
Investigation of the Adhesion Strength, Fracture Toughness, and Stability of M/Cr2N and M/V2N (M = Ti, Ru, Ni, Pd, Al, Ag, and Cu) Interfaces Based on First-Principles Calculations


Abstract
:1. Introduction
2. Mathematical and Computational Methods
2.1. Methodology
2.2. Bulk Properties
2.3. Surface Convergence Test
2.4. Surface Energy
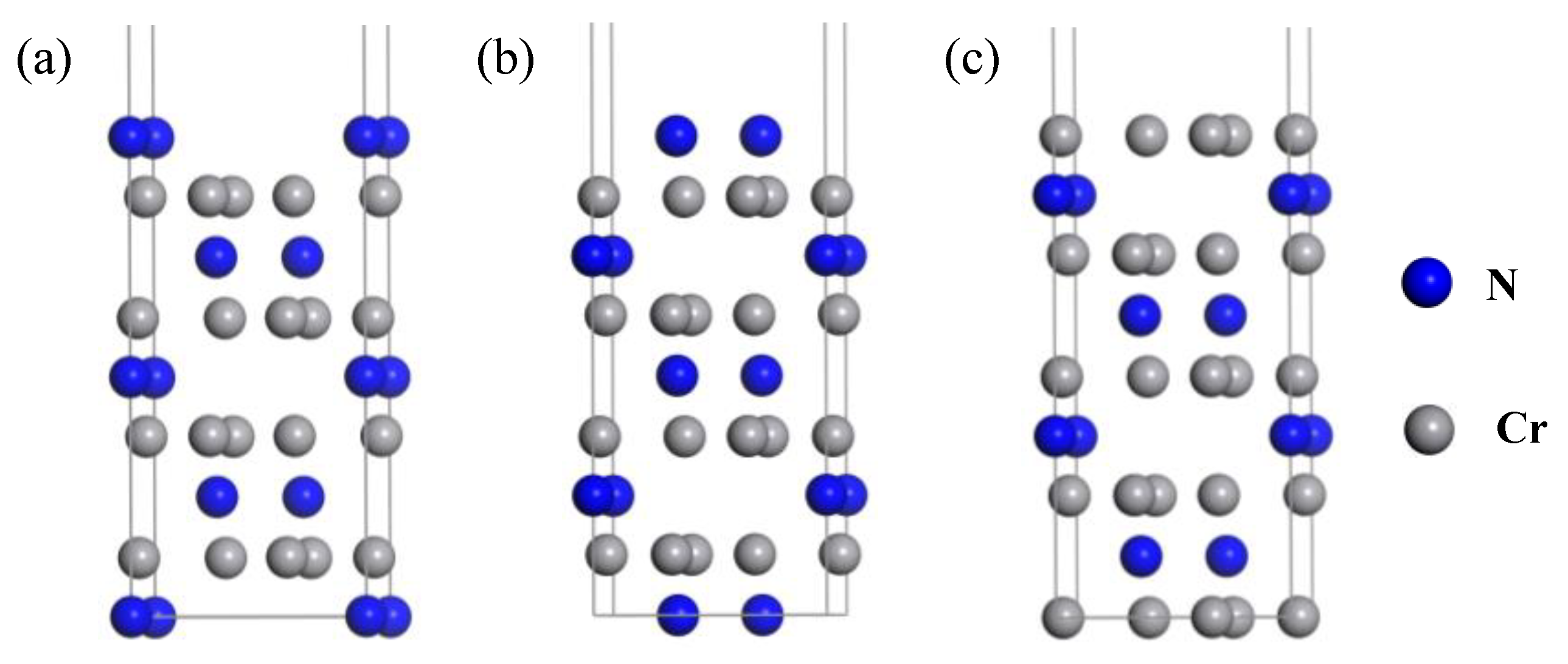
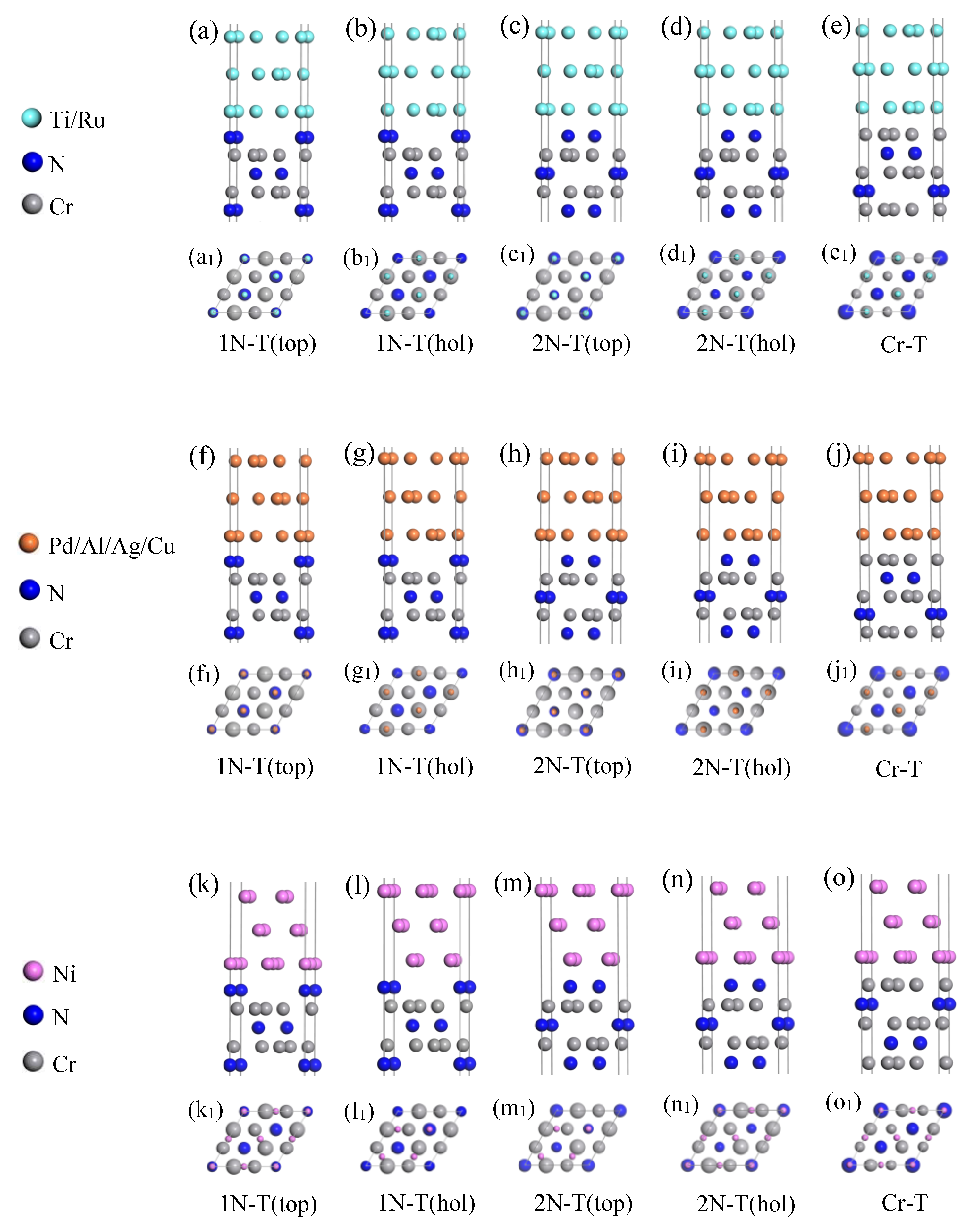
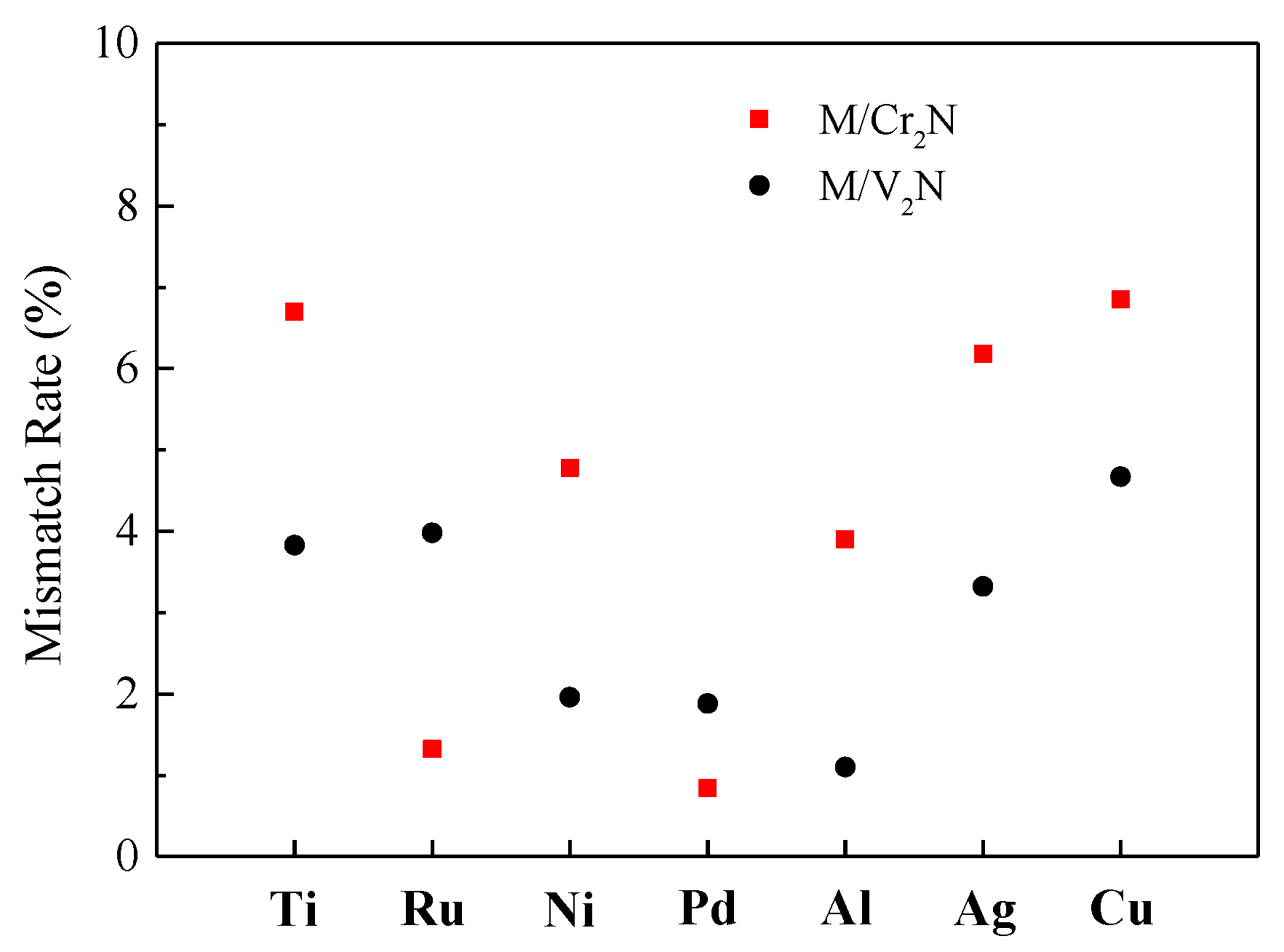
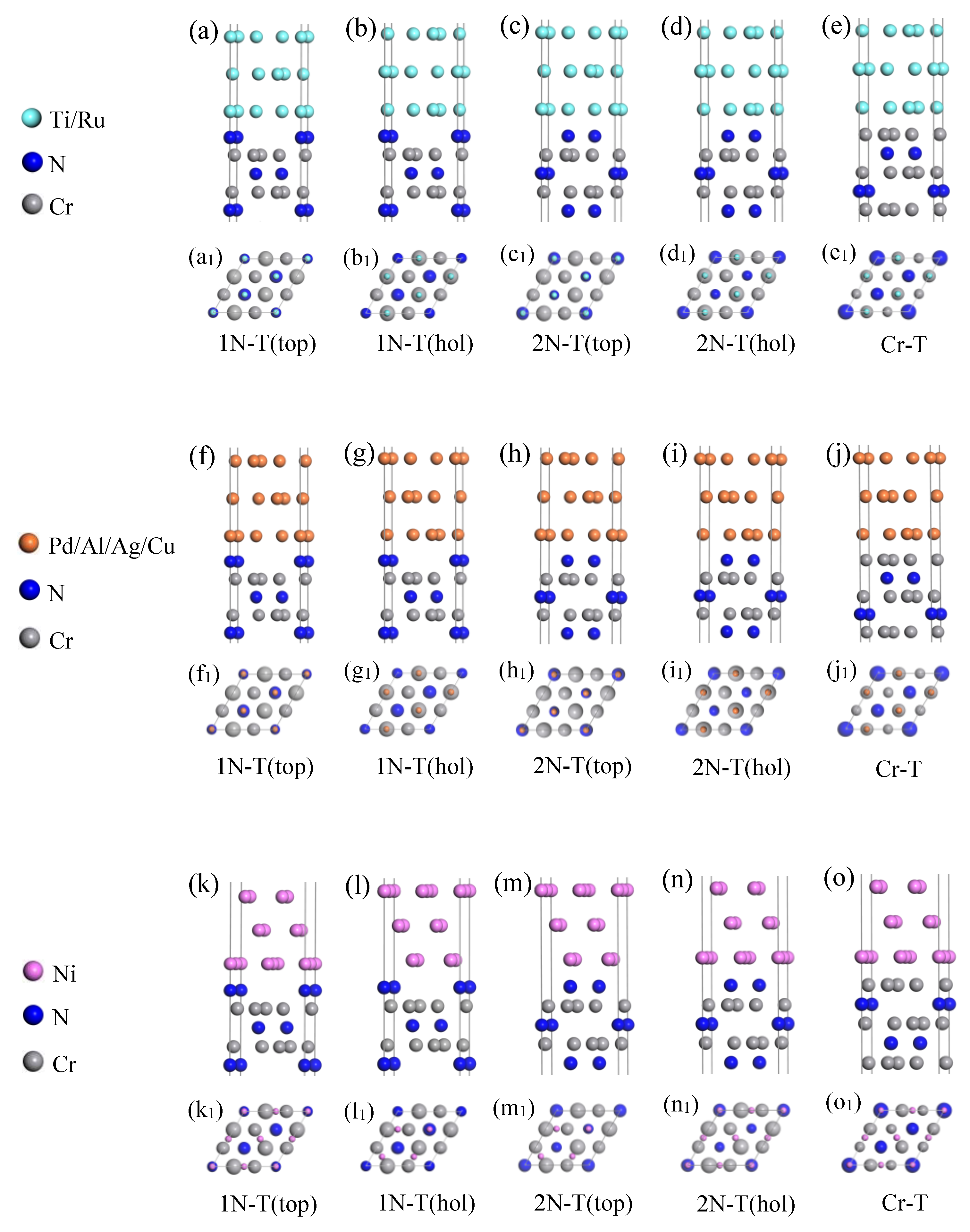
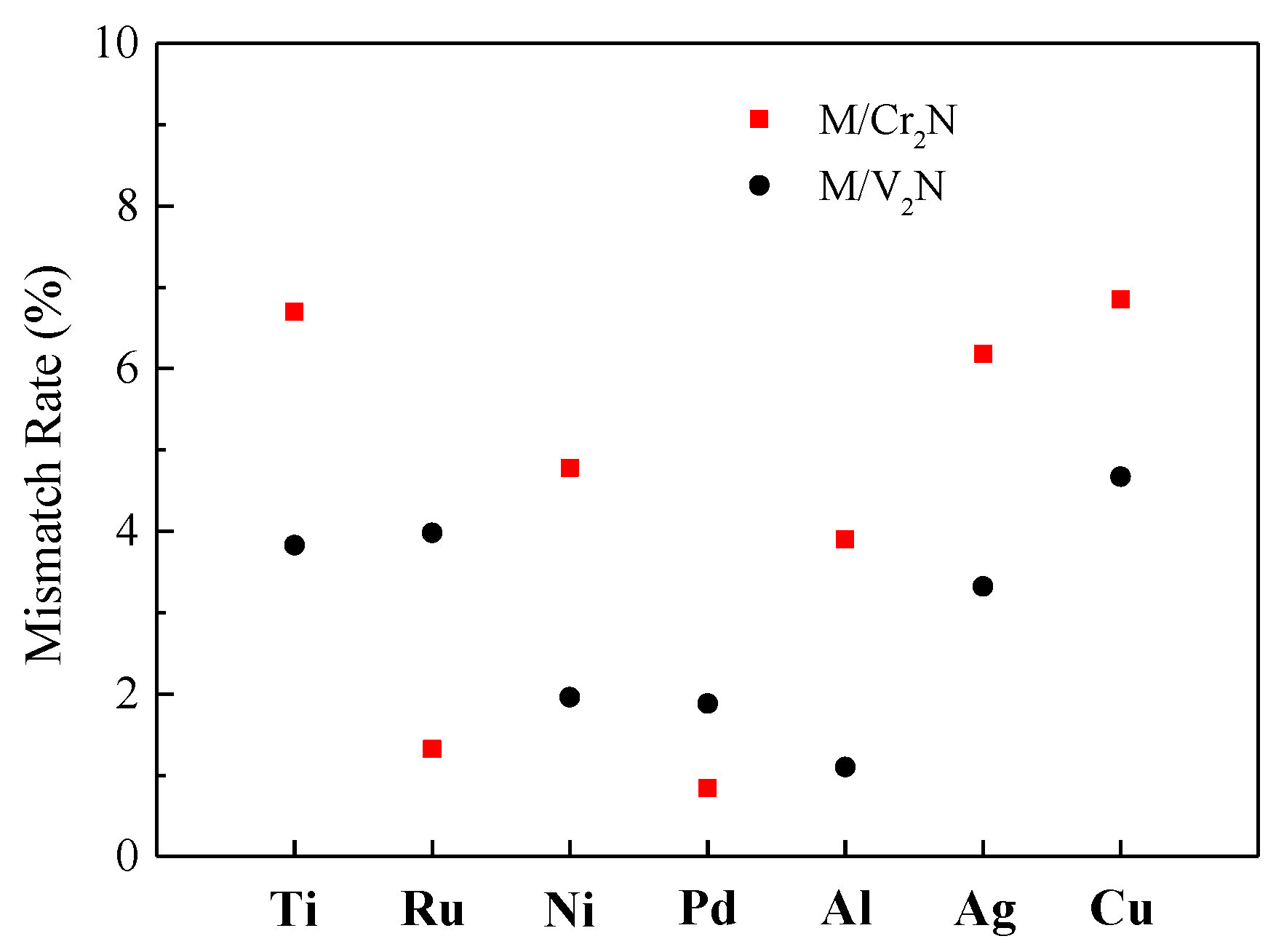
2.5. Interface Models
3. Results and Discussion
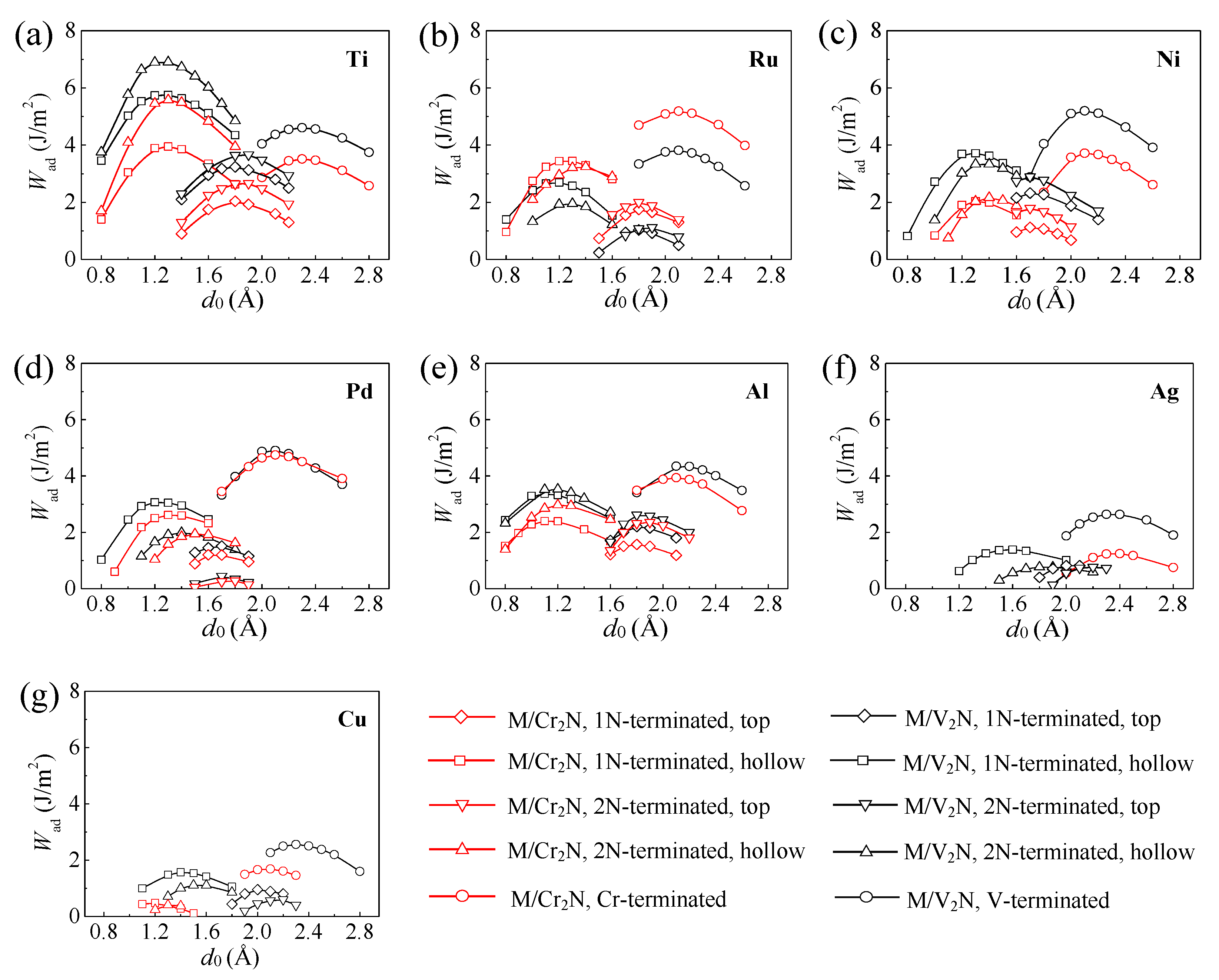
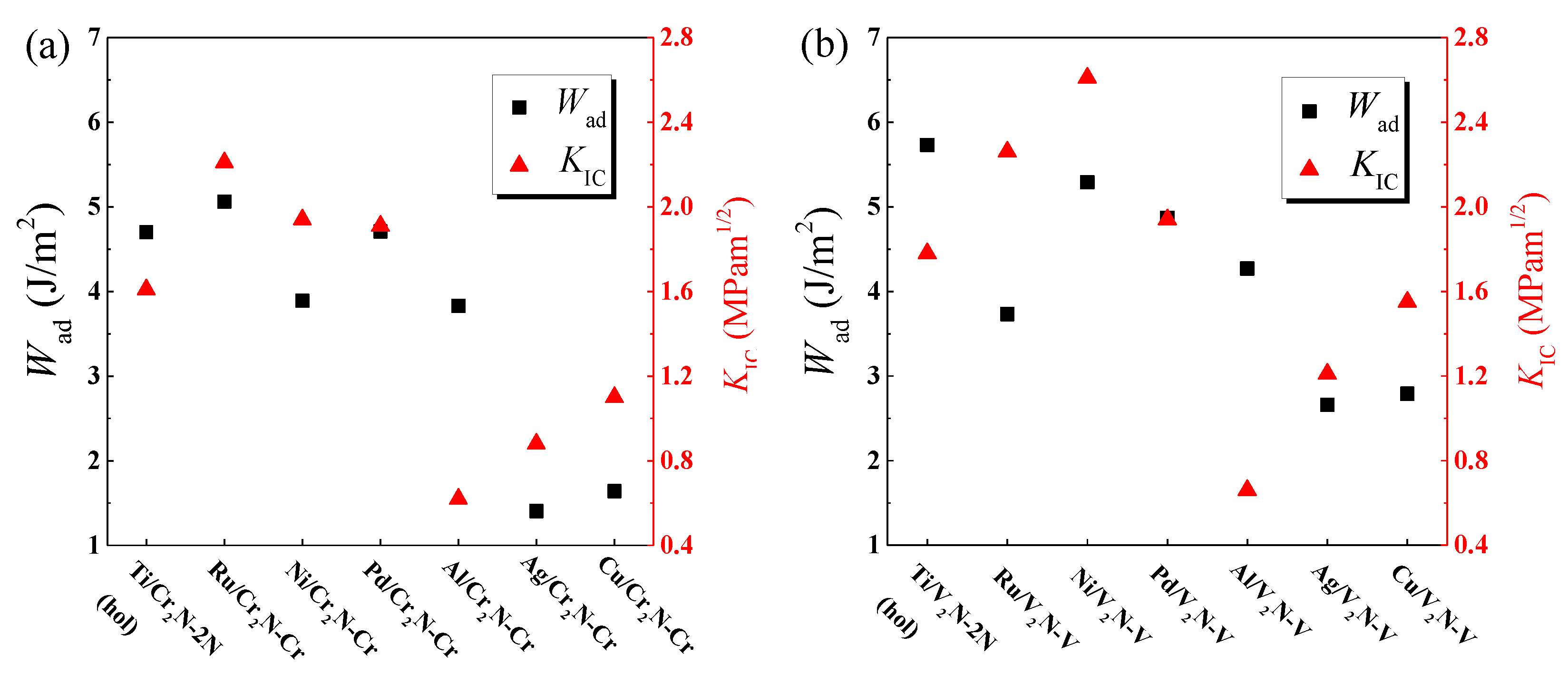
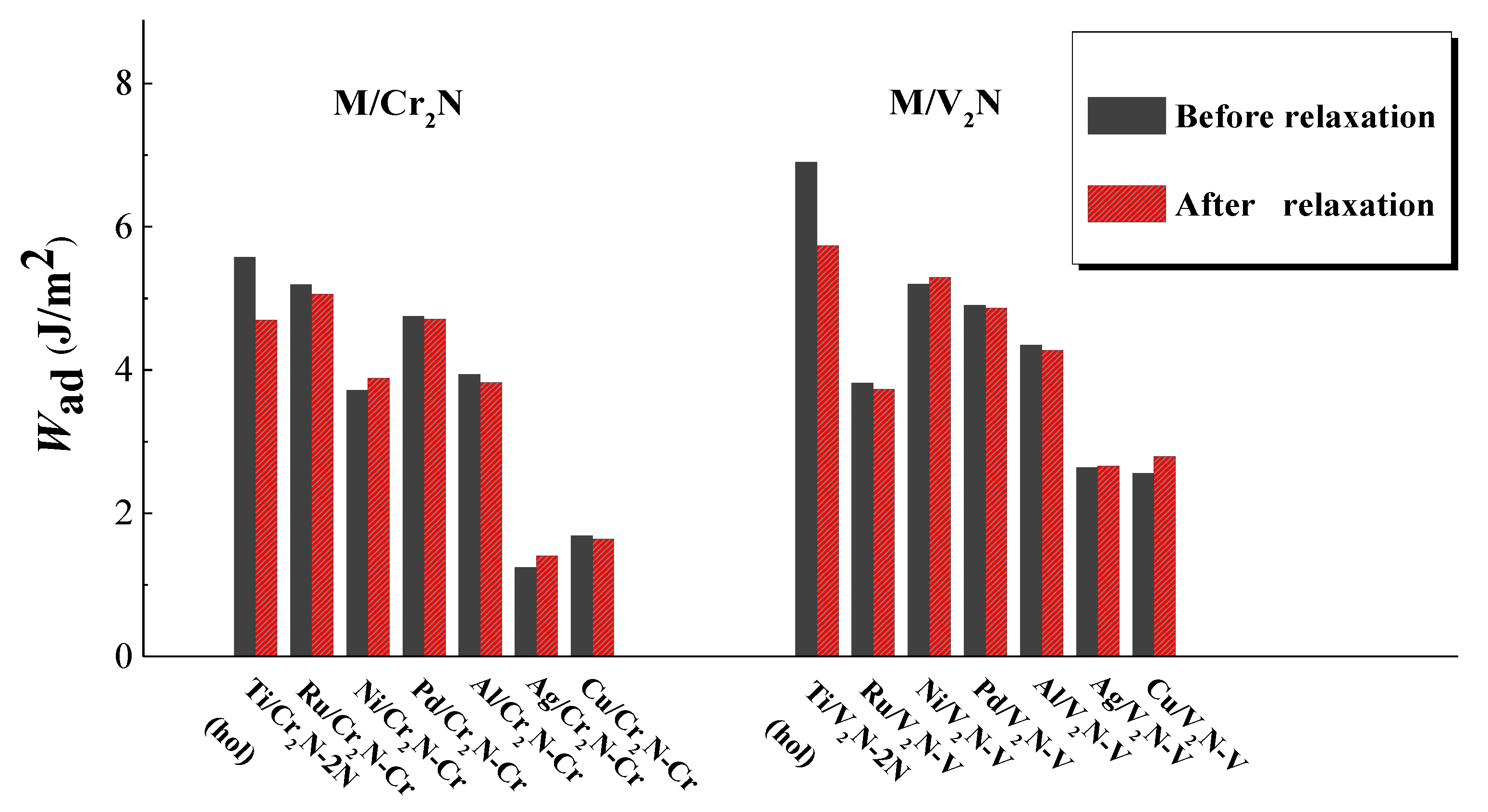
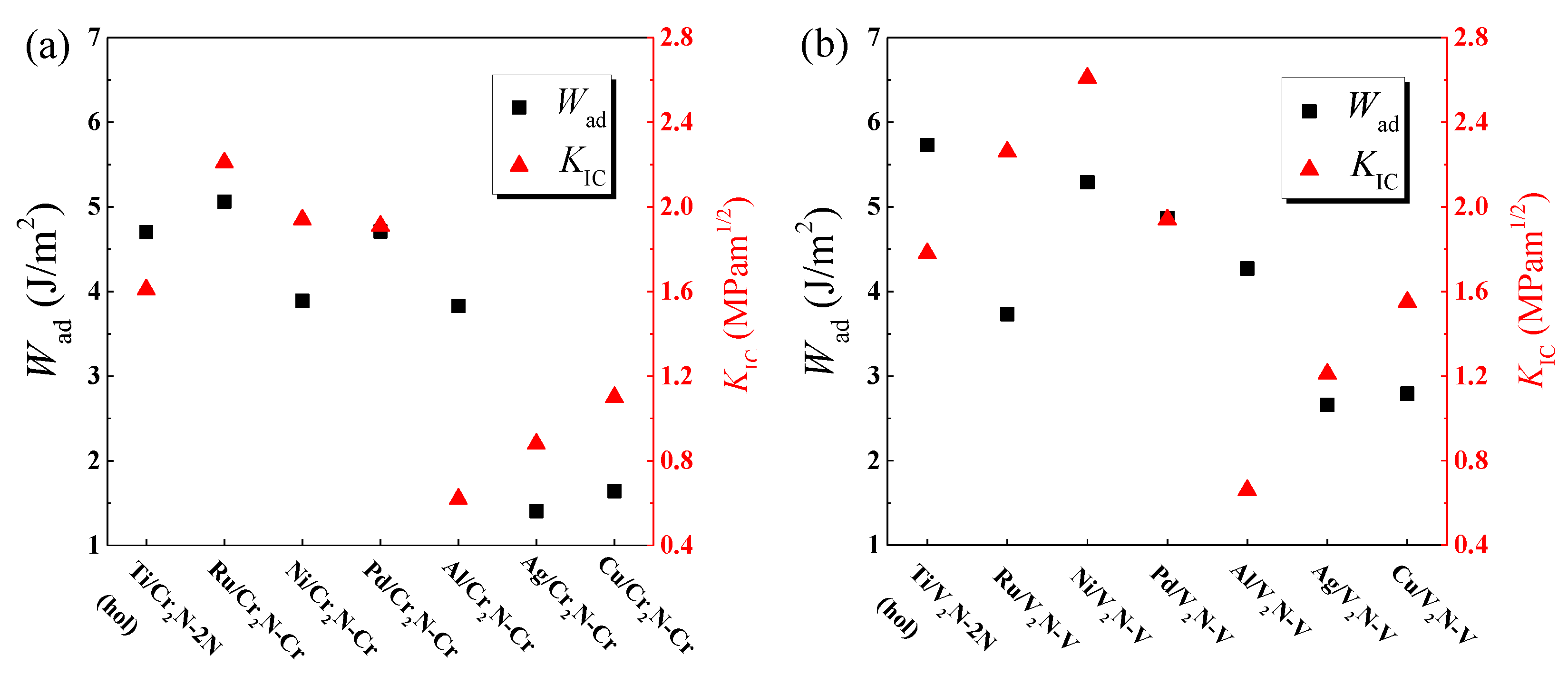
3.1. Work of Adhesion
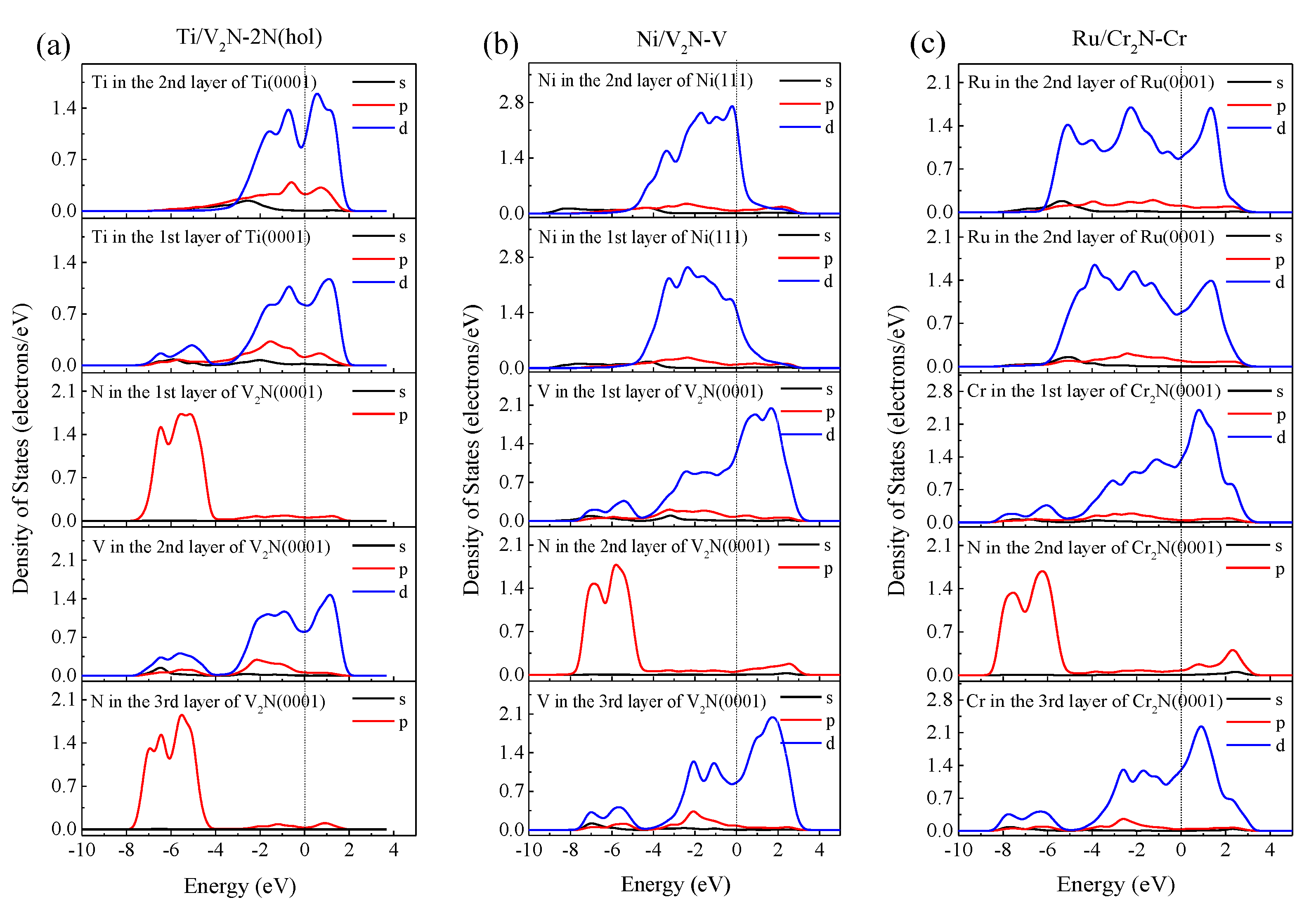
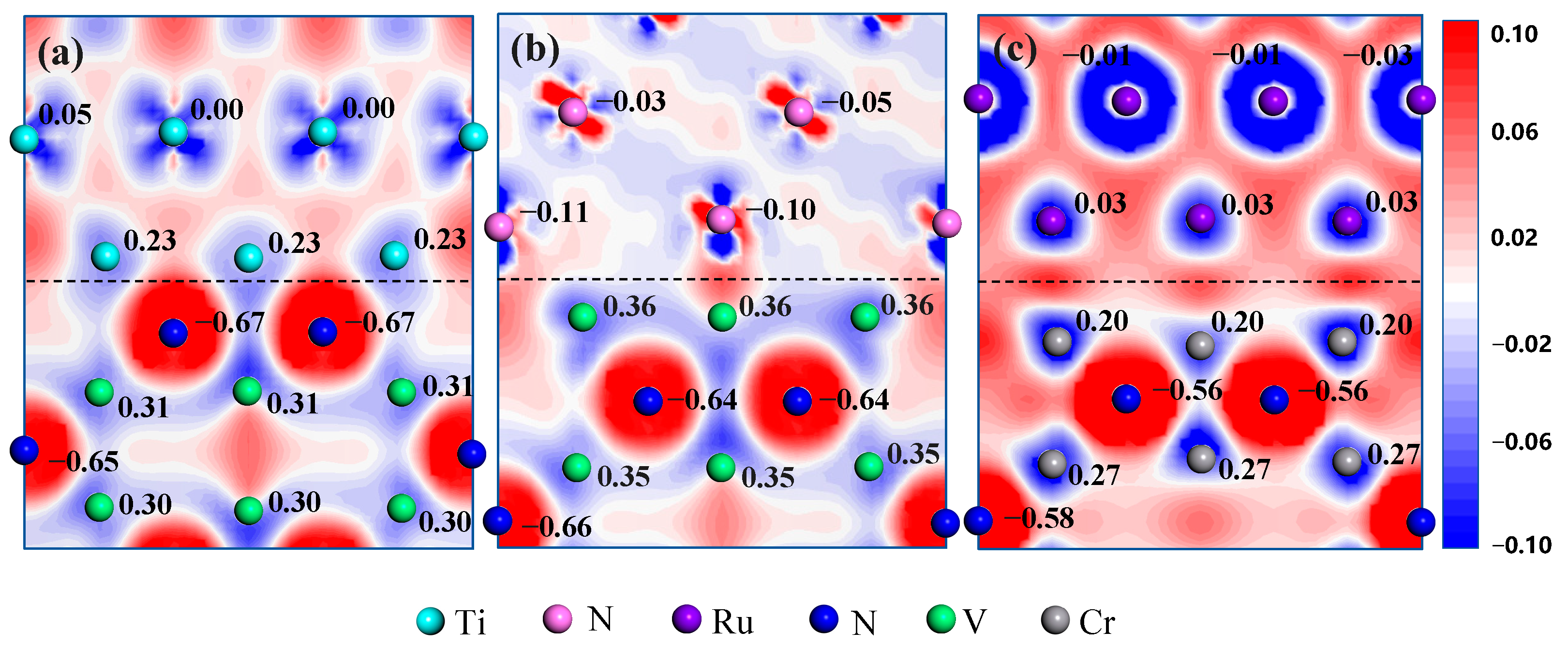
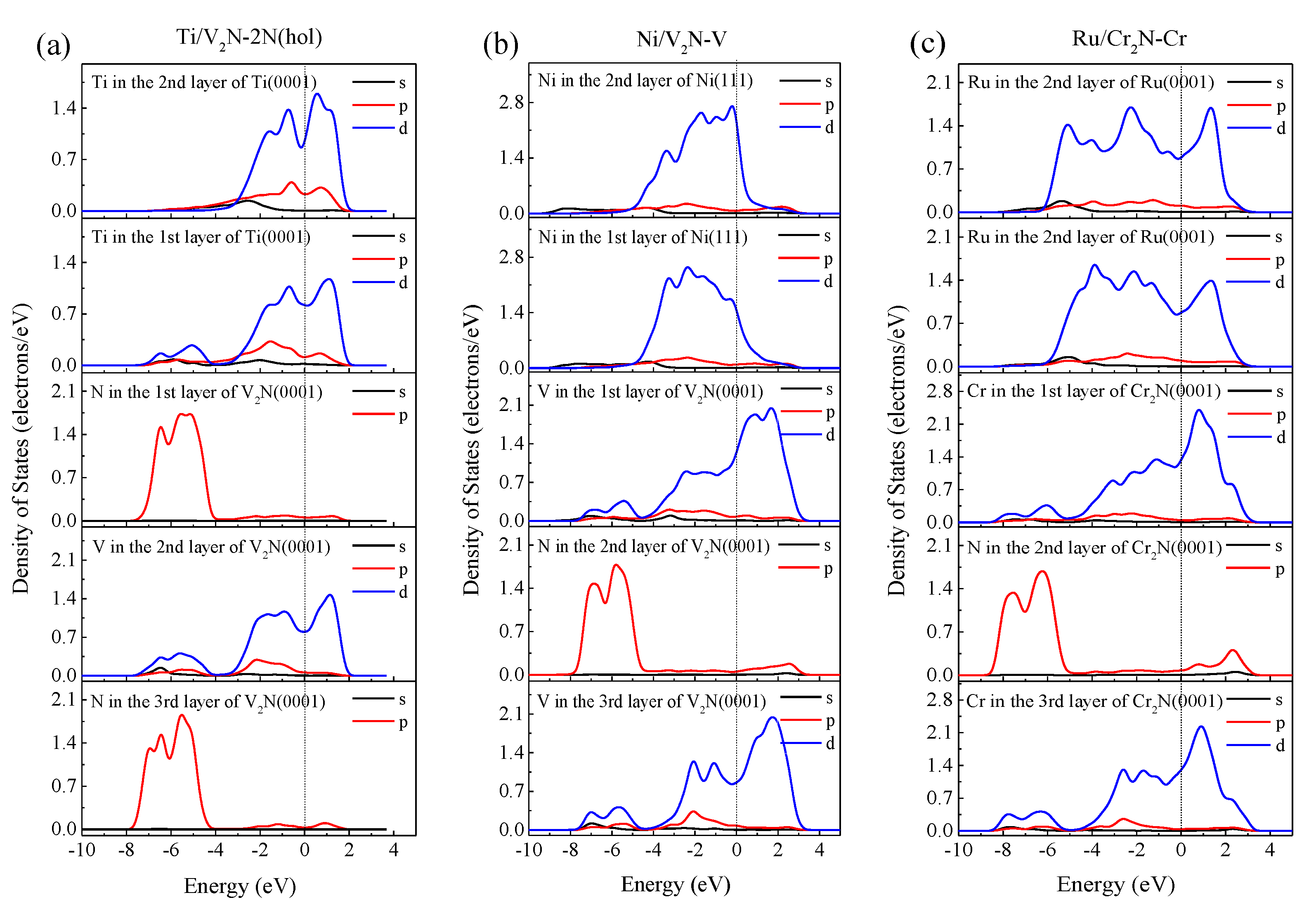
3.2. Electronic Structure
3.3. Interfacial Fracture Toughness
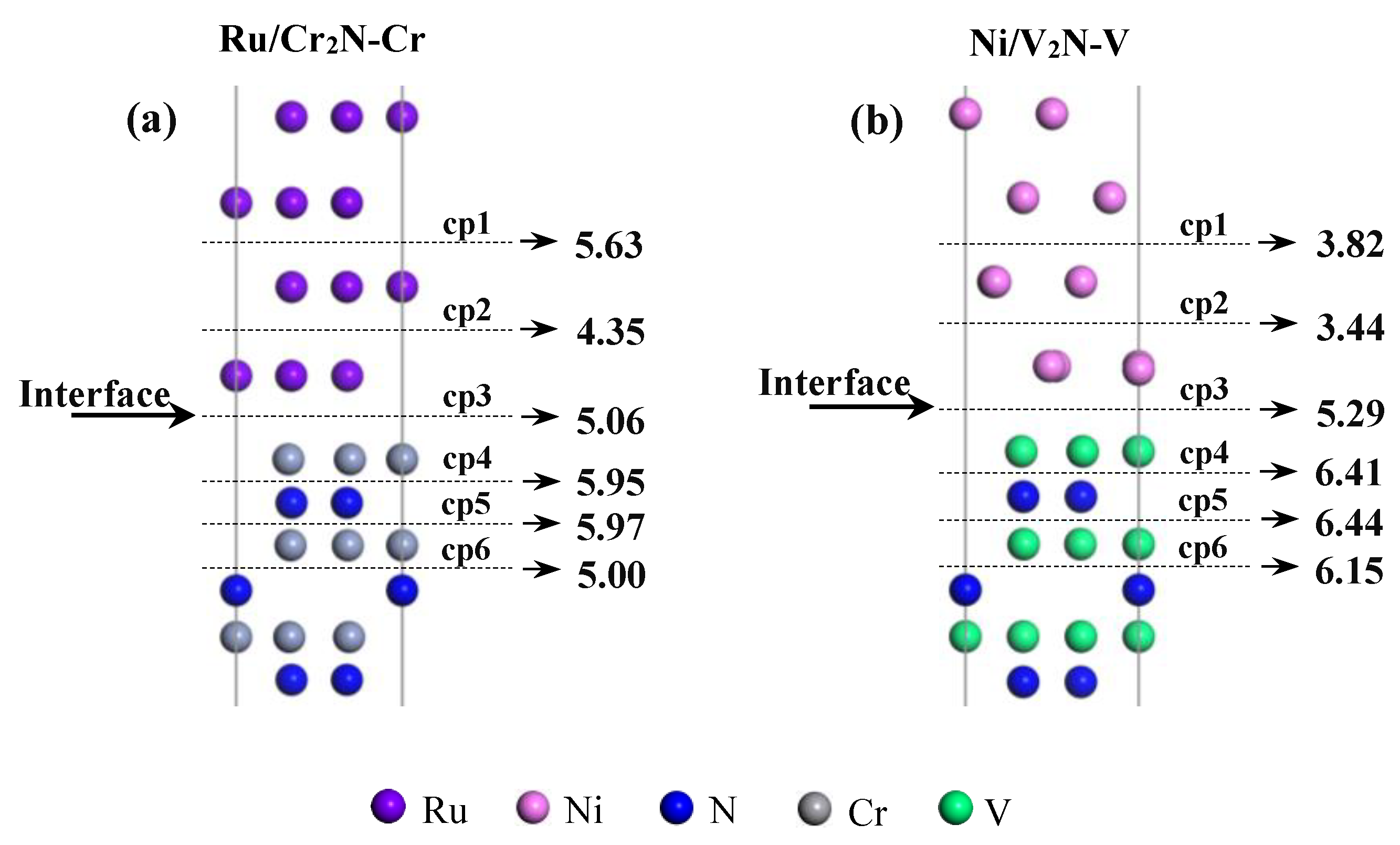
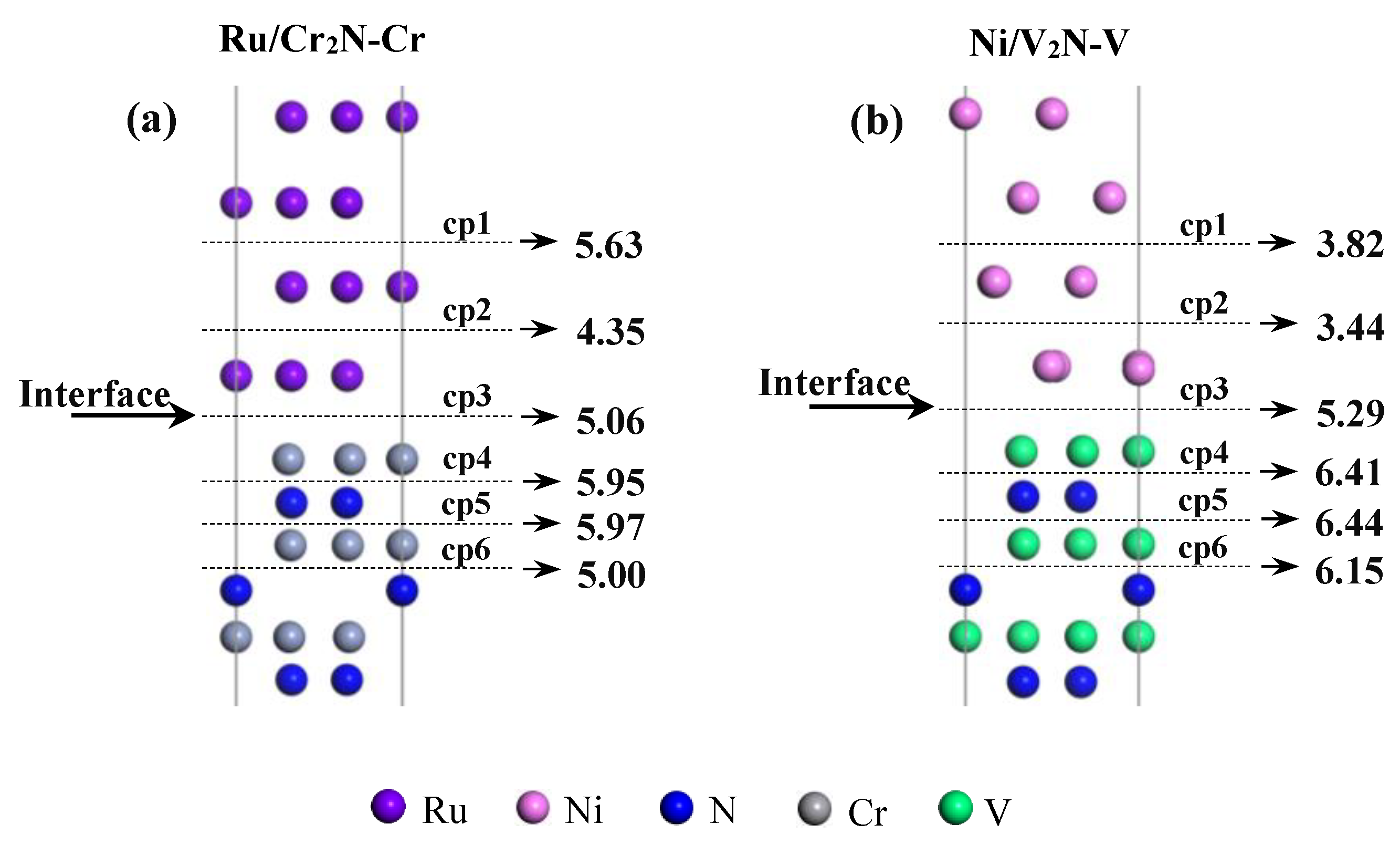
3.4. Interfacial Weak Point
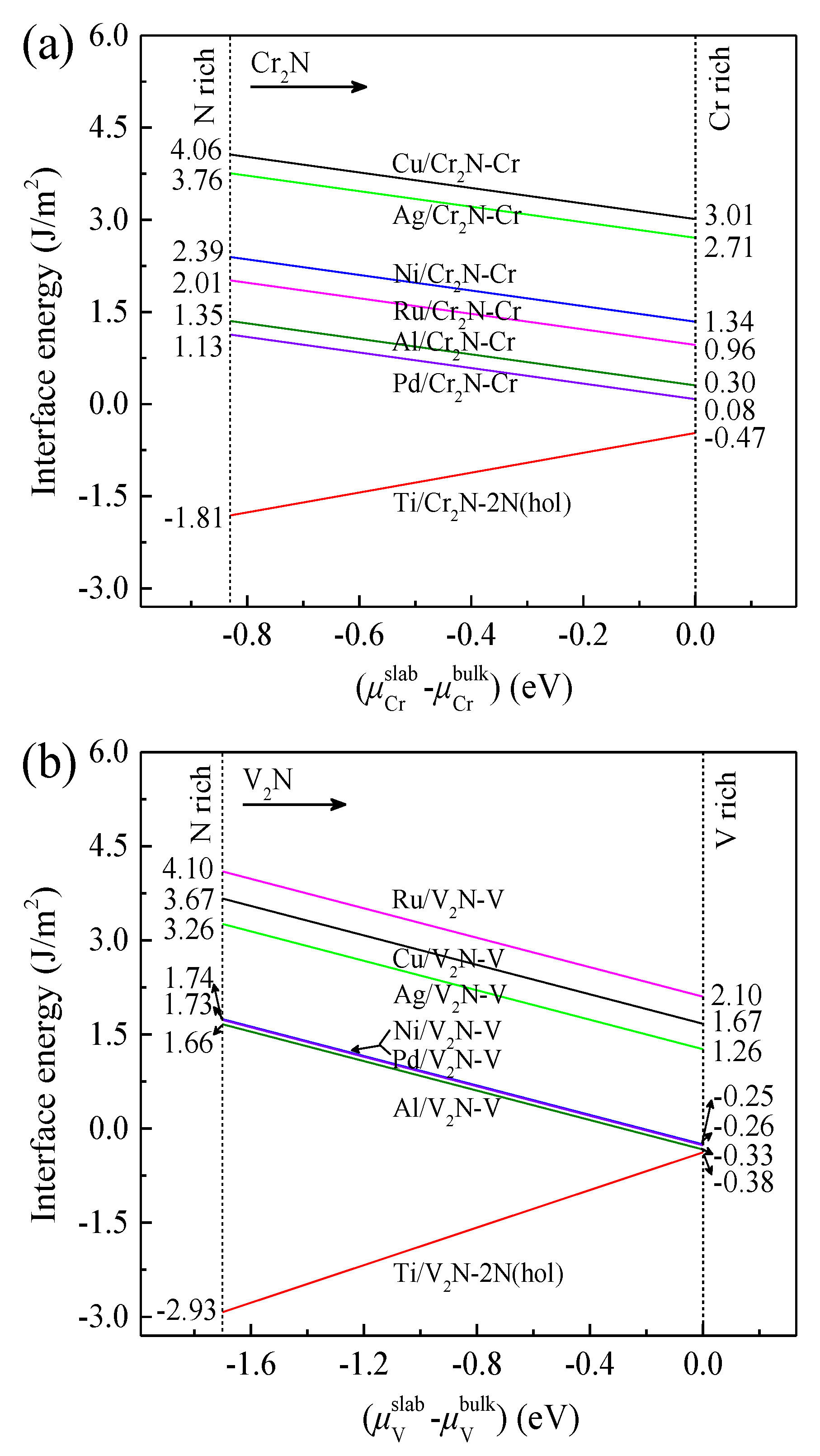
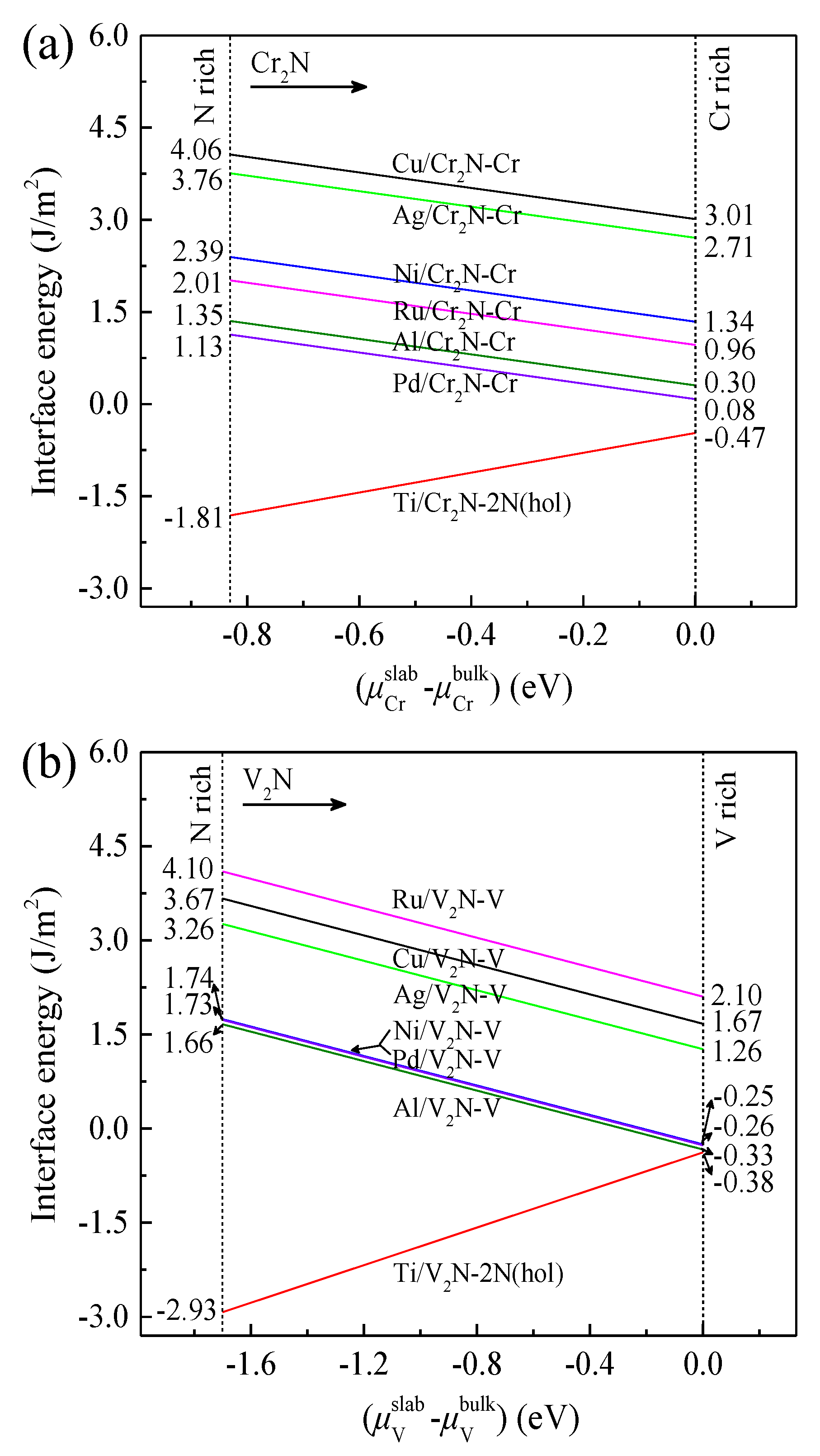
3.5. Interfacial Energy
4. Conclusions
- (1)
- The Ti/V2N-2N(hol), Ni/V2N-V, and Ru/Cr2N-Cr interface models exhibit the first, second, and third largest values of Wad, 5.73, 5.30, and 5.05 J/m2, among all the interface models considered, respectively.
- (2)
- The analyses of partial density of states (PDOS), charge density difference, and Mulliken charge are in good agreement with the observed trends in the Wad results.
- (3)
- The Ni/V2N-V, Ru/V2N-V, and Ru/Cr2N-Cr interface models exhibit the first, second, and third largest values of facture toughness, 2.61, 2.26, and 2.21 MPam1/2, among all the interface models considered, respectively.
- (4)
- The Ni/V2N-V interface model has a relatively low value range of γint from −0.25 to 1.74 J/m2, suggesting considerably high thermal stability compared to most of the other interface models.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, S.; Chen, L.; Wu, Y.; Hu, Y.; Liu, T.; Tang, K.; Wei, Q. Microstructure and corrosion resistance of Cr/Cr2N multilayer film deposited on the surface of depleted uranium. Corros. Sci. 2014, 82, 420–425. [Google Scholar] [CrossRef]
- Shuai, J.; Zuo, X.; Wang, Z.; Sun, L.; Chen, R.; Wang, L.; Wang, A.; Ke, P. Erosion behavior and failure mechanism of Ti/TiAlN multilayer coatings eroded by silica sand and glass beads. J. Mater. Sci. Technol. 2021, 80, 179–190. [Google Scholar] [CrossRef]
- Dang, C.; Li, J.; Wang, Y.; Chen, J. Structure, mechanical and tribological properties of self-toughening TiSiN/Ag multilayer coatings on Ti6Al4V prepared by arc ion plating. Appl. Surf. Sci. 2016, 386, 224–233. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, D.; Fu, Y.; Du, H. Toughening of hard nanostructural thin films: A critical review. Surf. Coat. Technol. 2005, 198, 2–8. [Google Scholar] [CrossRef]
- Shuai, J.; Zuo, X.; Wang, Z.; Guo, P.; Xu, B.; Zhou, J.; Wang, A.; Ke, P. Comparative study on crack resistance of TiAlN monolithic and Ti/TiAlN multilayer coatings. Ceram. Int. 2019, 46, 6672–6681. [Google Scholar] [CrossRef]
- Ranade, A.N.; Krishna, L.R.; Li, Z.; Wang, J.; Korach, C.S.; Chung, Y.-W. Relationship between hardness and fracture toughness in Ti–TiB2 nanocomposite coatings. Surf. Coat. Technol. 2012, 213, 26–32. [Google Scholar] [CrossRef]
- Daniel, R.; Meindlhumer, M.; Zalesak, J.; Sartory, B.; Zeilinger, A.; Mitterer, C.; Keckes, J. Fracture toughness enhancement of brittle nanostructured materials by spatial heterogeneity: A micromechanical proof for CrN/Cr and TiN/SiOx multilayers. Mater. Des. 2016, 104, 227–234. [Google Scholar] [CrossRef]
- Wieciński, P.; Smolik, J.; Garbacz, H.; Kurzydlowski, K. Failure and deformation mechanisms during indentation in nanostructured Cr/CrN multilayer coatings. Surf. Coat. Technol. 2014, 240, 23–31. [Google Scholar] [CrossRef]
- Wei, G.; Scharf, T.; Zhou, J.; Huang, F.; Weaver, M.; Barnard, J. Nanotribology studies of Cr, Cr2N and CrN thin films using constant and ramped load nanoscratch techniques. Surf. Coat. Technol. 2001, 146–147, 357–362. [Google Scholar] [CrossRef]
- Lin, J.; Sproul, W.D.; Moore, J.J.; Lee, S.; Myers, S. High rate deposition of thick CrN and Cr2N coatings using modulated pulse power (MPP) magnetron sputtering. Surf. Coat. Technol. 2011, 205, 3226–3234. [Google Scholar] [CrossRef]
- Sanjinés, R.; Hones, P.; Lévy, F. Hexagonal nitride coatings: Electronic and mechanical properties of V2N, Cr2N and δ-MoN. Thin Solid Film. 1998, 332, 225–229. [Google Scholar] [CrossRef]
- Guan, X.; Wang, Y.; Xue, Q.; Wang, L. Toward high load bearing capacity and corrosion resistance Cr/Cr2N nano-multilayer coatings against seawater attack. Surf. Coat. Technol. 2015, 282, 78–85. [Google Scholar] [CrossRef]
- Li, C.-L.; Wu, F.-B.; Lee, J.-W.; Tsai, Y.-Z.; Chang, L.-C. Characteristics of Cr2N/Cu multilayered thin films with different bilayer thickness. Surf. Coat. Technol. 2009, 204, 941–946. [Google Scholar] [CrossRef]
- Bílek, P.; Jurči, P.; Hudáková, M.; Pašák, M.; Kusý, M.; Bohovičová, J. Cr2N-7Ag nanocomposite thin films deposited on Vanadis 6 tool steel. Appl. Surf. Sci. 2014, 307, 13–19. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, L.C.; Gong, H.R.; Gong, X. Cohesion strength and fracture toughness of Mo-TiC interfaces. Surf. Coat. Technol. 2020, 382, 125158. [Google Scholar] [CrossRef]
- Zhong, W.; Wang, Z.; Gao, N.; Huang, L.-A.; Lin, Z.; Liu, Y.; Meng, F.; Deng, J.; Jin, S.; Zhang, Q.; et al. Coupled Vacancy Pairs in Ni-Doped CoSe for Improved Electrocatalytic Hydrogen Production Through Topochemical Deintercalation. Angew. Chem. Int. Ed. 2020, 59, 22743–22748. [Google Scholar] [CrossRef] [PubMed]
- Eglitis, R.; Rohlfing, M. First-principles calculations of the atomic and electronic structure of SrZrO3 and PbZrO3(001) and (011) surfaces. J. Phys. Condens. Matter 2010, 22, 415901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eglitis, R.; Popov, A.I. Systematic trends in (0 0 1) surface ab initio calculations of ABO3 perovskites. J. Saudi Chem. Soc. 2018, 22, 459–468. [Google Scholar] [CrossRef]
- Lin, Z.; Xiao, B.; Wang, Z.; Tao, W.; Shen, S.; Huang, L.; Zhang, J.; Meng, F.; Zhang, Q.; Gu, L.; et al. Planar-Coordination PdSe 2 Nanosheets as Highly Active Electrocatalyst for Hydrogen Evolution Reaction. Adv. Funct. Mater. 2021, 31, 2102321. [Google Scholar] [CrossRef]
- Zhuang, D.; Liu, Y.; Jin, N.; Li, J.; Cao, Z.; Zhang, J. First-principles calculations on FeWB bulk and FeWB(001)/α-Fe(111) interface. Mater. Res. Express 2021, 8, 046405. [Google Scholar] [CrossRef]
- Wang, Z.; Xiao, B.; Lin, Z.; Xu, Y.; Lin, Y.; Meng, F.; Zhang, Q.; Gu, L.; Fang, B.; Guo, S.; et al. PtSe2/Pt heterointerface with reduced coordination for boosted hydrogen evolution reaction. Angew. Chem. Int. Ed. 2021, 133, 23576–23581. [Google Scholar] [CrossRef]
- Li, J.; Cui, Y.; Zhang, M.; Zhao, J.; Luo, X. Thermodynamic evidence of α-Al heterogeneous nucleation on Al2MgC2 and the interfacial bonding mechanism: A first-principles study. J. Solid State Chem. 2020, 288, 121431. [Google Scholar] [CrossRef]
- He, C.; Cheng, M.; Zhang, M.; Zhang, W.X. Interfacial Stability and Electronic Properties of Ag/M (M = Ni, Cu, W, and Pd) and Cu/Cr Interfaces. J. Phys. Chem. C 2018, 122, 17928–17935. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, N.; Lian, L.; Cao, Z.; Zhuang, D. The behaviors of He atoms and vacancies at TiO/V interface: A first-principles study. Appl. Surf. Sci. 2020, 543, 148780. [Google Scholar] [CrossRef]
- Shao, W.; Liu, J.; Shi, Z.; Rao, L.; Xing, X.; Zhou, Y.; Yang, Q. Theoretical calculation of adhesion performance and mechanical properties of CrN/α-Fe interface. J. Alloys Compd. 2019, 810, 151921. [Google Scholar] [CrossRef]
- Wang, M.; Liu, G.; Luo, X.; Levchenko, V.A. Effect of interface orientation on the adhesion strength and fracture toughness of Ni/CrN interfaces by first-principles study. Mater. Res. Express 2021, 8, 096507. [Google Scholar] [CrossRef]
- Yadav, S.; Ramprasad, R.; Wang, J.; Misra, A.; Liu, X.-Y. First-principles study of Cu/TiN and Al/TiN interfaces: Weak versus strong interfaces. Model. Simul. Mater. Sci. Eng. 2014, 22, 035020. [Google Scholar] [CrossRef]
- Siegel, D.J.; Hector, L.G.; Adams, J.B. First-principles study of metal–carbide/nitride adhesion: Al/VC vs. Al/VN. Acta Mater. 2002, 50, 619–631. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, C.; Liu, J.; Li, Y.; Fang, X.; Li, J.; Han, P. First-Principles Study on the Structural Stability and Segregation Behavior of γ-Fe/Cr2N Interface with Alloying Additives M (M = Mn, V, Ti, Mo, and Ni). Metals 2016, 6, 156. [Google Scholar] [CrossRef]
- Wang, H.; Gao, X.; Yang, J.; Jia, Y.; Gong, J. First-Principles Study of Cr2N/γ-Fe Interface in High Nitrogen Steel. Mater. Trans. 2015, 56, 1047–1051. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B. 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Laasonen, K.; Pasquarello, A.; Car, R.; Lee, C.; Vanderbilt, D. Car-Parrinello molecular dynamics with Vanderbilt ultrasoft pseudopotentials. Phys. Rev. B. 1993, 47, 10142–10153. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Marquart, T.; Franzen, H.F. Structure refinement for Cr2N. J. Less Common Met. 1990, 158, L9–L10. [Google Scholar] [CrossRef]
- Yan, M.; Chen, H. Structural, elastic and electronic properties of Cr2N: A first-principles study. Comput. Mater. Sci. 2014, 88, 81–85. [Google Scholar] [CrossRef]
- Christensen, A.N.; Lebech, B. The structure of β-vanadium nitride. Acta Crystallogr. 1979, 35, 2677–2678. [Google Scholar] [CrossRef]
- Ma, S.; Liu, Y.; Ye, J.; Zhang, H.; Pang, J. Theoretical study on the elastic, electronic and thermodynamic properties of trigonal-type Cr2N under high pressures. Comput. Mater. Sci. 2014, 95, 620–625. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.; Feng, G.; Luo, X.; Sun, Q.; Jin, N. Adhesion and fracture toughness at α-Ti(0001)/TiC(111): A first-principles investigation. Appl. Surf. Sci. 2013, 286, 240–248. [Google Scholar] [CrossRef]
- Lugovskoy, A.V.; Belov, M.P.; Krasilnikov, O.; Vekilov, Y.K. Stability of the hcp Ruthenium at high pressures from first principles. J. Appl. Phys. 2014, 116, 103507. [Google Scholar] [CrossRef]
- Kim, D.; Shang, S.-L.; Liu, Z.-K. Effects of alloying elements on elastic properties of Ni by first-principles calculations. Comput. Mater. Sci. 2009, 47, 254–260. [Google Scholar] [CrossRef]
- Wolf, R.J.; Mansour, K.A.; Lee, M.W.; Ray, J.R. Temperature dependence of elastic constants of embedded-atom models of palladium. Phys. Rev. B 1992, 46, 8027–8035. [Google Scholar] [CrossRef]
- Zhang, M.; Shen, J.; He, J. Elastic constants of Al and TiN calculated by ab initio method. Nonferrous Met. Soc. China 2001, 11, 244–248. [Google Scholar]
- Zhang, K.; Pang, M.; Zhan, Y. Atomic structure and electronic properties of Ag(111)/TiC(111) interface: Insights from first-principles simulations. J. Phys. Chem. Solids 2019, 124, 212–220. [Google Scholar] [CrossRef]
- Zhu, Y.D.; Yan, M.F.; Zhang, Y.X.; Zhang, C.S. First-principles investigation of structural, mechanical and electronic properties for Cu–Ti intermetallics. Comput. Mater. Sci. 2016, 123, 70–78. [Google Scholar] [CrossRef]
- Li, J.; Zhang, M.; Zhou, Y.; Chen, G. First-principles study of Al/A13Ti heterogeneous nucleation interface. Appl. Surf. Sci. 2014, 307, 593–600. [Google Scholar] [CrossRef]
- Wu, Z.; Pang, M.; Zhan, Y.; Shu, S.; Xiong, L.; Li, Z. The bonding characteristics of the Cu(111)/WC(0001) interface: An insight from first-principle calculations. Vacuum 2021, 191, 110218. [Google Scholar] [CrossRef]
- Da Silva, J.L.F.; Stampfl, C.; Scheffler, M. Converged properties of clean metal surfaces by all-electron first-principles calculations. Surf. Sci. 2006, 600, 703–715. [Google Scholar] [CrossRef]
- Singh-Miller, N.E.; Marzari, N. Surface energies, work functions, and surface relaxations of low-index metallic surfaces from first principles. Phys. Rev. B 2009, 80, 235407. [Google Scholar] [CrossRef] [Green Version]
- Tyson, W.; Miller, W. Surface free energies of solid metals: Estimation from liquid surface tension measurements. Surf. Sci. 1977, 62, 267–276. [Google Scholar] [CrossRef]
- Liu, J.; Nolan, M. Coverage and Stability of NHx Terminated Cobalt and Ruthenium Surfaces: A First Principles Investigation. J. Phys. Chem. C 2019, 123, 25166–25175. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Huang, J.; Ye, Z.; Sun, X.; Chen, S.; Zhao, Y. First-principles calculations on Ni/W interfaces in Steel/Ni/W hot isostatic pressure diffusion bonding layer. Appl. Surf. Sci. 2019, 475, 906–916. [Google Scholar] [CrossRef]
- Liu, L.M.; Wang, S.Q.; Ye, H.Q. First-principles study of polar Al/TiN(111) interfaces. Acta Mater. 2004, 52, 3681–3688. [Google Scholar] [CrossRef]
- Li, J.; Cui, Y.; Chen, Y.; Lv, X.; Luo, X. Theoretical investigation on SiC(1 1 1)/Al4C3(0 0 0 1) interface using density functional theory calculations. Mater. Today Commun. 2019, 21, 100743. [Google Scholar] [CrossRef]
- Guo, W.; She, Z.; Xue, H.; Zhang, X. Study on the effect of Ti, Al, Cu, and Ag doping on the bonding properties of soldered β-Sn(100)/ZrO2(111) interface. Appl. Ceram. Technol. 2021, 18, 138–146. [Google Scholar] [CrossRef]
- Jiao, X.; Fu, W.; Shao, W.; Zhu, X.; Zhou, Y.; Xing, X.; Wang, Z.; Yang, Q. First-principles calculation on γ-Fe/La2O3 interface properties and austenite refinement mechanism by La2O3. Mater. Chem. Phys. 2020, 259, 124194. [Google Scholar] [CrossRef]
- Mori, T.; Fukuda, S.; Takemura, Y. Improvement of mechanical properties of Ti/TiN multilayer film deposited by sputtering. Surf. Coat. Technol. 2001, 140, 122–127. [Google Scholar] [CrossRef]
- Guo, W.; She, Z.; Xue, H.; Zhang, X. Effect of active Ti element on the bonding characteristic of the Ag(111)/α-Al2O3(0001) interface by using first principle calculation. Ceram. Int. 2020, 46, 5430–5435. [Google Scholar] [CrossRef]
- Mei, Z.-G.; Bhattacharya, S.; Yacout, A.M. First-principles study of fracture toughness enhancement in transition metal nitrides. Surf. Coat. Technol. 2018, 357, 903–909. [Google Scholar] [CrossRef]
- Chu, F.; Lei, M.; Maloy, S.; Petrovic, J.; Mitchell, T. Elastic properties of C40 transition metal disilicides. Acta Mater. 1996, 44, 3035–3048. [Google Scholar] [CrossRef]
- Chen, K.; Bielawski, M. Interfacial fracture toughness of transition metal nitrides. Surf. Coat. Technol. 2008, 203, 598–601. [Google Scholar] [CrossRef]
- Yu, R.; Song, H.; Zhang, X.-F.; Yang, P. Thermal Wetting of Platinum Nanocrystals on Silica Surface. J. Phys. Chem. B 2005, 109, 6940–6943. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, G.; Tan, X.; Wang, C.X.; Yang, G.W. Physical and chemical origin of size-dependent spontaneous interfacial alloying of core–shell nanostructures. Chem. Phys. Lett. 2006, 420, 65–70. [Google Scholar] [CrossRef]
- Bauer, E. Epitaxy of metals on metals. Appl. Surf. Sci. 1982, 11–12, 479–494. [Google Scholar] [CrossRef]
- Christensen, M.; Dudiy, S.; Wahnström, G. First-principles simulations of metal-ceramic interface adhesion: Co/WC versus Co/TiC. Phys. Rev. B 2002, 65, 045408. [Google Scholar] [CrossRef]
- Lu, S.; Hu, Q.-M.; Punkkinen, M.P.J.; Johansson, B.; Vitos, L. First-principles study of fcc-Ag/bcc-Fe interfaces. Phys. Rev. B 2013, 87, 224104. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.; Zhang, W. Stoichiometric interfaces of Al and Ag with Al2O3. Acta Mater. 2000, 48, 4395–4403. [Google Scholar] [CrossRef]
- Timchenko, N.A.; Zubavichus, Y.V.; Krysina, O.V.; Kuznetsov, S.I. Local structure of titanium nitride-based coatings. J. Surf. Investig. X-ray Synchrotron Neutron Tech. 2016, 10, 425–428. [Google Scholar] [CrossRef]
- Lee, B.-J. Predictive Analysis of Ti/AlN Interfacial Reaction Using Diffusion Simulation. Scr. Mater. 1998, 38, 499–507. [Google Scholar] [CrossRef]
- Herzler, J.; Leiberich, R.; Mick, H.-J.; Roth, P. Shock tube study of the formation of TiN molecules and particles. Nanostruct. Mater. 1998, 10, 1161–1171. [Google Scholar] [CrossRef]
- Hwang, S.; Laursen, A.B.; Porter, S.H.; Hongbin, Y.; Li, M.; Manichev, V.; Calvinho, K.U.; Amarasinghe, V.; Greenblatt, M.; Garfunkel, E.; et al. Garfunkel, Titanium Nitride As a Conducting Interfacial Layer between Hydrogen Evolution Catalysts and Silicon Photocathodes for Stable Solar-to-Hydrogen Water Splitting Devices. In ECS Meeting Abstracts; IOP Publishing: Bristol, UK, 2018. [Google Scholar]
- Faran, E.; Gotman, I.; Gutmanas, E.Y. Experimental study of the reaction zone at boron nitride ceramic–Ti metal interface. Mater. Sci. Eng. A 2000, 288, 66–74. [Google Scholar] [CrossRef]
- Morgiel, J.; Benkö, E. Microstructure of boron nitride sintered with titanium. Mater. Lett. 1995, 25, 49–52. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Space Group | Data Source | a | c | B | C11 | C12 | C13 | C33 | C44 |
|---|---|---|---|---|---|---|---|---|---|---|
| Cr2N | P1m (162) | This work | 4.773 | 4.406 | 288.9 | 476.7 | 157.5 | 230.6 | 409.0 | 169.2 |
| Ref. [37] | 4.773 | 4.406 | 289.0 | 477.0 | 158.0 | 231.0 | 410.0 | 171.0 | ||
| V2N | P1m (162) | This work | 4.905 | 4.543 | 264.9 | 454.1 | 162.2 | 177.7 | 444.7 | 153.7 |
| Ref. [36] | 4.917 | 4.568 | — | — | — | — | — | — | ||
| Ti | P63/mmc (194) | This work | 2.940 | 4.649 | 111.6 | 174.5 | 83.9 | 76.2 | 182.5 | 43.6 |
| Ref. [38] | 2.941 | 4.647 | 112.6 | 170.3 | 92.3 | 70.5 | 206.2 | 43.3 | ||
| Ru | P63/mmc (194) | This work | 2.719 | 4.283 | 313.2 | 577.0 | 182.4 | 160.7 | 660.1 | 190.1 |
| Ref. [39] | 2.720 | 4.292 | 329.9 | 577.4 | 176.7 | 170.9 | 644.2 | 190.4 | ||
| Ni | Fmm (225) | This work | 3.536 | — | 200.4 | 281.8 | 159.7 | — | — | 130.5 |
| Ref. [40] | 3.522 | — | 198.0 | 281.0 | 157.0 | — | — | 131.0 | ||
| Pd | Fmm (225) | This work | 3.930 | — | 183.4 | 224.2 | 163.0 | — | — | 73.0 |
| Ref. [41] | 3.890 | — | 195.0 | 230.0 | 177.0 | — | — | 78.1 | ||
| Al | Fmm (225) | This work | 4.049 | — | 76.1 | 130.5 | 48.8 | — | — | 8.7 |
| Ref. [42] | 4.000 | — | 73.4 | 123.8 | 48.6 | — | — | 21.4 | ||
| Ag | Fmm (225) | This work | 4.138 | — | 98.0 | 114.9 | 89.6 | — | — | 54.5 |
| Ref. [43] | 4.138 | — | 98.3 | 117.1 | 88.9 | — | — | 55.6 | ||
| Cu | Fmm (225) | This work | 3.630 | — | 134.7 | 178.2 | 112.9 | — | — | 86.6 |
| Ref. [44] | 3.631 | — | 137.4 | 162.1 | 125.1 | — | — | 84.1 |
| Slab | Terminate | Interlayer | Atom Layers of Slab | Slab | Terminate | Interlayer | Atom Layers of Slab | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 9 | 11 | 7 | 9 | 11 | ||||||
| Cr2N(0001) | 1N | 12 | −7.5 | −8.0 | −7.5 | V2N(0001) | 1N | 12 | −14.7 | −12.9 | −13.8 |
| 23 | −8.9 | −10.8 | −9.8 | 23 | −6.3 | −5.1 | −4.9 | ||||
| 34 | 2.9 | 4.9 | 4.3 | 34 | −1.4 | −1.7 | −1.4 | ||||
| 45 | −2.3 | −0.6 | 45 | 1.3 | 0.4 | ||||||
| 56 | −2.5 | 56 | −0.4 | ||||||||
| 2N | 12 | −15.2 | −16.3 | −16.2 | 2N | 12 | −27.0 | −26.1 | −25.4 | ||
| 23 | −11.5 | −10.8 | −9.9 | 23 | −0.3 | −1.1 | 0.2 | ||||
| 34 | 8.9 | 9.0 | 8.8 | 34 | 0.1 | −0.8 | 0.1 | ||||
| 45 | −0.9 | −1.3 | 45 | −0.2 | −0.2 | ||||||
| 56 | 1.5 | 56 | −0.6 | ||||||||
| Cr | 12 | −15.9 | −21.9 | −20.5 | V | 12 | −6.2 | −5.4 | −5.6 | ||
| 23 | 1.9 | 1.6 | 2.7 | 23 | −0.3 | 0.6 | 0.2 | ||||
| 34 | 13.5 | 11.9 | 11.4 | 34 | 1.6 | 0.6 | 0.6 | ||||
| 45 | 2.7 | 3.6 | 45 | 1.4 | 1.0 | ||||||
| 56 | 1.8 | 56 | 0 | ||||||||
| Metal Slab | Surface Energy (J/m2) | ||
|---|---|---|---|
| This Work | Other Calculations | Experiment | |
| Ti(0001) | 1.99 | 1.99 [47], 1.96 [48] | 1.99 [49] |
| Ru(0001) | 2.70 | 2.31 [50] | 3.04 [49] |
| Ni(111) | 1.91 | 1.93 [23], 1.92 [51] | 2.38 [49] |
| Pd(111) | 1.47 | 1.54 [23], 1.31 [48] | 2.00 [49] |
| Al(111) | 0.81 | 0.829 [45], 0.83 [52] | 1.14 [49] |
| Ag(111) | 0.79 | 0.80 [43], 0.75 [23], | 1.25 [49] |
| Cu(111) | 1.33 | 1.322 [46], 1.41 [47] | 1.79 [49] |
| M/Cr2N | Interfacial Orientation Relationship |
|---|---|
| M1/Cr2N (M1 = Ti and Ru) | (0001)M1[20]M1//(0001)C2N[100]C2N |
| M2/Cr2N (M2 = Pd, Al, Ag, and Cu) | (111)M2[10]M2//(0001)C2N[100]C2N |
| Ni/Cr2N | (111)Ni[2]Ni//(0001)C2N[100]C2N |
| Lattice | Phase | S11 | S12 | S13 | S33 | S44 | E0001 | E111 |
|---|---|---|---|---|---|---|---|---|
| hcp-TMN | Cr2N | 0.00292 | −0.00024 | −0.00151 | 0.00415 | 0.00596 | 241.20 | — |
| V2N | 0.00282 | −0.00071 | −0.00084 | 0.00292 | 0.00669 | 342.18 | — | |
| hcp-M | Ti | 0.00809 | −0.00296 | −0.00214 | 0.00727 | 0.02292 | 137.55 | — |
| Ru | 0.00200 | −0.00053 | −0.00036 | 0.00169 | 0.00526 | 592.10 | — | |
| fcc-M | Pd | 0.01150 | −0.00484 | — | — | 0.01370 | — | 193.35 |
| Al | 0.00962 | −0.00262 | — | — | 0.11494 | — | 25.14 | |
| Ag | 0.02756 | −0.01208 | — | — | 0.01835 | — | 137.93 | |
| Cu | 0.01103 | −0.00428 | — | — | 0.01155 | — | 213.89 | |
| Ni | 0.00601 | −0.00218 | — | — | 0.00766 | — | 321.70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Liu, G.; Huang, M.; Fu, Y.; Lin, C.; Wu, J.; Levchenko, V.A. Investigation of the Adhesion Strength, Fracture Toughness, and Stability of M/Cr2N and M/V2N (M = Ti, Ru, Ni, Pd, Al, Ag, and Cu) Interfaces Based on First-Principles Calculations. Coatings 2022, 12, 66. https://doi.org/10.3390/coatings12010066
Wang M, Liu G, Huang M, Fu Y, Lin C, Wu J, Levchenko VA. Investigation of the Adhesion Strength, Fracture Toughness, and Stability of M/Cr2N and M/V2N (M = Ti, Ru, Ni, Pd, Al, Ag, and Cu) Interfaces Based on First-Principles Calculations. Coatings. 2022; 12(1):66. https://doi.org/10.3390/coatings12010066
Chicago/Turabian StyleWang, Miao, Gang Liu, Min Huang, Yabo Fu, Changhong Lin, Jianbo Wu, and Vladimir A. Levchenko. 2022. "Investigation of the Adhesion Strength, Fracture Toughness, and Stability of M/Cr2N and M/V2N (M = Ti, Ru, Ni, Pd, Al, Ag, and Cu) Interfaces Based on First-Principles Calculations" Coatings 12, no. 1: 66. https://doi.org/10.3390/coatings12010066
APA StyleWang, M., Liu, G., Huang, M., Fu, Y., Lin, C., Wu, J., & Levchenko, V. A. (2022). Investigation of the Adhesion Strength, Fracture Toughness, and Stability of M/Cr2N and M/V2N (M = Ti, Ru, Ni, Pd, Al, Ag, and Cu) Interfaces Based on First-Principles Calculations. Coatings, 12(1), 66. https://doi.org/10.3390/coatings12010066