google Scholar]
- Davis, J.H. Task-specific ionic liquids. Chem. Lett. 2004, 33, 1072–1077. [Google Scholar] [CrossRef]
- Lee, S.G. Functionalized imidazolium salts for task-specific ionic liquids and their applications. Chem. Commun. 2006, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Giernoth, R. Task-specific ionic liquids. Angew. Chem. Int. Ed. 2010, 49, 2834–2839. [Google Scholar] [CrossRef] [PubMed]
- Graenacher, C. Cellulose solution. US Patent 1943176, 1934. [Google Scholar]
- Swatloski, R.P.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Dissolution of cellose with ionic liquids. J. Am. Chem. Soc. 2002, 124, 4974–4975. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska, M.E.; Bogel-Łukasik, E.; Bogel-Łukasik, R. Solubility of carbohydrates in ionic liquids. Energy Fuels 2010, 24, 737–745. [Google Scholar] [CrossRef]
- Wang, W.T.; Zhu, J.; Wang, X.L.; Huang, Y.; Wang, Y.Z. Dissolution behavior of chitin in ionic liquids. J. Macromol. Sci. Part B Phys. 2010, 49, 528–541. [Google Scholar] [CrossRef]
- Jaworska, M.M.; Kozlecki, T.; Gorak, A. Review of the application of ionic liquids as solvents for chitin. J. Polym. Eng. 2012, 32, 67–69. [Google Scholar] [CrossRef]
- Wu, Y.; Sasaki, T.; Irie, S.; Sakurai, K. A novel biomass-ionic liquid platform for the utilization of native chitin. Polymer 2008, 49, 2321–2327. [Google Scholar] [CrossRef]
- Prasad, K.; Murakami, M.; Kaneko, Y.; Takada, A.; Nakamura, Y.; Kadokawa, J. Weak gel of chitin with ionic liquid, 1-allyl-3-methylimidazolium bromide. Int. J. Biol. Macromol. 2009, 45, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Lu, X.M.; Sun, N.; Rogers, R.D. Dissolution or extraction of crustacean shells using ionic liquids to obtain high molecular weight purified chitin and direct production of chitin films and fibers. Green Chem. 2010, 12, 968–971. [Google Scholar] [CrossRef]
- Zhao, D.B.; Fei, Z.F.; Geldbach, T.J.; Scopelliti, R.; Laurenczy, G.; Dyson, P.J. Allyl-functionalised ionic liquids: Synthesis, characterisation, and reactivity. Helv. Chim. Acta 2005, 88, 665–675. [Google Scholar] [CrossRef]
- Sharma, M.; Mukesh, C.; Mondal, D.; Prasad, K. Dissolution of a-chitin in deep eutectic solvents. RSC Adv. 2013, 3, 18149–18155. [Google Scholar] [CrossRef]
- Kadokawa, J.; Takegawa, A.; Mine, S.; Prasad, K. Preparation of chitin nanowhiskers using an ionic liquid and their composite materials with poly(vinyl alcohol). Carbohydr. Polym. 2011, 84, 1408–1412. [Google Scholar] [CrossRef]
- Kadokawa, J. Ionic liquid as useful media for dissolution, derivatization, and nanomaterial processing of chitin. Green Sustain. Chem. 2013, 3, 19–25. [Google Scholar] [CrossRef]
- Takada, A.; Kadokawa, J. Fabrication and characterization of polysaccharide ion gels with ionic liquids and their further conversion into value-added sustainable materials. Biomolecules 2015, 5, 244–262. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J. Fabrication of nanostructured and microstructured chitin materials through gelation with suitable dispersion media. RSC Adv. 2015, 5, 12736–12746. [Google Scholar] [CrossRef]
- Tajiri, R.; Setoguchi, T.; Wakizono, S.; Yamamoto, K.; Kadokawa, J. Preparation of self-assembled chitin nanofibers by regeneration from ion gels using calcium halide·dihydrate/methanol solutions. J. Biobased Mater. Bioenergy 2013, 7, 655–659. [Google Scholar] [CrossRef]
- Mukesh, C.; Mondal, D.; Sharma, M.; Prasad, K. Choline chloride-thiourea, a deep eutectic solvent for the production of chitin nanofibers. Carbohydr. Polym. 2014, 103, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Felix, J.M.; Gatenholm, P. The nature of adhesion in composites of modified cellulose fibers and polypropylene. J. Appl. Polym. Sci. 1991, 42, 609–620. [Google Scholar] [CrossRef]
- Valadez-Gonzalez, A.; Cervantes-Uc, J.M.; Olayo, R.; Herrera-Franco, P.J. Effect of fiber surface treatment on the fiber–matrix bond strength of natural fiber reinforced composites. Compos. Part B Eng. 1999, 30, 309–320. [Google Scholar] [CrossRef]
- Vautard, F.; Fioux, P.; Vidal, L.; Schultz, J.; Nardin, M.; Defoort, B. Influence of the carbon fiber surface properties on interfacial adhesion in carbon fiber–acrylate composites cured by electron beam. Compos. Part A Appl. Sci. Manuf. 2011, 42, 859–867. [Google Scholar] [CrossRef]
- Setoguchi, T.; Yamamoto, K.; Kadokawa, J. Preparation of chitin nanofiber-graft-poly(L-lactide-co-e-caprolactone) films by surface-initiated ring-opening graft copolymerization. Polymer 2012, 53, 4977–4982. [Google Scholar] [CrossRef]
- Albertsson, A.C.; Varma, I.K. Aliphatic polyesters: Synthesis, properties and applications. Adv. Polym. Sci. 2002, 157, 1–40. [Google Scholar]
- Seyednejad, H.; Ghassemi, A.H.; van Nostrum, C.F.; Vermonden, T.; Hennink, W.E. Functional aliphatic polyesters for biomedical and pharmaceutical applications. J. Control. Release 2011, 152, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.D.; Detrembleur, C.; Jérôme, R. Novel aliphatic polyesters based on functional cyclic (di)esters. Macromol. Rapid Commun. 2003, 24, 161–172. [Google Scholar] [CrossRef]
- Jérôme, C.; Lecomte, P. Recent advances in the synthesis of aliphatic polyesters by ring-opening polymerization. Adv. Drug Deliv. Rev. 2008, 60, 1056–1076. [Google Scholar] [CrossRef] [PubMed]
- Grijpma, D.W.; Pennings, A.J. Polymerization temperature effects on the properties of L-lactide and e-caprolactone copolymers. Polym. Bull. 1991, 25, 335–341. [Google Scholar] [CrossRef]
- Kricheldorf, H.R. Polypeptides and 100 years of chemistry of alpha-amino acid N-carboxyanhydrides. Angew. Chem. Int. Ed. 2006, 45, 5752–5784. [Google Scholar] [CrossRef] [PubMed]
- Deming, T.J. Synthetic polypeptides for biomedical applications. Prog. Polym. Sci. 2007, 32, 858–875. [Google Scholar] [CrossRef]
- Kadokawa, J.; Setoguchi, T.; Yamamoto, K. Preparation of highly flexible chitin nanofiber-graft-poly(g-L-glutamic acid) network film. Polym. Bull. 2013, 70, 3279–3289. [Google Scholar] [CrossRef]
- Phongying, S.; Aiba, S.; Chirachanchai, S. Direct chitosan nanoscaffold formation via chitin whiskers. Polymer 2007, 48, 393–400. [Google Scholar] [CrossRef]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-catalyzed living radical polymerization. Chem. Rev. 2001, 101, 3689–3745. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K. Atom transfer radical polymerization (ATRP): Current status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yoshida, S.; Mine, S.; Kadokawa, J. Synthesis of chitin-graft-polystyrene via atom transfer radical polymerization initiated from a chitin macroinitiator. Polym. Chem. 2013, 4, 3384–3389. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yoshida, S.; Kadokawa, J. Surface-initiated atom transfer radical polymerization from chitin nanofiber macroinitiator film. Carbohydr. Polym. 2014, 112, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Endo, R.; Yamamoto, K.; Kadokawa, J. Surface-initiated graft atom transfer radical polymerization of methyl methacrylate from chitin nanofiber macroinitiator under dispersion conditions. Fibers 2015, 3, 338–347. [Google Scholar] [CrossRef]






© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadokawa, J.-i. Preparation and Grafting Functionalization of Self-Assembled Chitin Nanofiber Film. Coatings 2016, 6, 27. https://doi.org/10.3390/coatings6030027
Kadokawa J-i. Preparation and Grafting Functionalization of Self-Assembled Chitin Nanofiber Film. Coatings. 2016; 6(3):27. https://doi.org/10.3390/coatings6030027
Chicago/Turabian StyleKadokawa, Jun-ichi. 2016. "Preparation and Grafting Functionalization of Self-Assembled Chitin Nanofiber Film" Coatings 6, no. 3: 27. https://doi.org/10.3390/coatings6030027
APA StyleKadokawa, J.-i. (2016). Preparation and Grafting Functionalization of Self-Assembled Chitin Nanofiber Film. Coatings, 6(3), 27. https://doi.org/10.3390/coatings6030027