
First-Principles Study on the Electronic Structure and Optical Properties of BiOIO3 Doped with As, Se, and Te

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
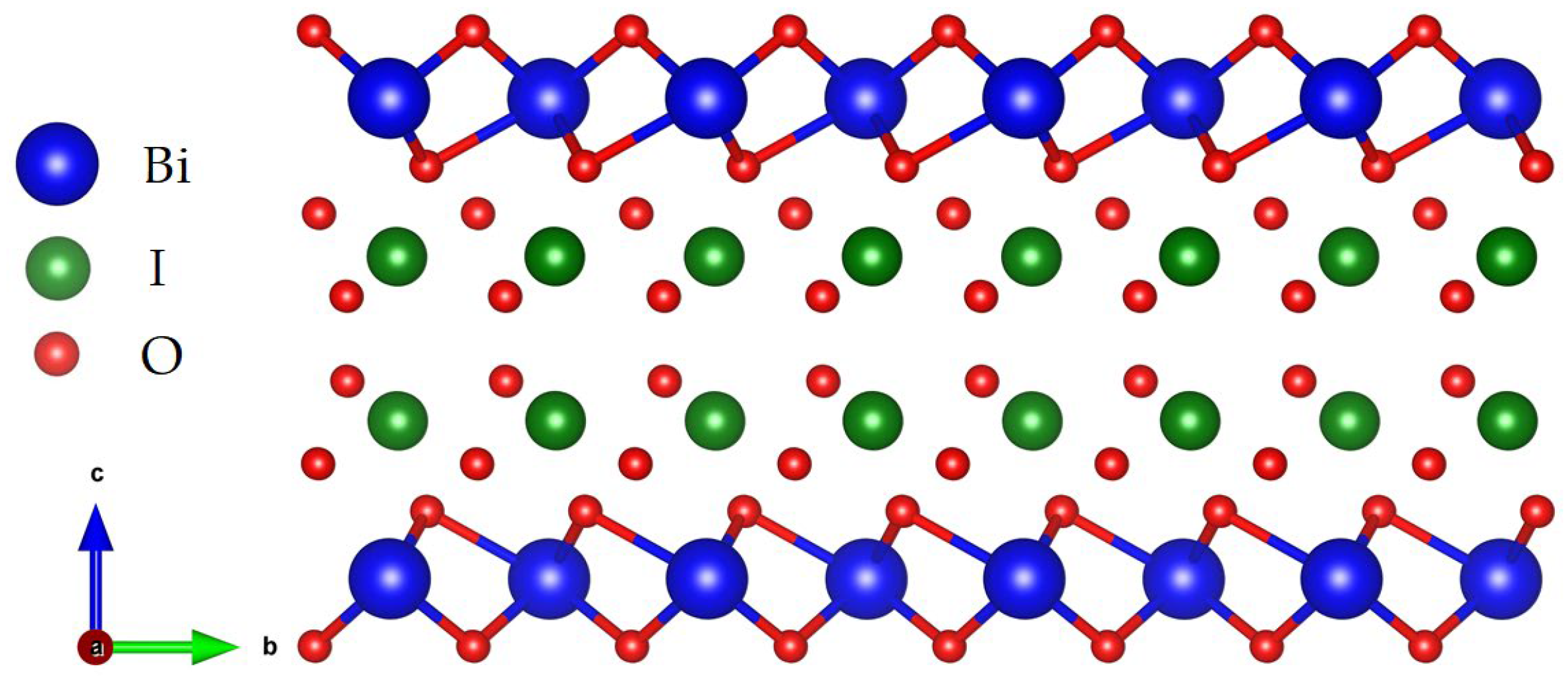
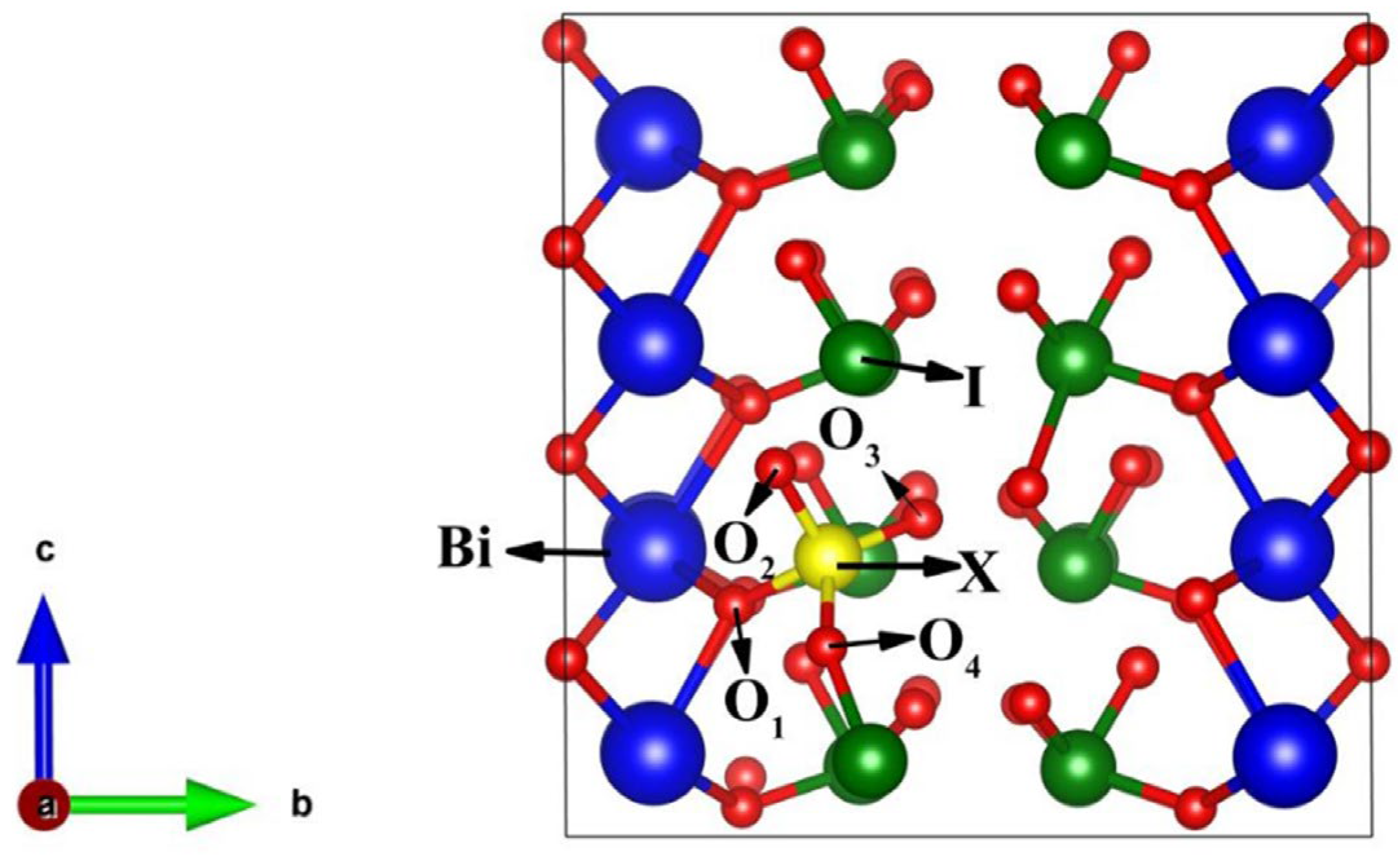
3.1. Geometric Optimization Results
3.2. Structural Stability of BiOIO3 and X-BiOIO3
3.3. Electronic Structure of BiOIO3 and X-BiOIO3
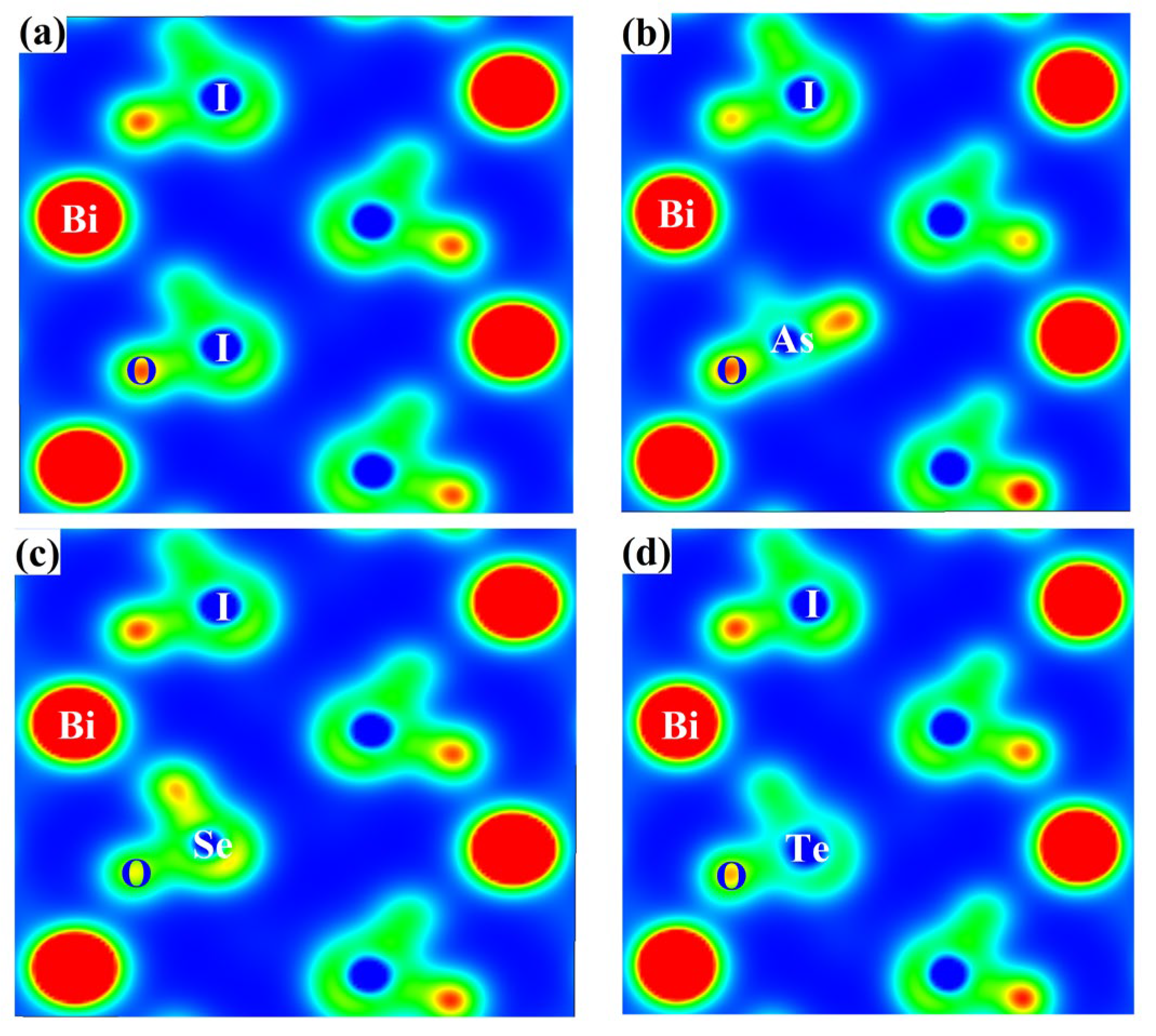
3.4. The Two-Dimensional Electron Density and Bader Charges of BiOIO3 and X-BiOIO3
3.5. Optical Properties of BiOIO3 and X-BiOIO3
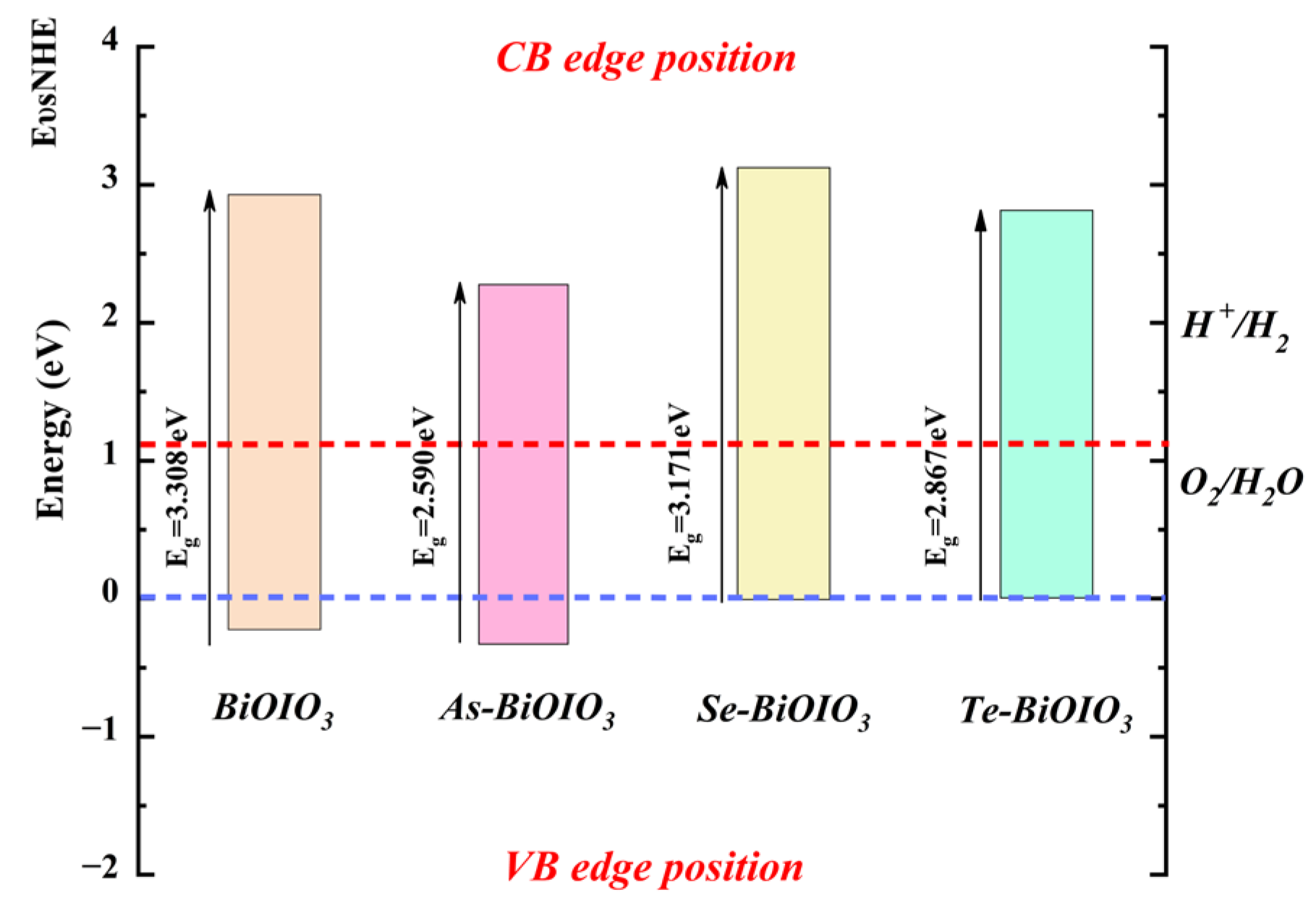
3.5.1. Alignment with Band Structure Edges
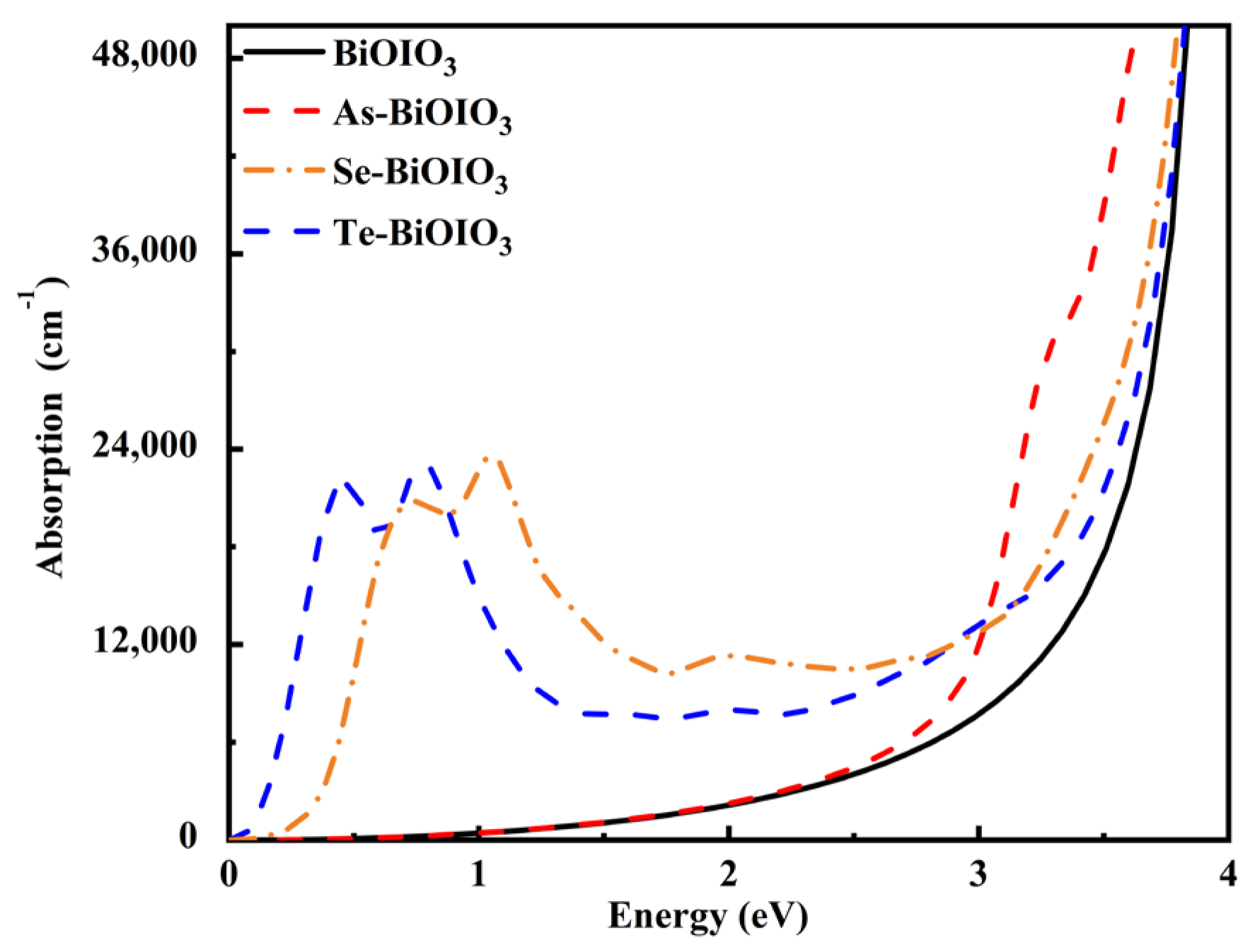
3.5.2. Absorption Spectrum
3.5.3. Dielectric Function
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Dasgupta, S. Visible light induced photocatalytic degradation of organic pollutants. J. Photochem. Photobiol. C Photochem. Rev. 2005, 6, 186–205. [Google Scholar] [CrossRef]
- Ma, S.; Yu, X.; Li, W.; Kong, J.; Long, D.; Bai, X. Bismuth-based photocatalysts for pollutant degradation and bacterial disinfection in sewage system: Classification, modification and mechanism. Environ. Res. 2024, 264, 120297. [Google Scholar] [CrossRef]
- El-Bahy, S.M.; Arshad, J.; Munir, S.; Chaudhary, K.; Alhashmialameer, D.; Eddy, D.R.; Warsi, M.F.; Shahid, M. Improved photocatalytic performance of a new silver doped BiSbO4 photocatalyst. Ceram. Int. 2022, 48, 23915. [Google Scholar] [CrossRef]
- Grosso, D.; Boissière, C.; Smarsly, B.; Brezesinski, T.; Pinna, N.; Albouy, P.A.; Amenitsch, H.; Antonietti, M.; Sanchez, C. Periodically ordered nanoscale islands and mesoporous films composed of nanocrystalline multimetallic oxides. Nat. Mater. 2004, 3, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Subramanian, B. A strategy to fabricate bismuth ferrite (BiFeO3) nanotubes from electrospun nanofibers and their Solar light-driven photocatalytic properties. RSC Adv. 2013, 3, 23737–23744. [Google Scholar] [CrossRef]
- Cui, D.H.; Zheng, Y.F.; Song, X.C. Hydrothermal synthesis, characterisation and photocatalytic properties of BiOIO3 nanoplatelets. J. Exp. Nanosci. 2016, 11, 1000–1010. [Google Scholar] [CrossRef]
- Li, J.; Xie, J.; Zhang, X.; Lu, E.; Cao, Y. The Solid-State Synthesis of BiOIO3 Nanoplates with Boosted Photocatalytic Degradation Ability for Organic Contaminants. Molecules 2023, 28, 3681. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Z.; Chen, T.Z.; Shui, J.Y.; Zhi, Y.Y.; Yun, Y. Ferroelectric polarization effect promoting the bulk charge separation for enhance the efficiency of photocatalytic degradation. Chem. Eng. J. 2021, 410, 128430. [Google Scholar]
- Huang, H.; Chen, F.; Reshak, A.H.; Auluck, S.; Zhang, Y. Insight into crystal-structure dependent charge separation and photo-redox catalysis: A combined experimental and theoretical study on Bi(IO3)3 and BiOIO3. Appl. Surf. Sci. 2018, 458, 129–138. [Google Scholar] [CrossRef]
- Dong, X.D.; Yao, G.Y.; Liu, Q.L.; Zhao, Q.M.; Zhao, Z.Y. Spontaneous polarization effect and photocatalytic activity of layered compound of BiOIO3. Inorg. Chem. 2019, 58, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Patiphatpanya, P.; Phuruangrat, A.; Thongtem, S.; Kungwankunakorn, S.; Thongtem, T. Effect of microwave power on phase, morphology, and photocatalytic properties of BiOIO3 nanostructure. J. Aust. Ceram. Soc. 2019, 55, 501–506. [Google Scholar] [CrossRef]
- Sun, Y.; Xiong, T.; Dong, F.; Huang, H.; Cen, W. Interlayer-I-doped BiOIO3 nanoplates with an optimized electronic structure for efficient visible light photocatalysis. Chem. Commun. 2016, 52, 8243–8246. [Google Scholar] [CrossRef]
- Nguyen, S.D.; Yeon, J.; Kim, S.; Halasyamani, P.S. BiO(IO3): A New Polar Iodate that Exhibits an Aurivillius-Type (Bi2O2)2+ Layer and a Large Shg Response. J. Am. Chem. Soc. 2011, 133, 12422–12425. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, Y.; Li, Y.; Huang, S.; Xu, Y.; Xu, H.; Li, H. Calcination synthesis of N-doped BiOIO3 with high Led-light-driven photocatalytic activity. Mater. Lett. 2019, 246, 219–222. [Google Scholar] [CrossRef]
- Liu, Y.L.; Zhu, Z.R.; Liu, Y.Q.; Jiang, W.; Yang, L.; Zi, J.X.; Shuai, Q.; Yu, B.W.Y.; Meng, J.B. First principles insight on enhanced photocatalytic performance of sulfur-doped bismuth oxide iodate. Mater. Sci. Semicond. Process 2023, 165, 2–7. [Google Scholar] [CrossRef]
- Huang, H.; Ou, H.; Feng, J.; Du, X.; Zhang, Y. Achieving highly promoted visible-light sensitive photocatalytic activity on BiOIO3 via facile iodine doping. Colloids Surf. A Physicochem. Eng. Asp. 2017, 518, 158–165. [Google Scholar] [CrossRef]
- Yi, W.; Tang, G.; Chen, X.; Yang, B.; Liu, X. qvasp: A flexible toolkit for VASP users in materials simulations. Comput. Phys. Commun. 2020, 257, 107535. [Google Scholar] [CrossRef]
- Debidatta, B.; Jisha, A.A.; Sharma, R.; Sanat, K.M.; Ekta, J. First Principles Study of New d0 Half-Metallic Ferromagnetism in CsBaC Ternary Half-Heusler Alloy. J. Supercond. Nov. Magn. 2022, 35, 3431–3437. [Google Scholar]
- Monkhorst, J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Wu, J.; Kai, X.; Qi, Z.L.; Chen, H.Q.; Xue, M.Q.; Hui, Z.; Yu, G.; Ping, H.; Liang, J.Z. Controlling dominantly reactive (010) facets and impurity level by in-situ reduction of BiOIO3 for enhancing photocatalytic activity. Appl. Catal. B Environ. 2018, 232, 136–144. [Google Scholar] [CrossRef]
- Arora, A.; Nandi, P.; De Sarkar, A. Ferroelectricity-controlled magnetic ordering and spin photocurrent in NiCl2/GeS multiferroic heterostructures. J. Phys. Condens. Matter 2024, 36, 445301. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liu, G.; Li, X.; Wang, H.; Zhang, G. First-principles study of strain on BN-doped arsenene. J. Mol. Model. 2022, 28, 190. [Google Scholar] [CrossRef]
- Shahriar, R.; Hoque, K.S.; Tristant, D.; Zubair, A. Vacancy induced magnetism and electronic structure modification in monolayer hexagonal boron arsenide: A first-principles study. Appl. Surf. Sci. 2022, 600, 154053. [Google Scholar] [CrossRef]
- Qi, S.; Wu, S.; Zhang, Y.; Guan, L.; Zhang, K. Construction and first-principles analysis of BiOI and Ni doped MoS2 Z-type heterojunctions. J. Solid State Chem. 2024, 335, 124658. [Google Scholar] [CrossRef]
- Zhou, J.; Li, D.; Zhao, W.; Jing, B.; Ao, Z.; An, T. First-Principles Evaluation of Volatile Organic Compounds Degradation in Z-Scheme Photocatalytic Systems: MXene and Graphitic-CN Heterostructures. ACS Appl. Mater. Interfaces 2021, 13, 23843–23852. [Google Scholar] [CrossRef] [PubMed]
- Ghaithan, H.M.; Alahmed, Z.A.; Qaid, S.M.H.; Aldwayyan, A.S. Structural, Electronic, and Optical Properties of CsPb(Br1−XClx)3 Perovskite: First-Principles Study with PBE-GGA and mBJ-GGA Methods. Materials 2020, 13, 4944. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Feng, X.; Teng, D.; Xu, J.; Cao, S.; Rydosz, A.; Kong, J.; Gao, F. Effect of Intrinsic Nb5+ Vacancy On Dielectric and Polarization Behaviors of KSr2Nb5O15: First-Principles Investigation. Phys. Status Solidi (B)-Basic Solid State Phys. 2021, 259, 2100488. [Google Scholar] [CrossRef]
- Zhao, X.; Dai, M.; Lang, F.M.; Zhao, C.; Chen, Q.Y.; Zhang, L.L.; Huang, Y.N.; Lu, H.M.; Qin, C.X. Investigating the Impact of Stress on the Optical Properties of GaN-MX2 (M=Mo, W; X=S, Se) Heterojunctions Using the First Principles. Catalysts 2024, 14, 732. [Google Scholar] [CrossRef]
- Masihi, A.; Naseri, M.; Fatahi, N. A first-principles study of the electronic and optical properties of monolayer α-PbO. Chem. Phys. Lett. 2019, 721, 27–32. [Google Scholar] [CrossRef]
- Ling, S.Y.; Bang, L.D.; Yu, X.H. Optical properties of arsenene nanoribbons: A first principle study. Mater. Sci. Semicond. Process 2021, 136, 106139. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | a/nm | b/nm | c/nm | V/nm3 |
|---|---|---|---|---|
| BiOIO3 (Experiment) [21] | 0.5658 | 1.1039 | 0.5748 | / |
| BiOIO3 (This work) | 0.5712 | 1.1263 | 0.5788 | 1.4898 |
| As-BiOIO3 | 0.5698 | 1.1311 | 0.5779 | 1.4899 |
| Se-BiOIO3 | 0.5706 | 1.1237 | 0.5780 | 1.4826 |
| Te-BiOIO3 | 0.5695 | 1.1272 | 0.5775 | 1.4830 |
| Model | E/eV | Eb/eV | Ef/eV |
|---|---|---|---|
| BiOIO3 | −477.2118 | / | / |
| As-BiOIO3 | −479.9295 | −0.3186 | −4.9405 |
| Se-BiOIO3 | −479.3817 | −0.3183 | −2.1449 |
| Te-BiOIO3 | −479.9295 | −0.3185 | −2.6956 |
| Model | The Charge of O Atoms | ||||
|---|---|---|---|---|---|
| O1 | O2 | O3 | O4 | Average | |
| BiOIO3 | 6.8260 | 6.9150 | 6.9662 | 6.9119 | 6.9048 |
| As-BiOIO3 | 7.0762 | 7.0254 | 6.9087 | 7.1356 | 7.0364 |
| Se-BiOIO3 | 7.0203 | 7.0499 | 6.9736 | 7.0125 | 7.0141 |
| Te-BiOIO3 | 7.0304 | 7.1611 | 7.0102 | 7.1411 | 7.0857 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lang, F.; Wen, X.; Liu, J.; Huang, Y.; Zhang, L.; Lu, H.; Jiang, K.; Zhang, B. First-Principles Study on the Electronic Structure and Optical Properties of BiOIO3 Doped with As, Se, and Te. Coatings 2025, 15, 111. https://doi.org/10.3390/coatings15010111
Lang F, Wen X, Liu J, Huang Y, Zhang L, Lu H, Jiang K, Zhang B. First-Principles Study on the Electronic Structure and Optical Properties of BiOIO3 Doped with As, Se, and Te. Coatings. 2025; 15(1):111. https://doi.org/10.3390/coatings15010111
Chicago/Turabian StyleLang, Fumei, Xue Wen, Jibo Liu, Yineng Huang, Lili Zhang, Haiming Lu, Kaiye Jiang, and Baohua Zhang. 2025. "First-Principles Study on the Electronic Structure and Optical Properties of BiOIO3 Doped with As, Se, and Te" Coatings 15, no. 1: 111. https://doi.org/10.3390/coatings15010111
APA StyleLang, F., Wen, X., Liu, J., Huang, Y., Zhang, L., Lu, H., Jiang, K., & Zhang, B. (2025). First-Principles Study on the Electronic Structure and Optical Properties of BiOIO3 Doped with As, Se, and Te. Coatings, 15(1), 111. https://doi.org/10.3390/coatings15010111