
Metagenomics of the Surface of an Architectural Heritage Site: A Case Study of the Ji Family’s Residence in the Southeast of Shanxi Province, China

 ,
,  , ,
, ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Literature Review: Progress in Research on Microorganisms in Cultural Heritage
3. Materials and Methods
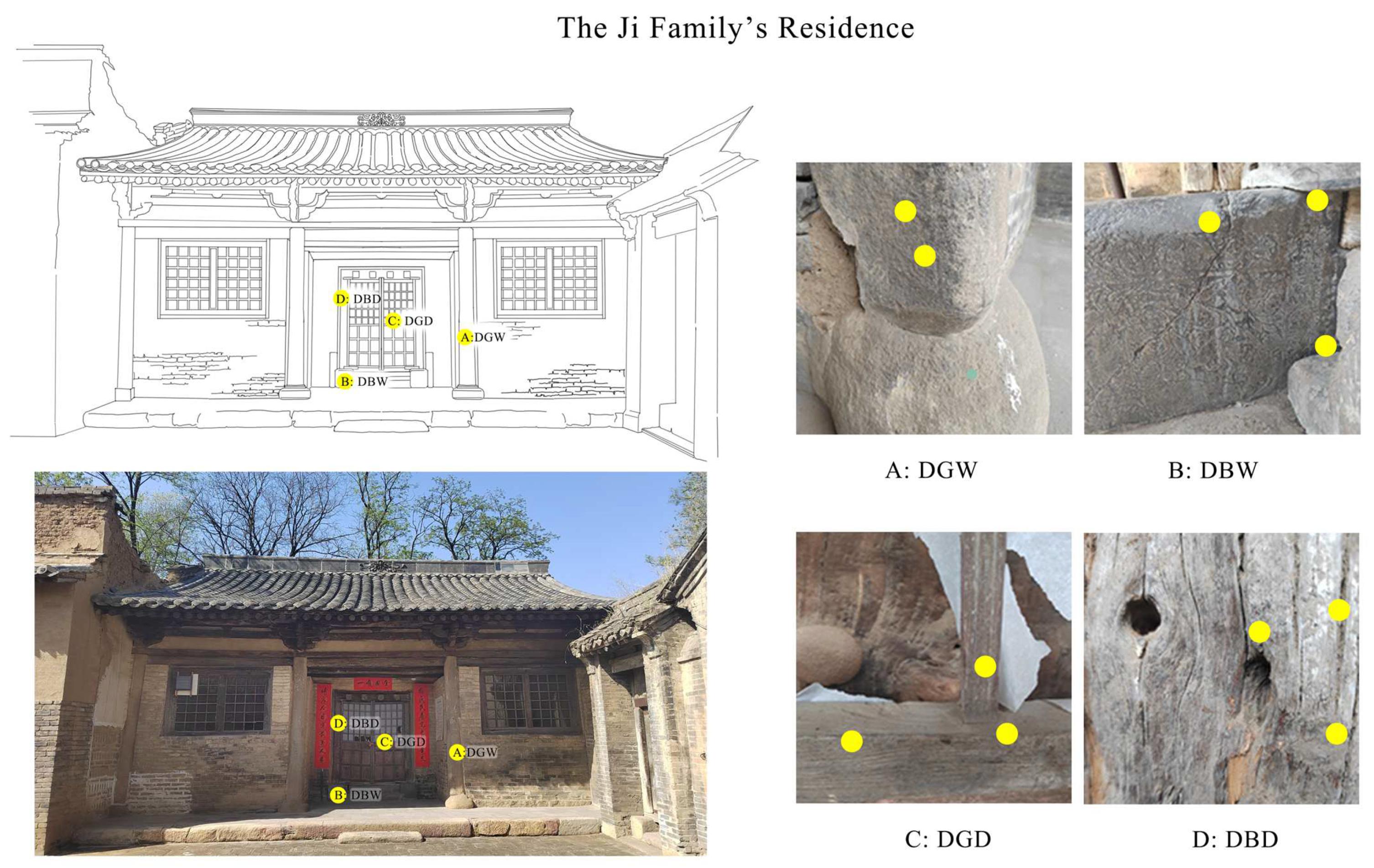
3.1. Site Description and Sampling
3.1.1. Research Objects: The Ji Family’s Residential Houses
3.1.2. Sampling
3.2. DNA Extraction, Library Construction, and Next-Generation Sequencing
3.3. Metagenome Assembly, Taxonomy, and Functional Annotation
4. Results
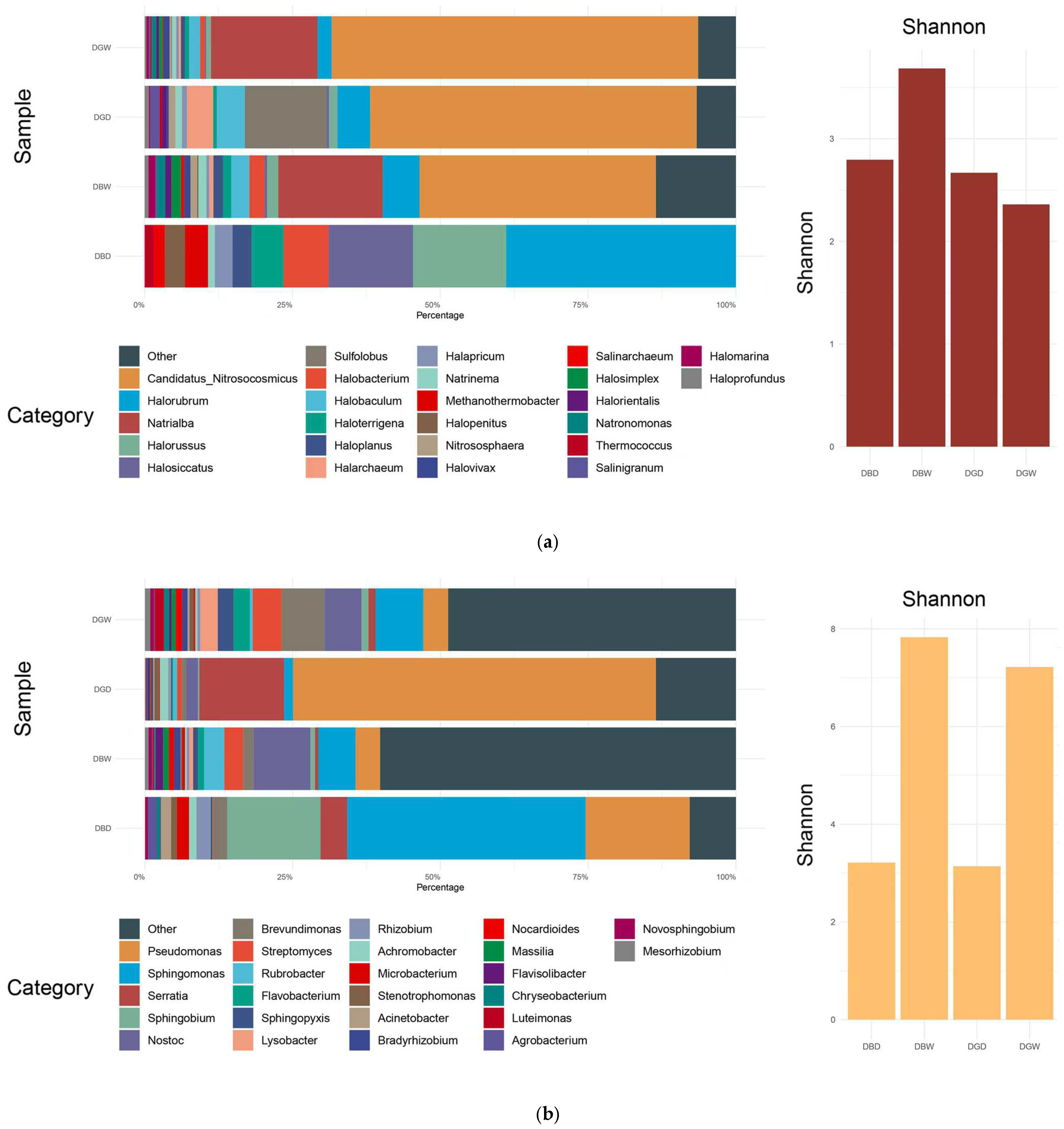
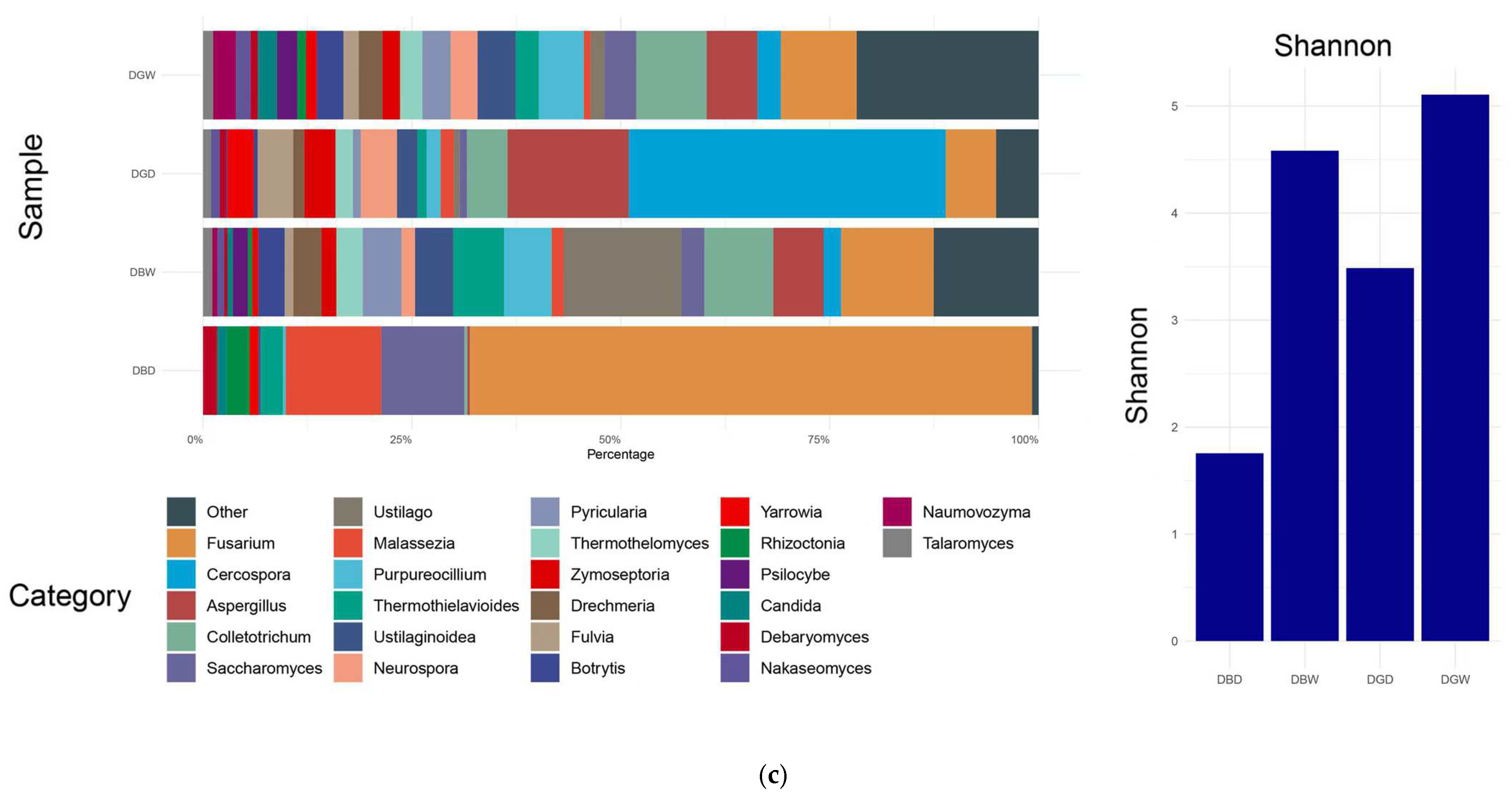
4.1. Microbial Community Composition
4.2. Microbial Diversity
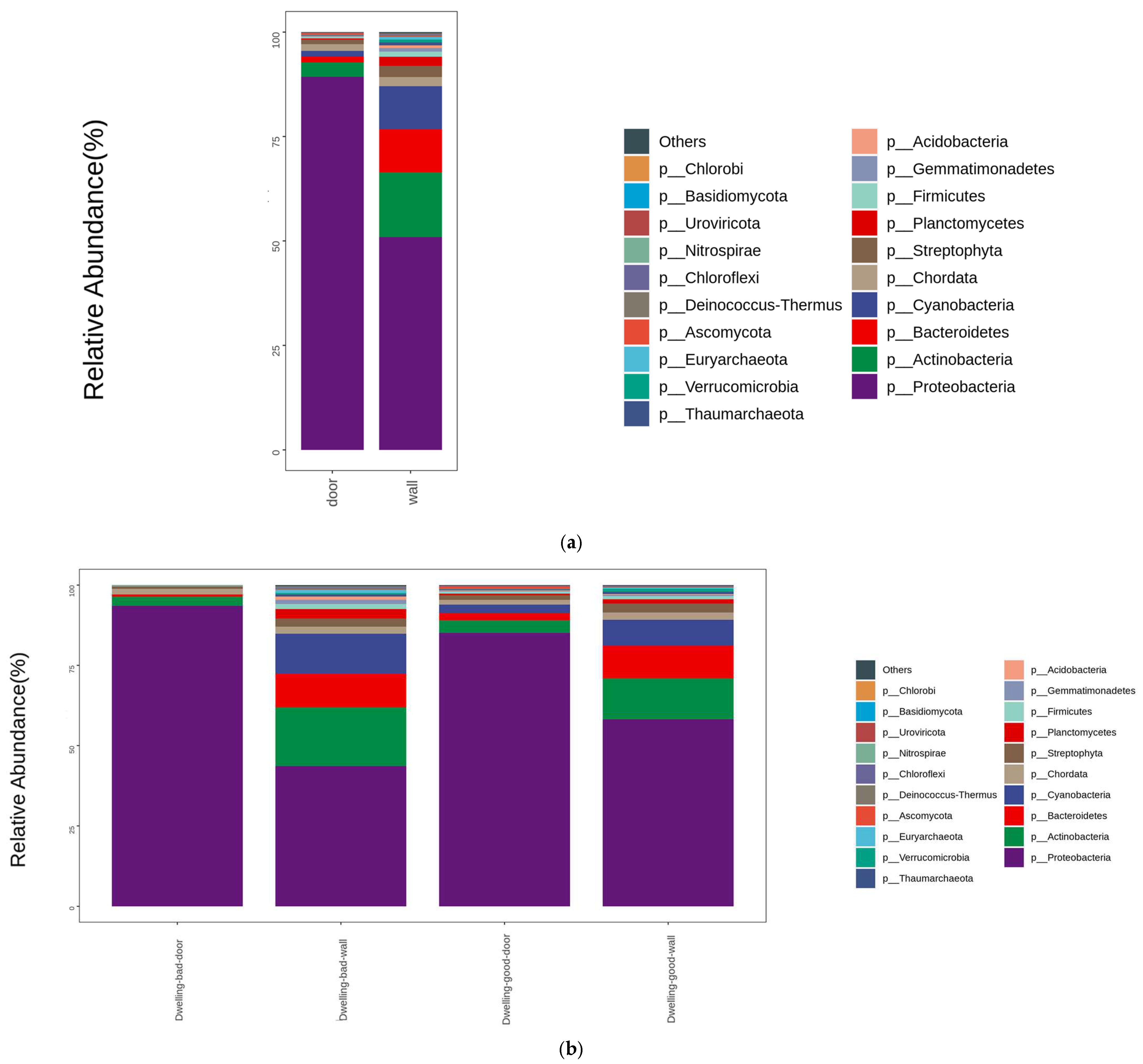
4.3. Composition of Dwelling Microbial Communities in Phylum
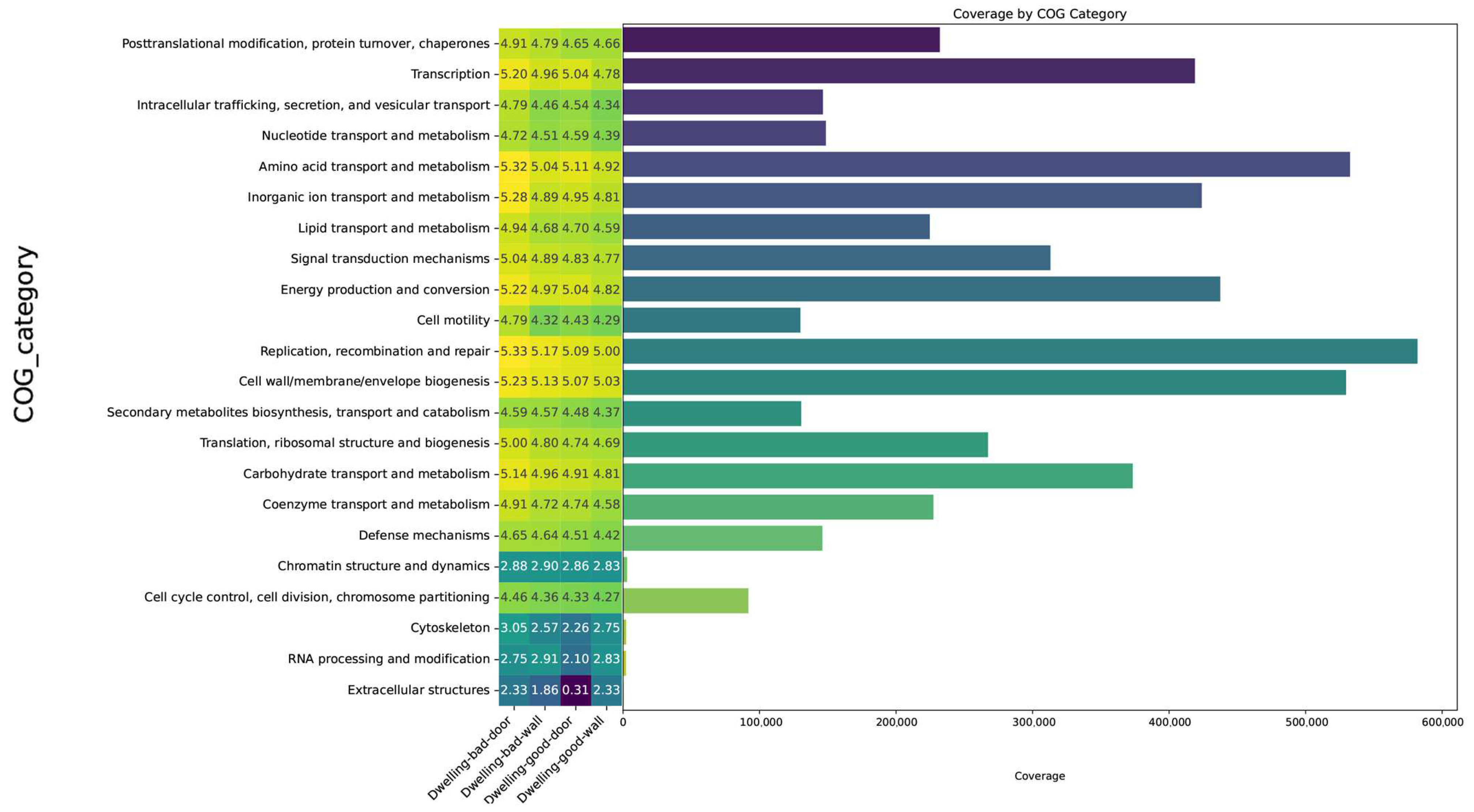
4.4. Microbial Functions Annotated by Cluster of Orthologous Groups of Proteins (COGs)
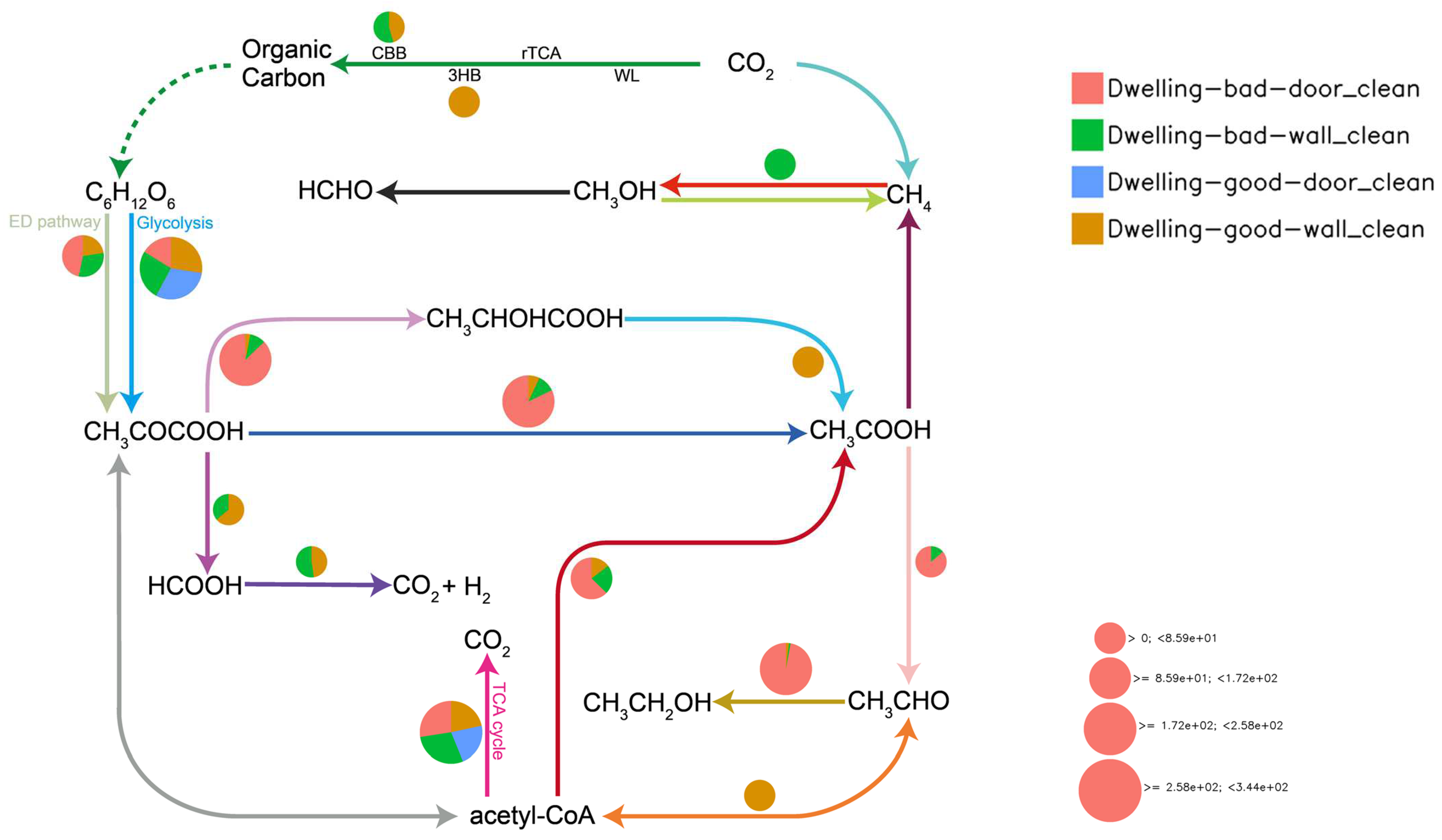
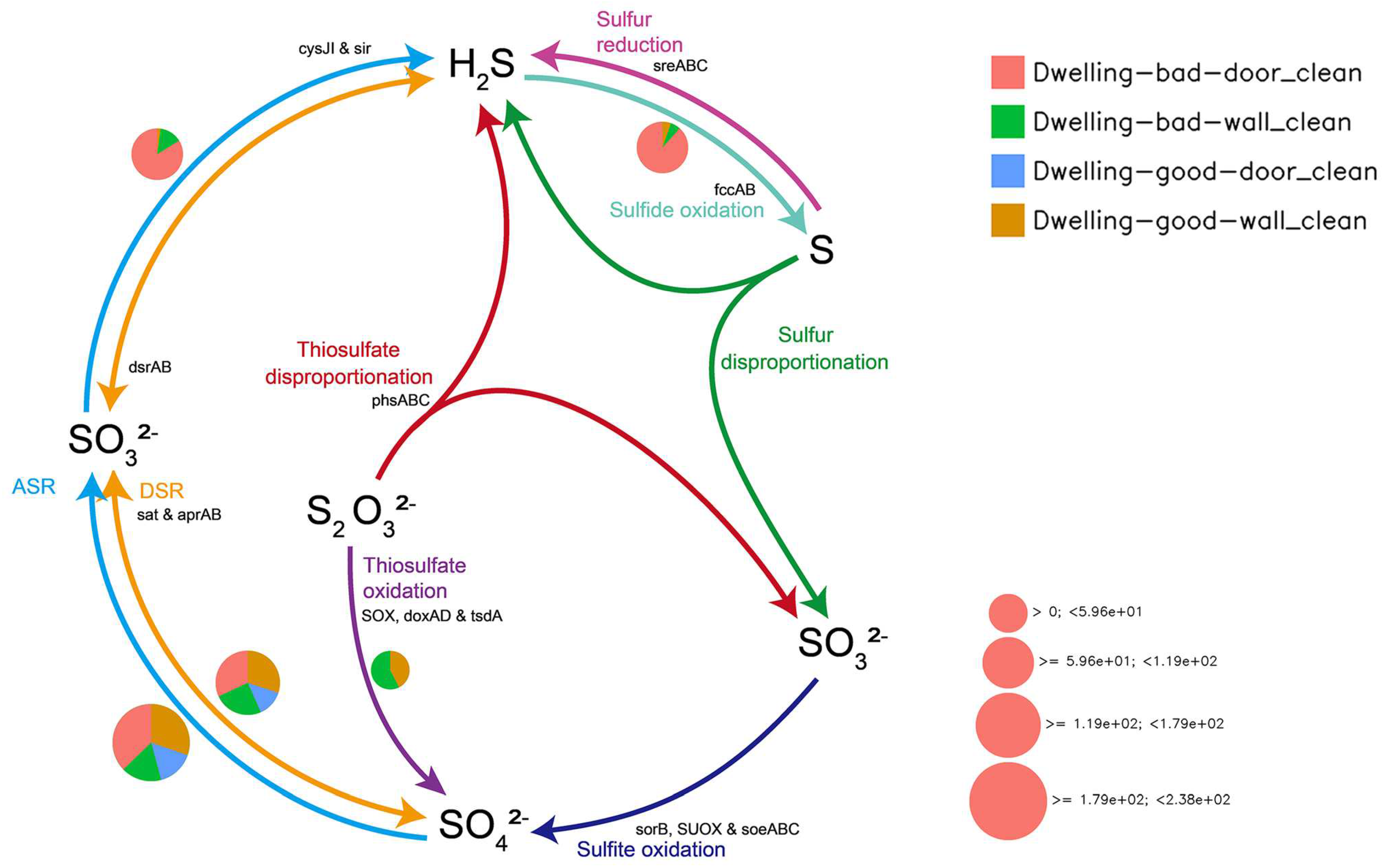
4.5. Metabolic Mapping of Geomicrobiological Cycles
4.5.1. Cabon Cycling
4.5.2. Nitrogen Cycling
4.5.3. Sulfur Cycle
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. The Location Analysis of Ji Family’s Residential Houses

Appendix B. Site Climate Analysis

References
- Ma, Y.; Liu, M. Research on the construction characteristics of Yuan Dynasty dwellings in Jincheng region: A case study of the main house of the Ji family residence in Xiyaotou, Gaoping. China Natl. Exhib. 2024, 92–94. Available online: https://chn.oversea.cnki.net/KCMS/detail/detail.aspx?dbcode=CJFD&dbname=CJFDLAST2024&filename=MZBL202409031&uniplatform=OVERSEA&v=fhcxrMb_7QFoNsQO47XaFf2g5ELvlN-Z8qaYQJJDFwiK0YpkA2aW7RQgomKa3MuM (accessed on 16 October 2024).
- Liu, Q.; Liu, Y.; Zeng, K.; Yang, F.; Zhu, H.; Liu, Q. Advanced design of Chinese traditional materials for the conservation of historic stone buildings. J. Archaeol. Sci. 2011, 38, 1896–1900. [Google Scholar] [CrossRef]
- Huo, Q.; Cheng, X.; Du, W.; Zhang, H.; Han, R. Remote sensing evaluation and monitoring of spatial and temporal changes in ecological environmental quality in coal mining-intensive cities. Appl. Sci. 2024, 14, 8814. [Google Scholar] [CrossRef]
- Suihko, M.L.; Alakomi, H.L.; Gorbushina, A.; Fortune, I.; Marquardt, J.; Saarela, M. Characterization of aerobic bacterial and fungal microbiota on surfaces of historic Scottish monuments. Syst. Appl. Microbiol. 2007, 30, 494–508. [Google Scholar] [CrossRef]
- Sanmartín, P.; DeAraujo, A.; Vasanthakumar, A. Melding the old with the new: Trends in methods used to identify, monitor, and control microorganisms on cultural heritage materials. Microb. Ecol. 2018, 76, 64–80. [Google Scholar] [CrossRef]
- Tong, Y.; Zhu, X.; Wang, C. Discussion on protection, exploitation and utilization of Chinese traditional civilian house before Yuan Dynasty—A case study of Gaoping Jishi Civilian House. In Proceedings of the 2nd International Conference on Electronic & Mechanical Engineering and Information Technology (EMEIT 2012), Shenyang, China, 7 September 2012; Atlantis Press: Amsterdam, The Netherlands, 2012; pp. 366–369. [Google Scholar] [CrossRef]
- People’s Daily Online—Shanxi Channel. “Emperor Yan’s Hometown Ancient Rhyme Changping” Series (6)|Gaoping Ji’s Folk Residence: The Earliest Wooden Structure Folk House Found in the Country. In People’s Daily Online; 27 October 2021. Available online: http://www.sxgp.gov.cn/zjgp/dmgp_423/202110/t20211027_1486099.shtml (accessed on 16 October 2024).
- Stanaszek-Tomal, E. Environmental factors causing the development of microorganisms on the surfaces of national cultural monuments made of mineral building materials. Coatings 2020, 10, 1203. [Google Scholar] [CrossRef]
- Kembel, S.W.; Jones, E.; Kline, J.; Northcutt, D.; Stenson, J.; Womack, A.M.; Bohannan, B.J.M.; Brown, G.Z.; Green, J.L. Architectural design influences the diversity and structure of the built environment microbiome. ISME J. 2012, 6, 1469–1479. [Google Scholar] [CrossRef]
- Verdier, T.; Coutand, M.; Bertron, A.; Roques, C. A review of indoor microbial growth across building materials and sampling and analysis methods. Build. Environ. 2014, 80, 136–149. [Google Scholar] [CrossRef]
- Adams, R.I.; Bhangar, S.; Dannemiller, K.C.; Eisen, J.A.; Fierer, N.; Gilbert, J.A.; Green, J.L.; Marr, L.C.; Miller, S.L.; Siegel, J.A.; et al. Ten questions concerning the microbiomes of buildings. Build. Environ. 2016, 109, 224–234. [Google Scholar] [CrossRef]
- Brown, G.Z.; Kline, J.; Mhuireach, G.; Northcutt, D.; Stenson, J. Making microbiology of the built environment relevant to design. Microbiome 2016, 4, 6. [Google Scholar] [CrossRef]
- Leary, D.H.; Li, R.W.; Hamdan, L.J.; Hervey IV, W.J.; Lebedev, N.; Wang, Z.; Deschamps, J.R.; Kusterbeck, A.W.; Vora, G.J. Integrated metagenomic and metaproteomic analyses of marine biofilm communities. Biofouling 2014, 30, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.J.; Wilkins, D.; Long, E.; Evans, F.; DeMaere, M.Z.; Raftery, M.J.; Cavicchioli, R. The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ. Microbiol. 2013, 15, 1302–1317. [Google Scholar] [CrossRef]
- Grob, C.; Taubert, M.; Howat, A.M.; Burns, O.J.; Dixon, J.L.; Richnow, H.H.; Jehmlich, N.; von Bergen, M.; Chen, Y.; Murrell, J.C. Combining metagenomics with metaproteomics and stable isotope probing reveals metabolic pathways used by a naturally occurring marine methylotroph. Environ. Microbiol. 2015, 17, 4007–4018. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Ren, H.; Lu, Y.; Fang, C.; Hou, G.; Yang, Z.; Chen, B.; Yang, F.; Zhao, Y.; Shi, Z.; et al. Distinct gut metagenomics and metaproteomics signatures in prediabetics and treatment-naïve type 2 diabetics. EBioMedicine 2019, 47, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Guirro, M.; Costa, A.; Gual-Grau, A.; Mayneris-Perxachs, J.; Torrell, H.; Herrero, P.; Canela, N.; Arola, L. Multi-omics approach to elucidate the gut microbiota activity: Metaproteomics and metagenomics connection. Electrophoresis 2018, 39, 1692–1701. [Google Scholar] [CrossRef]
- Hassa, J.; Maus, I.; Off, S.; Pühler, A.; Scherer, P.; Klocke, M.; Schlüter, A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl. Microbiol. Biotechnol. 2018, 102, 5045–5063. [Google Scholar] [CrossRef]
- An, X.; Chen, Y.; Chen, G.; Feng, L.; Zhang, Q. Integrated metagenomic and metaproteomic analyses reveal potential degradation mechanism of azo dye-Direct Black G by thermophilic microflora. Ecotoxicol. Environ. Saf. 2020, 196, 110557. [Google Scholar] [CrossRef]
- Piñar, G.; Poyntner, C.; Tafer, H.; Sterflinger, K. A time travel story: Metagenomic analyses decipher the unknown geographical shift and the storage history of possibly smuggled antique marble statues. Ann. Microbiol. 2019, 69, 1001–1021. [Google Scholar] [CrossRef]
- Wu, F.; Ding, X.; Zhang, Y.; Gu, J.-D.; Liu, X.; Guo, Q.; Li, J.; Feng, H. Metagenomic and metaproteomic insights into the microbiome and the key geobiochemical potentials on the sandstone of rock-hewn Beishiku Temple in Northwest China. Sci. Total Environ. 2023, 893, 164616. [Google Scholar] [CrossRef]
- Marvasi, M.; Cavalieri, D.; Mastromei, G.; Casaccia, A.; Perito, B. Omics technologies for an in-depth investigation of biodeterioration of cultural heritage. Int. Biodeterior. Biodegrad. 2019, 144, 104736. [Google Scholar] [CrossRef]
- Saridaki, A.; Katsivela, E.; Glytsos, T.; Tsiamis, G.; Violaki, E.; Kaloutsakis, A.; Kalogerakis, N.; Lazaridis, M. Identification of bacterial communities on different surface materials of museum artefacts using high throughput sequencing. J. Cult. Herit. 2022, 54, 44–52. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, J.; Chen, R.; Coleine, C.; Liu, W.; Delgado-Baquerizo, M.; Feng, Y. Unearthing the global patterns of cultural heritage microbiome for conservation. Int. Biodeterior. Biodegrad. 2024, 190, 105784. [Google Scholar] [CrossRef]
- Wei, W. A Study on the Xiagetuoyuan Courtyard of Yuan Dynasty Dwellings. World Cult. Relics. 2016, 29–31+47. Available online: https://chn.oversea.cnki.net/KCMS/detail/detail.aspx?dbcode=CJFD&dbname=CJFDLAST2016&filename=WWJK201604009&uniplatform=OVERSEA&v=EHxyZp54jL2Bqgwe7KLEguQy4TsyALks28poRufdb5h3pCn31tIjEnzm7Sn9Pnwa (accessed on 25 October 2024).
- Zhang, G. Yuan Dynasty dwelling in Gaoping County: Ji residence. Cult. Relics Q. 1993, 29–33. Available online: https://chn.oversea.cnki.net/KCMS/detail/detail.aspx?dbcode=CJFD&dbname=CJFD9093&filename=WWJK199303003&uniplatform=OVERSEA&v=uIjqNsxATUWM36aj3W6NNgUb2ZFTWj2DnCxVuwcge1HmYNPOBFq_zRdqIYoJbNjs (accessed on 16 October 2024).
- Eriksson, K.E.L.; Blanchette, R.A.; Ander, P. Microbial and Enzymatic Degradation of Wood and Wood Components; Springer: Berlin/Heidelberg, Germany, 1991; Volume 23, p. 1333. [Google Scholar] [CrossRef]
- Wang, B.; Qi, M.; Ma, Y.; Zhang, B.; Hu, Y. Microbiome diversity and cellulose decomposition processes by microorganisms on the ancient wooden seawall of Qiantang River of Hangzhou, China. Microb. Ecol. 2023, 86, 2109–2119. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Xue, C.-X.; Lin, H.; Zhu, X.-Y.; Liu, J.; Zhang, Y.; Rowley, G.; Todd, J.D.; Li, M.; Zhang, X.-H. DiTing: A pipeline to infer and compare biogeochemical pathways from metagenomic and metatranscriptomic data. Front. Microbiol. 2021, 12, 698286. [Google Scholar] [CrossRef]
- Tláskal, V.; Brabcová, V.; Baldrian, P. Complementary roles of wood-inhabiting fungi and bacteria facilitate deadwood decomposition. Msystems 2021, 6, 10–1128. [Google Scholar] [CrossRef]
- He, J.; Zhang, N.; Shao, Y. Deciphering environmental resistome and mobilome risks on the stone monument: A reservoir of antimicrobial resistance genes. Sci. Total Environ. 2022, 838, 156443. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.D.; Roman, M.; Esselman, T.; Mitchell, R. The role of microbial biofilms in deterioration of space station candidate materials. Int. Biodeterior. Biodegrad. 1998, 41, 25–33. [Google Scholar] [CrossRef]
- Liu, X.; Koestler, R.J.; Warscheid, T.; Katayama, Y.; Gu, J.D. Microbial deterioration and sustainable conservation of stone monuments and buildings. Nat. Sustain. 2020, 3, 991–1004. [Google Scholar] [CrossRef]
- Liu, X.; Qian, Y.; Wu, F.; Wang, Y.; Wang, W.; Gu, J.D. Biofilms on stone monuments: Biodeterioration or bioprotection? Trends Microbiol. 2022, 30, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Park, J.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef]
- Galloway, J.N.; Dentener, F.J.; Capone, D.G.; Boyer, E.W.; Howarth, R.W.; Seitzinger, S.P.; Asner, G.P.; Cleveland, C.C.; Green, P.A.; Holland, E.A.; et al. Nitrogen cycles: Past, present, and future. Biogeochemistry 2004, 70, 153–226. [Google Scholar] [CrossRef]
- Lehtovirta-Morley, L.E.; Stoecker, K.; Vilcinskas, A.; Prosser, J.I.; Nicol, G.W. Cultivation of an obligate acidophilic ammonia oxidizer from a nitrifying acid soil. Proc. Natl. Acad. Sci. USA 2011, 108, 15892–15897. [Google Scholar] [CrossRef]
- Orellana, L.H.; Rodriguez-R, L.M.; Higgins, S.; Chee-Sanford, J.C.; Sanford, R.A.; Ritalahti, K.M.; Löffler, F.E.; Konstantinidis, K.T. Detecting nitrous oxide reductase (nosZ) genes in soil metagenomes: Method development and implications for the nitrogen cycle. MBio 2014, 5, 10–1128. [Google Scholar] [CrossRef]
- Ding, X.; Lan, W.; Wu, J.; Hong, Y.; Li, Y.; Ge, Q.; Urzì, C.; Katayama, Y.; Gu, J.-D. Microbiome and nitrate removal processes by microorganisms on the ancient Preah Vihear temple of Cambodia revealed by metagenomics and N-15 isotope analyses. Appl. Microbiol. Biotechnol. 2020, 104, 9823–9837. [Google Scholar] [CrossRef]
- Kountz, D.J.; Balskus, E.P. Leveraging microbial genomes and genomic context for chemical discovery. Acc. Chem. Res. 2021, 54, 2788–2797. [Google Scholar] [CrossRef]
- Prosser, J.I.; Nicol, G.W. Archaeal and bacterial ammonia-oxidisers in soil: The quest for niche specialisation and differentiation. Trends Microbiol. 2012, 20, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Wei, S.P.; Zheng, Y.; Choi, E.; Li, X.; Johnston, J.; Wan, X.; Abrahamson, B.; Flinkstrom, Z.; Wang, B.; et al. Ammonia-oxidizing bacteria and archaea exhibit differential nitrogen source preferences. Nat. Microbiol. 2024, 9, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Ni, G.; Leung, P.M.; Daebeler, A.; Guo, J.; Hu, S.; Cook, P.; Nicol, G.W.; Daims, H.; Greening, C. Nitrification in acidic and alkaline environments. Essays Biochem. 2023, 67, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.G.; Bardischewsky, F.; Rother, D.; Quentmeier, A.; Fischer, J. Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 2005, 8, 253–259. [Google Scholar] [CrossRef]
- Huber, B.; Herzog, B.; Drewes, J.E.; Koch, K.; Müller, E. Characterization of sulfur oxidizing bacteria related to biogenic sulfuric acid corrosion in sludge digesters. BMC Microbiol. 2016, 16, 153. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; Nelson, D.C. Sulfide oxidation in marine sediments: Geochemistry meets microbiology. Geol. Soc. Am. Spec. Pap. 2004, 379, 63–81. [Google Scholar] [CrossRef]
- Ghosh, W.; Dam, B. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 2009, 33, 999–1043. [Google Scholar] [CrossRef]
- Hensen, D.; Sperling, D.; Trüper, H.G.; Brune, D.C. Thiosulfate oxidation in the phototrophic sulfur bacterium Allochromatium vinosum. Arch. Microbiol. 2006, 185, 364–373. [Google Scholar] [CrossRef]
- Curson, A.R.; Todd, J.D.; Sullivan, M.J.; Johnston, A.W. Catabolism of dimethylsulphoniopropionate: Microorganisms, enzymes and genes. Nat. Rev. Microbiol. 2011, 9, 849–859. [Google Scholar] [CrossRef]
- Seymour, J.R.; Simó, R.; Ahmed, T.; Stocker, R. Chemoattraction to dimethylsulfoniopropionate throughout the marine microbial food web. Science 2010, 329, 342–345. [Google Scholar] [CrossRef]
- Meng, H.; Katayama, Y.; Gu, J.D. More wide occurrence and dominance of ammonia-oxidizing archaea than bacteria at three Angkor sandstone temples of Bayon, Phnom Krom and Wat Athvea in Cambodia. Int. Biodeterior. Biodegrad. 2017, 117, 78–88. [Google Scholar] [CrossRef]
- Gaylarde, C.; Morton, L. Deteriogenic biofilms on buildings and their control: A review. Biofouling 1999, 14, 59–74. [Google Scholar] [CrossRef]
- Chimienti, G.; Piredda, R.; Pepe, G.; van der Werf, I.D.; Sabbatini, L.; Crecchio, C.; Ricciuti, P.; D’Erchia, A.M.; Manzari, C.; Pesole, G. Profile of microbial communities on carbonate stones of the medieval church of San Leonardo di Siponto (Italy) by Illumina-based deep sequencing. Appl. Microbiol. Biotechnol. 2016, 100, 8537–8548. [Google Scholar] [CrossRef]
- David, S.R.; Jaouen, A.; Ihiawakrim, D.; Geoffroy, V.A. Biodeterioration of asbestos cement by siderophore-producing Pseudomonas. J. Hazard. Mater. 2021, 403, 123699. [Google Scholar] [CrossRef]
- Stocks-Fischer, S.; Galinat, J.; Bang, S. Microbiological precipitation of CaCO3. Soil Biol. Biochem. 1999, 31, 1563–1571. [Google Scholar] [CrossRef]
- Hobmeier, K.; Goëss, M.C.; Sehr, C.; Schwaminger, S.; Berensmeier, S.; Kremling, A.; Kunte, H.J.; Pflüger-Grau, K.; Marin-Sanguino, A. Anaplerotic Pathways in Halomonas elongata: The Role of the Sodium Gradient. Front. Microbiol. 2020, 11, 561800. [Google Scholar] [CrossRef]
- Leys, N.; Ryngaert, A.; Springael, D. Occurrence and phylogenetic diversity of Sphingomonas in soils contaminated with polycyclic aromatic hydrocarbons. Appl. Environ. Microbiol. 2004, 70, 1944–1955. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Lan, W.; Yan, A.; Li, Y.; Katayama, Y.; Gu, J.-D. Microbiome characteristics and the key biochemical reactions identified on stone world cultural heritage under different climate conditions. J. Environ. Manag. 2022, 302, 114041. [Google Scholar] [CrossRef]
- Zanardini, E.; May, E.; Purdy, K.J.; Murrell, J.C. Nutrient cycling potential within microbial communities on culturally important stoneworks. Environ. Microbiol. Rep. 2019, 11, 147–154. [Google Scholar] [CrossRef]
- Aho, A.; DeMartini, N.; Murzin, D. Pyrolysis of pine and gasification of pine chars—Influence of organically bound metals. Bioresour. Technolgy 2013, 128, 22–29. [Google Scholar] [CrossRef]
- Turrini, P.; Chebbi, A.; Riggio, F.P. The geomicrobiology of limestone, sulfuric acid speleogenetic, and volcanic caves: Basic concepts and future perspectives. Front. Microbiol. 2024, 15, 1370520. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, A.; Li, X.S.; Katayama, Y. Mycobacteria Isolated from Angkor Monument Sandstones Grow Chemolithoautotrophically by Oxidizing Elemental Sulfur. Front. Microbiol. 2011, 2, 104. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Jiang, Z.; Liu, H.; Zhou, D.; Sanchez-Silva, M. Microbiologically induced deterioration of concrete: A review. Braz. J. Microbiol. 2013, 44, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhao, F.; Li, Y.; Zhang, J.; Feng, Y. Variations in the bacterial community compositions at different sites in the tomb of Emperor Yang of the Sui Dynasty. Microbiol. Res. 2017, 196, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Sui Pheng, L. Construction of dwellings and structures in ancient China. Struct. Surv. 2001, 19, 262–274. [Google Scholar] [CrossRef]
- Hasegawa, N.; Sugiyama, M.; Igarashi, K. Acetylxylan esterase is the key to the host specialization of wood-decay fungi predicted by random forest machine-learning algorithm. J. Wood Sci. 2024, 70, 44. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhao, M.; Jiang, J.; Chen, Y.; Chen, H.; Zheng, L.; Chen, H.; Wu, Y. Metagenomics of the Surface of an Architectural Heritage Site: A Case Study of the Ji Family’s Residence in the Southeast of Shanxi Province, China. Coatings 2025, 15, 337. https://doi.org/10.3390/coatings15030337
Li Y, Zhao M, Jiang J, Chen Y, Chen H, Zheng L, Chen H, Wu Y. Metagenomics of the Surface of an Architectural Heritage Site: A Case Study of the Ji Family’s Residence in the Southeast of Shanxi Province, China. Coatings. 2025; 15(3):337. https://doi.org/10.3390/coatings15030337
Chicago/Turabian StyleLi, Yanyu, Mingyi Zhao, Jinyan Jiang, Yile Chen, Haojie Chen, Liang Zheng, Huanhuan Chen, and Yue Wu. 2025. "Metagenomics of the Surface of an Architectural Heritage Site: A Case Study of the Ji Family’s Residence in the Southeast of Shanxi Province, China" Coatings 15, no. 3: 337. https://doi.org/10.3390/coatings15030337
APA StyleLi, Y., Zhao, M., Jiang, J., Chen, Y., Chen, H., Zheng, L., Chen, H., & Wu, Y. (2025). Metagenomics of the Surface of an Architectural Heritage Site: A Case Study of the Ji Family’s Residence in the Southeast of Shanxi Province, China. Coatings, 15(3), 337. https://doi.org/10.3390/coatings15030337