Abstract
Increasing demands for environmental accountability and energy efficiency in industrial practice necessitates significant modification(s) of existing technologies and development of new ones to meet the stringent sustainability demands of the future. Generally, development of required new technologies and appropriate modifications of existing ones need to be premised on in-depth appreciation of existing technologies, their limitations, and desired ideal products or processes. In the light of these, published literature mostly in the past 30 years on the sealing process; the second highest energy consuming step in aluminum anodization and a step with significant environmental impacts has been critical reviewed in this systematic review. Emphasis have been placed on the need to reduce both the energy input in the anodization process and environmental implications. The implications of the nano-porous structure of the anodic oxide on mass transport and chemical reactivity of relevant species during the sealing process is highlighted with a focus on exploiting these peculiarities, in improving the quality of sealed products. In addition, perspective is provided on plausible approaches and important factors to be considered in developing sealing procedures that can minimize the energy input and environmental impact of the sealing step, and ensure a more sustainable aluminum anodization process/industry.
1. Introduction
The aluminum anodization process is an energy intensive process. Besides the anodization step, the sealing step when it involves high temperature sealing in hot water (often at temperatures ≥ 95 °C) is arguably the second highest energy consuming step. The sealing step is an important step employed in the aluminum anodization process principally to preserve the aesthetics [1] and improve the corrosion resistance of the anodized aluminum by ensuring that the pores of the porous oxide layer are sealed [2,3]. Historically, sealing had been predominantly carried out in deionized/distilled water at high temperatures (close to 100 °C); a practice that is still popular today. The efficacy of hot-water sealing treatment is based on its ability to promote hydration of the porous aluminum oxide and barrier layers, producing a crystalline hydrate phase (boehmite) which fills the pores [4]. Hydrothermal sealing of anodized aluminum surface in deionized boiling water (T > 95 °C) is reported to proceed at rates in the range of about 2 min/μm [5,6], which translates to long sealing times, typically several minutes. Consequently, the high energy requirement of maintaining the sealing bath at temperatures ≥ 95 °C over several minutes, and the high-water quality requirement of the hydrothermal sealing process have jointly driven research efforts towards the development of mid-temperature and room temperature sealing methods. Furthermore, the applicability of another hitherto popular sealing process; chromate sealing, is currently limited to essential parts in the aerospace industry due to toxicological, health, and environmental implications traced to Cr(VI) employed in the process [7,8,9,10,11,12,13,14,15,16]. On the other hand, the advantages of another industrially utilized sealing process; the nickel fluoride (cold) sealing process is limited by the toxicity of nickel salts which narrows its range of application, and introduces added costs due to post-sealing wastewater treatments and management [17,18,19,20]. Further efforts at sealing anodized aluminum at temperatures lower than that used in hydrothermal (high temperature) sealing, have led to much variety in the chemical constitution and operating temperatures of sealing baths [21]. On the basis of temperature at which the sealing step is carried out, sealing can be classified into three major categories; high temperature, mid-temperature, and room-temperature or cold sealing. In this work sealing at temperatures from 0 to 40 °C is classified as low temperature sealing, from ≥ 40 °C to 70 °C as intermediate or mid temperature sealing, and sealing at temperatures > 70 °C as high temperature sealing.
2. Scope and Methodology of the Review
This systematic review was carried out as a contribution to the quest for post-sealing anodized aluminum with qualities superior to that obtained by hot-water sealing but at temperatures much lower than that used for hot-water sealing and without the use of toxic compounds. In this regard a lot of research efforts have been reported in the last 30 years on alternative and less energy consuming sealing procedures. Focused on the objectives of the sealing step in aluminum anodization, these literature reports and some relevant much older reports are reviewed with respect to their respective potentials to ensure reduction in the energy input in the anodization process and environmental implications. In pursuing the objectives of this study, much consideration was given to the combination of present and anticipated regulatory constraints and operational demands in the aerospace industry, which informs the need for development of energy efficient sealing procedures that are not based on the use of chromium, nickel, or cobalt, but yet possesses the active corrosion protection ability characteristic of chromium sealing. In addition, perspective is provided on plausible approaches to minimizing the energy and environmental impact of the sealing step and ensure a more sustainable aluminum anodization process.
For this systematic review, a search was made using the bibliographic search engine, Google Scholar, using the terms; “sealing of anodized aluminum”, “chromate sealing”, “rare earth sealing of anodized aluminum”, “manganese toxicity”, “nickel toxicity to humans”, “molybdenum OR molybdate toxicity”, “vanadium OR vanadate toxicity”, and “tungsten OR tungstate toxicity”. Only materials published in peer-reviewed academic journals, books, conference papers, theses and dissertations, technical reports, and patents as at November 30, 2019 were considered. Relevant references from these works of similar pedigree were also consulted and considered.
3. Summary of Current State of Art in Sealing Anodized Aluminum
Table 1, presents information from the search on recent reports on alternative sealing methods reported for anodized aluminum alloys, the bath compositions, operating temperature(s) and major findings. For further information from much earlier reports on the sealing process the review by Hao and Cheng [22] and the even earlier report by Wood [23] are recommended.

Table 1.
Table of reported sealing procedures, sealing bath conditions and major findings.
4. Structure and Composition of Anodized Oxide Layer on Anodized Aluminum and Implications for Sealing
Prior to discussing the mechanism of the sealing step in different sealing methods, it will be vital to highlight important features of the post-anodizing but unsealed anodic oxide layer, particularly the structure and composition of the porous anodic oxide layer. In terms of structure, the anodic oxide layer formed on aluminum after anodization is comprised of two layers; a thicker and porous outer layer (measured in µm) and a much thinner but compact inner layer, the barrier layer (measured in nm). A surface view of anodized aluminum surface prior to sealing showed tubular shaped pores more or less normal to the interface with diameter reported in the range of 100–200 Ǻ (10–20 nm) for sulfuric acid (17.5% v/v at 19 ± 1 °C) anodized 99.99% aluminum sample [59,60,61]. Keller et al. [62] and Hunter and Fowle [63] in their respective studies of the structural features of anodized oxide coatings on aluminum had concluded that while the pore size is much dependent on the electrolyte used and independent of anodization voltage, the pore wall thickness and the barrier layer thickness are primary functions of anodization voltage, and are less affected by the electrolyte used. Figure 1 illustrates the structure of the anodized layer on aluminum prior to sealing and changes that occur during sealing.
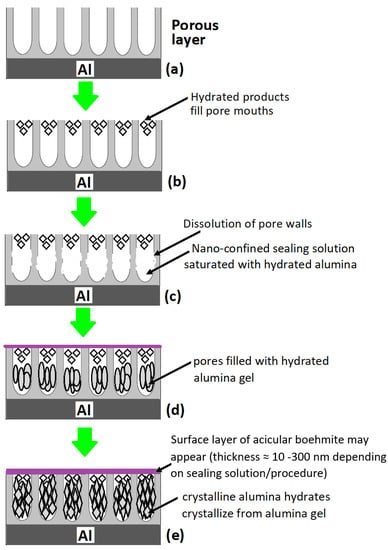
Figure 1.
Structure of the anodized layer on aluminum prior to sealing and changes that occur during sealing; (a) anodized aluminum prior to sealing, (b) pore mouths filled with hydrated products, (c) dissolution of pore walls, (d) precipitation of hydrated alumina gel in pores and emergence of surface layer of acicular boehmite, and (e) crystallization of crystalline alumina from alumina gel.
The chemical and phase composition of the anodized layer on aluminum has been a subject of much interest [64,65,66,67,68,69,70,71,72]. From analysis of these numerous reports on the subject, there appears to be a consensus that the porous anodized layer on aluminum prior to sealing is predominantly comprised of amorphous alumina [64,69,73,74], some boehmite (AlOOH) [66,75,76], and aluminum oxide compounds whose actual compositions are determined by the bath electrolyte anion chemistry. The structure of amorphous alumina which constitute much of the anodic layer has been studied extensively using a variety of techniques suitable for probing structural details of non-crystalline and poorly crystalline materials. However, while some workers [77,78] have reported that Al cation in anodized layer are coordinated to oxygen anions in two ways (tetrahedral (AlO4) and octahedral (AlO6) coordination), other researchers [79,80,81,82] have reported 3 types of co-ordination of Al cations with oxygen anions (tetrahedral (AlO4), pentahedral (AlO4), and octahedral (AlO6) coordination). Hashimoto et al. [82] recently investigated the local structure around aluminum atoms in anodic alumina using nuclear magnetic resonance spectroscopy, critiqued earlier studies on the subject matter, and reported that aluminum cations coordinated with oxygen anions in three types of co-ordination; AlO4 (tetra-coordination), AlO5 (penta-coordination), and AlO6 (octahedral-coordination), and that penta-coordination was predominant while the proportion of octahedrally coordinated (AlO6) units decreased with removal of physisorbed water. They [82] also evaluated the range of ratios of each mode of coordination to be 30%–40% for AlO4, 50%–60% for AlO5, and 4%–15% for AlO6. The roles that surface coordination plays in precipitation and dissolution of mineral phases had been discussed elsewhere [83,84,85,86]. The three types of aluminum cation coordination with oxygen anions in the porous anodized aluminum oxide layer is schematically presented in Figure 2.
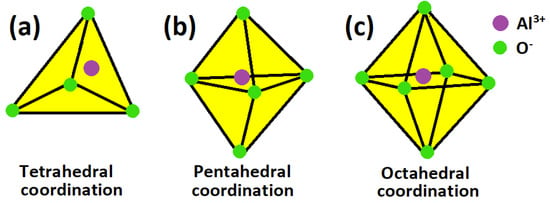
Figure 2.
Schematic illustration of the three ways in which aluminum cations coordinate with oxygen anions in the porous anodized aluminum oxide layer (octahedral coordination is reported [78,87] to be enhanced by sealing at the expense of tetrahedral coordination).
Rocca et al. [78,87] investigated the structural evolutions and chemical reactivity of nano-porous anodic alumina films and their influences on the sealing process and concluded that current perception of the anodic film as composed of amorphous alumina is an over-simplification of the reality, and proposed a description of the anodic alumina layer that recognizes the important role(s) of incorporated electrolyte anion(s), which was the sulphate anion in their study. Their [78,87] description of the structure of the anodic oxide on aluminum is, an oxide structure composed of 2/3 of its aluminum in tetrahedral coordination (AlO4), 1/3 in octahedral coordination (AlO6), and incorporated electrolyte species; sulphate species. Baker and Pearson [88] described the anodic oxide on aluminum as consisting of close-packed oxide crystallites of crystallite radius in the range of 2.4 nm, significantly similar to Murphy’s description [89] of the structure of the porous anodic oxide layer on aluminum.
From literature reports [69,90,91,92] it is inferred that typical electric field strengths in the anodic oxide film during anodization which act on electrolyte species and influence their incorporation into the anodic oxide layer is in the range of 106–108 V cm−1. Aluminum sulphate (Al2(SO4)3), is reported to present in porous oxide layers of aluminum anodized in sulfuric acid [93]. The degree of electrolyte anion (SO42−) incorporation into anodic aluminum oxide films have been correlated to the anodization potential [94], and can be further correlated to the electric field strengths in the anodic oxide film during anodization, as the electric field strength has a dependency on the applied anodization potential. Lee et al. [94] had reported about 88% higher anionic impurities (mostly SO42−) in hard anodized anodic aluminum oxide samples (formation voltage = 35 to 55 V), compared to mild anodized anodic aluminum oxide samples (formation voltage = 25 V), and explained the observation using the high-field conduction theory. For porous oxide layers on aluminum anodized in phosphoric acid, the presence of AlPO4 [77,95] or Al2PO4(OH)3 [68], Al2O3·17P0.072 [69], and (Al2O2.88)100(PO4)8(OH)0.1H2O [96] have been reported. With respect to the presentation of electrolyte species incorporated into porous anodic oxides layers in aluminum anodized in baths containing carboxylic acids like oxalic acid there is apparently no consensus. Whereas Fukuda [97,98] suggest that the oxalate anions (HC2O4−) undergo de-protonation under the effect of the operative electric field to yield (C2O42−) which most probably is incorporated into the anodic alumina layer, Yamamoto and Baba [99] are of the opinion that though C2O42− might be incorporated in the anodic layer at some point most probably as oxalate-aluminate species, these undergo decomposition processes followed by polymerization in which radical species generated under the operative electric field play are presumed to play important roles. In the light of these, it is obvious that both barrier type and porous-type anodic films formed in aluminum are not entirely composed of alumina but are "contaminated" by up-take of electrolyte species [100]. Surganov et al. [101] employed the combination of Auger electron spectroscopy (AES) and Rutherford backscattering spectroscopy (RBS) in studies an anodic oxides of aluminum anodized in oxalic acid (C2H2O4) solution and reported the presence of significant carbon concentration in the oxide layer, with a maximum at the surface and a slow decrease through the oxide, but no carbon signal was detected in the thin barrier layer next to the metal. Fukuda [97] studied the role of oxalate ion in the formation of porous anodic oxide films in aluminum anodized in oxalic acid and reported that the concentration of C2O4 in the anodized layer varied with depth, and anodization time, and increased with increase in the oxalic acid concentration in the anodizing bath and/or on lowering the bath temperature. Based on the concentration of C2O4 with respect to depth (depth profile) he concluded that the anodic oxide layer can be divided into three parts; an outer layer rich in carbon (high C2O4 content ≈ 9%), an intermediate layer in which the C2O4 content reaches a maximum (≈11%), and an inner layer with very little C2O4 content, as the electrolyte species content decreased rapidly with depth in this inner region. This depth profile reported for carbon in oxide layers on aluminum anodized in oxalic acid is quite similar to that reported by Thanh et al. [102] for phosphorus in phosphoric acid anodized aluminum. With respect to anodization time, Fukuda [97] reported that the C2O4 content of the anodic oxide was small at the onset of anodization (≈3.1% after 10 seconds of anodization) reached a maximum after 60 s of anodization (≈7.5%), and then decreased quite slightly to an almost constant value (≈7.2 %) from 3 to 30 minutes of anodization. Thanh et al. [102] compared the depth profiles of phosphorus and sulfur in anodic oxide films formed in phosphoric acid with that formed in sulfuric acid and reported that phosphorus presents largely in the oxide region next to the oxide- electrolyte interface and decreases gradually, moving towards the oxide-metal interface with its maximum concentration at ≈1/3 of the film thickness from the oxide metal interface (i.e., ≈2/3 away from the electrolyte-oxide interface (Figure 3a). On the other hand, the sulfur profile presented a bell-shaped profile, increasing steeply from the electrolyte-oxide interface, remains constant in the middle of the oxide film, and then decreases about as steeply towards the oxide-metal interface (Figure 3b), which led them to their obvious conclusion that the distribution of electrolyte species is more uniform in oxide films formed in sulfuric acid than those formed in phosphoric acid [102].
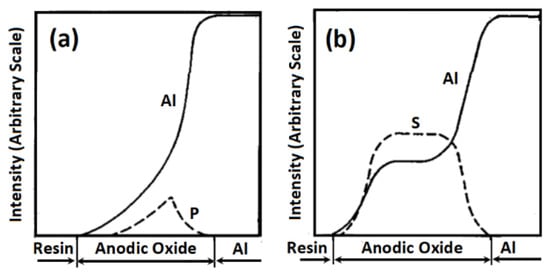
Figure 3.
Phosphorus and sulfur profiles for anodic oxide films formed in (a) phosphoric acid bath, and (b) sulfuric acid bath. Reproduced with permission from The Electrochemical Society of Japan from Ref [102].
Having thus established the different trends in the distribution of the respective common electrolyte species incorporated into porous anodic oxide films on aluminum with respect to depth (elemental depth profile information), the trends in the reported total content(s) of the respective electrolyte species shall be reviewed. Fukuda and Fukushima [103,104] reported that SO4 contents was in the range of 12.8% to 14.0% in anodic oxide films they formed on aluminum in sulfuric acid baths at 1.97 A/dm2, and that the sulphate content of the films was almost independent of the sulphate concentration in the bath. From reports by several other authors [69,79,103,104,105,106,107,108,109] it can be inferred that sulfur content as SO4 in anodic porous oxide films on sulfuric acid anodized aluminum can vary from 10 to 17% depending on anodization conditions. Farnan et al. [80,81] had reported average phosphorus content of 4 wt.% for anodic oxide films on aluminum anodized in phosphoric acid, while Bocchetta et al. [74] reported about 10 at. % phosphorus content for anodic oxide films on aluminum anodized in phosphoric acid. Wernick et al. [2] reported that anodic oxide layers on aluminum anodized in phosphoric acid usually have a phosphate content in the range of 6–8 wt.% phosphate. Reported oxalate incorporation into porous anodic films on aluminum anodized in the presence of oxalic acid/oxalate ion is in the range of 2–7 wt.% oxalate [70,80,107]. Generally, the level of contamination of alumina by anodizing bath anions is reported to vary according to the sequence; chromic < oxalic < phosphoric < sulfuric [110].
Tracer studies [69,111,112,113,114] on anodic films formed on aluminum in different electrolytes have enabled better determination of the nature/presentation of the electrolyte species incorporated in the anodic films, the nature of their respective mobilities in the anodic film and the resultant differences in their depth profiles into the anodic oxide layer. Thompson [69] had reported that boron, tungsten and phosphorus species incorporated into anodic oxide layers on aluminum anodized in borate, tungstate and phosphate electrolytes respectively, permeated the anodic layer to different depths. He [69] determined that from the film–electrolyte interface, W species present only in the very outer film regions (very little penetration into anodic layer), B species present over about 0.4 of the film thickness, while P species showed very marked penetration into the anodic film presenting in over two-thirds of the film thickness (Figure 4). These observations were attributed to the differences in mobilities of the different electrolyte species in anodic oxide layer on aluminum under the influence of the operative electric field, in which W species are outwardly mobile, B species immobile, while P species are inwardly mobile in anodic alumina, respectively [69]. The use of Rutherford Back Scattering Spectroscopy (RBS) and related nuclear methods was reported [69] to enable further determination that W is present as an outwardly mobile cation (WO22+ and/or W6+), P as an inwardly mobile anion ((PO4)3−), while B is present as neutral species [115] which might explain its immobility under the operative electric field. Based on similarities in the depth profiles of carbon in anodic oxide films on aluminum anodized in oxalic acid reported by Fukuda [92], and the depth profile reported for phosphorus in anodic oxide films on aluminum anodized in phosphoric [69,102], it might be concluded that the oxalate ion (C2O42−) like the phosphate ion (PO43−) are largely unaffected by the high field, and thus migrate inwards into the anodic oxide layer from the electrolyte-oxide interface. These reports of differences in the extent of incorporation of electrolyte species into the anodic oxide layer formed on aluminum might have important implications on development of procedures for obtaining sealed aluminum products suited for applications in which good adhesion properties post-anodizing and sealing of products is a requirement; as in certain materials for the aerospace and automobile industries [116,117,118]. High temperature sealing (with boiling water) [119,120,121,122,123,124,125,126] and the presence of a “sealing oxide layer” (top-layer over the anodic oxide film) [127] over the pores of the porous anodic oxide layer on aluminum have been reported to exert deleterious effects on adhesion properties [121,128] in anodized aluminum products that might need to be coated or adhesively bonded to other materials. In the light of these, we postulate that by careful choice and inclusion of preferentially outwardly mobile species/cation(s)) like tungsten species (Figure 4) which consequently present only in the very outer regions of the film, and are able to exert inhibitive effects on the growth of significant “sealing oxide layer” on the sealed anodic oxide layer but no significant deleterious effects on pore sealing, improvements in adhesion and abrasion properties of sealed aluminum products might be obtained without sacrificing corrosion resistance. Boiling water treatment on aluminum alloy prior to structural adhesive bonding has been reported [129,130,131,132] to lead to improvements in the fatigue life and bond strength, durability and moisture stability of aluminum alloys adhesively bonded with epoxy adhesives. These improvements have been largely attributed to the formation of a highly porous structured pseudo-boehmite layer at the aluminum surface [133]. It has been demonstrated that pseudo-boehmite (which accounts for the sealing effect in sealed anodic aluminum oxide) can be formed on aluminum surfaces in warm water at such low temperatures as 40–50 °C [129,134,135]. Abrahami et al. [123] studied the effect of the surface chemistry of the anodic oxide formed on aluminum on adhesive bonding with epoxy, and based on their correlation of the mechanical performance of different joints to calculations of the relative amounts of O2−,OH−, PO43−, and SO42− species on the surface from measured X-ray photoelectron spectroscopy (XPS) data, observed that wet adhesive strength increases with the hydroxyl concentration at the aluminum oxide surface, and concluded that interfacial bonding with the epoxy is established through surface hydroxyls. Their [123] conclusion is consistent with results of an earlier theoretical work [136]. In the light of this, it is plausible that increasing the proportion of hydroxyls on the surface of sealed anodized aluminum cam improve adhesion properties.
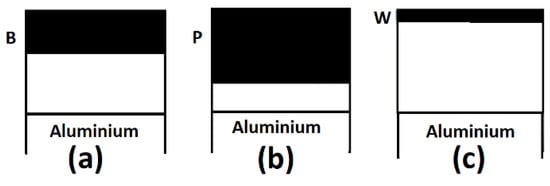
Figure 4.
Schematic diagrams of sections of barrier films formed on aluminum in: (a) borate; (b) phosphate; and (c) tungstate electrolytes, adapted and reproduced with permission of Elsevier from Ref [69].
Thompson [69] had made an important postulation that differences in the mobility and presentation of the electrolyte species is linked to their stability (binding energies) under the influence of the operative electric field(s), and that the relative mobilities of cations within the anodic film roughly correlate with the single M–O bond energies. By comparison of the energies required to break the single X–O bonds (X = metal or non-metal) in electrolyte species most likely to be present in the anodizing bath (Table 2) with those of B (B–O), P (P–O), and W (W–O) with known behaviors in the predominantly alumina layer on anodized aluminum under anodization conditions using Al–O bond energies as a baseline, vital insights can be obtained on the probable migration/mobility behavior of electrolyte species that might be introduced. Such insight will be vital in developing the ability to deliberately manipulate the depth profile of incorporated species in anodic oxide films on aluminum surface which might be advantageous.
Table 2.
Binding energies of relevant bonds in the anodization bath.
Having thus highlighted the peculiarities of the structure and composition of the anodized oxide layer on aluminum which goes into the sealing bath, the various mechanism(s) involved in the most widely used sealing methods shall be briefly reviewed.
5. Mechanisms of the Sealing Step in Various Sealing Methods for Anodized Aluminum
Generally, sealing of the pores in anodized aluminum occurs by some degree of pore wall dissolution coupled with formation/precipitation of phases that fill the pores (Figure 1), but the actual mechanism by which this occur vary with each sealing method, the sealing temperature, and the constitution of the sealing bath. For brevity detailed discussions of the mechanism(s) of each sealing method is omitted. Particular focus is on highlighting the uniqueness of the sealing agents in dichromate sealing (its relevant characteristics) using that for hydrothermal sealing as a baseline, to give insights on plausible requirements and routes to developing sealing methods that can meet the composite performance benchmarks obtained with chromate sealing in the aerospace industry [16]. However, the highlights of the mechanism of each sealing method and the precipitated agent(s) responsible for the reported sealing effects are highlighted in Table 3. For further reading on the mechanism of each of these sealing methods the references in Table 3 and several other reports and reviews by earlier workers are recommended [4,22,23,26,56,88,138,139,140,141,142,143,144].
Table 3.
Table of different proposed sealing mechanism for anodized aluminum.
In hydrothermal sealing (boiling deionized water), the sealing phenomena is generally attributed to conversion of the oxyhydroxide to aluminum trihydroxide which having a higher specific volume than the aluminum oxyhydroxide occupies a greater volume, and thus ensures closure of the pores [148,149,153]. According to Tomashov and Tyukina [146,154] chromate sealing process of anodic alumina commences with chromate absorption unto alumina surface, followed by formation of either aluminum oxydichromate or aluminum oxychromate depending on pH, and then subsequent hydration of the coating due to reaction with water; the rate of hydration increasing with increase in pH of the studied potassium dichromate solution (of pH 3.7) up to pH 10, above which rapid dissolution of the oxide film commences.
An alternative theory to explain hydrothermal sealing of anodized aluminum was propounded by Murphy [155] to be based formation of inert hydrated oxide skin over the internal pore surfaces due to anion exchange processes between the absorbed and/or incorporated electrolyte anions and the sealing media. On the basis of experimental proofs of initiation period(s) prior to observation of sealing effects [156], influence of anions, and their propensity to adsorb unto alumina on the sealing process [157,158] which are expected from analysis of this theory, Diggle et al. [4] had concluded that Murphy’s model is operative at the commencement of the sealing process while the pore blocking mechanism predominates after this period.
For ambient temperature sealing of porous anodic oxide films on aluminum using nickel fluoride solutions, Kalantary et al. [26] had proposed a mechanism in which entry of fluoride ions into the pores initiates a place exchange mechanism that causes sufficient shift in the local pH for precipitation of nickel ions as nickel hydroxide. The precipitated nickel hydroxide then ensures sealing of the film by blocking the pore mouths [26]. According to Kalantary et al. [26] the ageing effect which is prominent in nickel sealing is due to diffusion of water into the film, in a slow process that leads to film hydration, and consequently further and general blocking of pores.
In recent reports, Rocca et al. [78,87] on the basis of results from their studies on the chemical reactivity of nano-porous alumina and their description of nano-porous anodic alumina consistent with its reactivity as an amorphous structured oxide with around 60% of Al3+ cations in the AlO4 configuration (tetrahedrally coordinated), 40% in the AlO6 configuration (octahedrally coordinated), and containing around 5 wt.% sulphate anions that are homogeneously distributed, attributed the “sealing effect” on anodic alumina in aqueous media to incorporation of water in the nanostructure(s), which then induces transformation of aluminum coordination in the amorphous oxide from tetrahedral coordination (disappearance of AlO4 clusters) to octahedral coordination (increase in AlO6 clusters), and the release of sulphate species by hydrolysis (Equations (1) and (2)) [78,87]. Figure 1 illustrates the structure of the anodized layer on aluminum after the sealing process.
6. Factors Affecting Sealing and How these Factors Might be Exploited in Developing Better Sealing Procedures
Since from the review of the mechanism(s) of the sealing process above it is quite obvious that some dissolution of the pore walls is necessary to generate materials of higher specific volume to fill the pores under the sealing conditions, appreciation of factors that might influence alumina dissolution and/or boehmite precipitation will be vital to developing new sealing procedures that enhance energy efficiency of the sealing process. Wei et al. [19] had demonstrated that greatest influence on sealed film quality is exerted by temperature, followed by the pH of the sealing media, then the presence of salts, surfactants, and complexants, respectively, (i.e., temperature > pH > salt mixture > surfactant > complexant). Sheasby and Bancroft [159] had reported that pH drops in the hot water sealing bath below 5·5 (recommended pH 5.5 to 6.5) leads to inhibited formation of boehmite during sealing, and resultant poor sealing outcomes. To obviate the wide pH fluctuations in the sealing bath due to among other factors residual acid carried over from the anodizing bath, they [159] demonstrated that addition of 0.1% to 1% ammonium acetate to the hot water sealing bath exerted pH buffering effects without compromising sealing quality, in addition to enhancing the resilience of the sealing bath/process to impurities (e.g., Cu).
Okada et al. [160] studied boehmite (γ-AlOOH) precipitation from solutions of aluminum nitrate and sodium aluminate under varying conditions and concluded that in the temperature range between 22 °C and 80 °C gels precipitated below pH 7, crystalline boehmite in the pH range 7 to 10, while and bayerite (Al(OH)3) formed at pH ≥ 11 (Figure 5). Though this report does not provide much information on the kinetics of boehmite precipitation, it indicated that boehmite precipitation can be achieved at temperatures lower than that currently used in hydrothermal sealing in boiling deionized water. This report is corroborated by the later report of Lee et al. [161] in their study that whilst bayerite (Al(OH)3) was formed at pH >10.6, pseudo-boehmite was formed in the pH range 7.6 to 9.6. Furthermore, Alwitt [162] had reported that pseudo-boehmite film growth on aluminum is feasible on aluminum immersed in water in the temperature range 50–100 °C in a process that is initially determined by the nucleation and growth of hydrolysis sites on the amorphous oxide surface, and afterwards by solid state diffusion through the already formed pseudo-boehmite layer, and that at temperatures as low as 40 °C pseudo-boehmite is able to grow concurrently with bayerite, though bayerite crystallization ultimately become predominant. Although Ito et al. [163] report that boehmite (Al2O3·H2O) formation is favored over bayerite (Al2O3·3H2O) formation on aluminum exposed to water at temperatures higher than 80 °C other reports [164,165,166,167] appear to confirm that boehmite can be formed on aluminum at temperatures ≥ 55 °C. From these reports, it can be inferred that by adjusting the pH of the sealing solution towards mildly alkaline pH values (pH ≥ 8) boehmite precipitation might be likely at temperatures significantly lower (50–60 °C) than that currently employed in hydrothermal sealing (>90 °C), albeit sealing time might need to be increased to compensate for the slower kinetics at lower temperatures. The enhanced sealing effect(s) observed in sealing solution for anodized aluminum containing Ce(III) species have been attributed to its role in catalyzing hydrothermal transformation of the anodic oxide to more stable morphological form(s) [37,168].
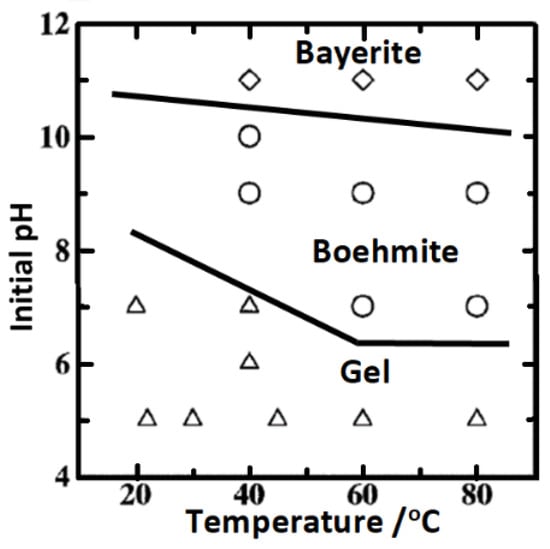
Figure 5.
Initial pH–temperature stability field map of products produced by precipitation method, reproduced with permission by Elsevier from Ref [160].
7. Physical and Chemical Changes during the Sealing Step in Aluminum Anodizing
Hot water sealing of anodized aluminum is reported to result in ≈100-fold improvement in the corrosion resistance of the sealed films compared to anodized but unsealed films [169]. Sealing of the anodic oxide layer in anodized aluminum involves a progression of series and varieties of physical and chemical changes [2,3,127]. However, whereas some of these changes are universal irrespective of the sealing method employed others are closely related to the particular sealing method employed. Understanding of these physical and chemical changes and development of tools and procedures to monitor their progression during the sealing process can provide a means of monitoring the sealing process, and the seal quality of the products from the process. It is important to note that in some applications, a high-quality of sealing might not be desirable as high-quality sealing drastically reduces the porosity. A high degree of sealing is reported to yield a smoother anodized aluminum surface which results in lowering of the contact area between anodized aluminum and primers or other coatings due to reduction in mechanical interlocking effect, which consequently results in poor adhesion [118]. Wood [23] had reported that major changes to anodic alumina due to sealing are reductions in porosity, change(s) in crystal structure, and decrease in ability to absorb dyes. According to González et al. [170] sealing of the anodic oxide film on aluminum generally results in the formation of four distinct layers; an outer surface layer principally consisting of well-crystallized boehmite/pseudo-boehmite, an intermediate layer composed of hydrated oxide(s), an inner layer that retains the original cellular structure, but with partially dissolved pore walls and partly or completely filled with aluminum hydroxide gel, and a significantly thin barrier-type layer that demarcates the porous layer from the metallic aluminum substrate.
Dorsey [171] studied structural transitions in anodic oxide layers on anodized aluminum and from analysis of IR-data for sealed and unsealed anodic oxide films on aluminum demonstrated that sealing causes removal of some of the incorporated electrolyte anions and their probable replacement with hydroxyl group(s); an idea earlier expressed by Murphy [155]. Baker and Pearson [88] employed wide-line nuclear magnetic resonance (NMR) measurements in their studies and reported that the anodic oxide layer on aluminum anodized in 15% H2SO4 at 25 °C and 129.17 A m−2 for 1 h and then sealed for 30 min in boiling water at pH 6 contained 11%–13% H2O as OH groups and an additional 1%–4% as physically adsorbed water. Furthermore, they [88] reported that 46% of this water content of sealed anodic oxide layer on anodized aluminum is on the surface, and that the surface area of the anodized layer reduced from 20 m2/g (post-anodization) to 5 m2/g after sealing, and thus regarded sealing to be a predominantly surface reaction on the crystallites that make up the coating structure. From analysis of their data they [88] concluded that more than half of the combined water in sealed anodic oxide is resident within the first two oxide ion layers of the surface, and that the major pore surface is almost completely covered by OH, while the remaining hydroxyl groups are distributed over crystallite surfaces of the pore walls, and inside the crystallites that they postulated to comprise the anodic oxide layer. Thompson et al. [172] studied morphological changes on porous anodic films formed on aluminum in phosphoric acid (with its unique much larger cell and pore diameters) due to long term (up to 72 h) sealing in hot water and stated that sealing apparently involves three distinct but overlapping steps; material precipitation in the pores, particularly near the outer film surface, formation of crystalline material at the oxide layer surface, and further changes that occur deeper which probably involves the oxide micro-crystallites aggregation and redistribution of porosity. From their observations they [172] concluded that the major morphological changes due to sealing occurs in the outer 2–3 µm of the anodic oxide film, pore closure commences at the outer surface and proceeds towards the entire depth, and that an intermediate layer is formed between the outer surface crystals and the filled pores. Murphy et al. [58] studied sealing of porous anodic layer on aluminum using different sealing methods and concluded that sealing increases the degree of hydration throughout the oxide layer irrespective of sealing method but the degree of hydration with nickel acetate sealing is much higher than that obtained by dichromate sealing. Patermarakis and Papandreadis [173] carried out prolonged sealing treatment of porous anodic films on aluminum in water at 100 °C for 4.5 h, calculated the quantity of H2O retained, correlated it to the pore void volume of dry films, and reported that complete blocking of the pore mouths significantly retard hydration process(es), and suggested that the formation of a gas phase is a likely factor contributing to inhibition of hydration processes. They [173] concluded that many structural features are involved in the mechanism of oxide hydration and pore closure. González et al. [31] made a comparative study of the behavior of properly sealed, poorly sealed and unsealed anodized specimens that were allowed to age for periods ranging from 1 month to 20 years under moderately aggressive atmospheres, and postulated that under these conditions there is a competition between deteriorative processes and auto sealing processes, and that the long held complex mechanism for sealing need to be modified to account for observed changes in the anodic films during ageing. The changes observed during the ageing step were postulated [31] to apparently involve the loss of inter-crystalline water, and very slow transformation of initially kinetically favored hydrated alumina species into more thermodynamically stable allotropic species. From their results they [31] concluded that partial initial sealing (e.g., for time durations as short as 1 min) apparently accelerates the autosealing process during ageing, and that for unsealed and poorly sealed anodic films in moderately aggressive atmospheres, the very significant effects of sealing/autosealing process is masked by deteriorative effects of aggressive species until the sealing processes become predominant. They [31] also concluded that mass gain in unsealed anodic films is the first step of the autosealing process, and is due to gradual absorption of water until the pores are saturated, resulting in the mass gained becoming equivalent to that gained during sealing. In a later work, González et al. [174] studied changes in cold sealed aluminum oxide films (sealed in cold nickel fluoride solutions) and reported that fluorine and nickel concentrate exclusively in the outer layers of cold sealed anodic films (near the mouths of the pores). They [174] also reported significant mass changes in cold sealed aluminum oxide films exposed to highly wet and extremely dry atmospheres attributed to water absorption (which can reach up to 30 mg/dm2), and that closure of the pore mouths is not completed in the sealing bath, but outside the sealing bath during ageing. Thus, it was established that changes continue in sealed anodic oxide films on aluminum after the sealing step is concluded [31,174]. In yet another publication, González et al. [170] studied post-sealing changes in anodic oxide films formed on aluminum by monitoring the different/successive stages of the sealing and auto-sealing processes (proposed to involve, “pore plugging, pore wall dissolution, precipitation, crystallization, agglomeration of hydrated alumina and formation of the intermediate layer”), the sealing quality of the anodic layers, the degree of sealing and aging and reported that auto-sealing process of anodic films can continued to evolve for decades. In addition, in contrast to their observations that very short time sealing resulted in significant increase in the specific surface areas, they [170] demonstrated that generally on the long-run, the specific surface area reduced with progression of the sealing and auto-sealing process; (specific surface area ≈36 m2/g) for pore wall dissolution and pore widening > (specific surface area ≈18 m2/g) during partial filling of pores and pore plugging > (specific surface area ≈ 2 m2/g) during precipitation-recrystallization and formation of the intermediate layer > (specific surface area < 0.5 m2/g) during ageing and long duration auto-sealing that results in the agglomeration and formation of larger crystals at the expense of smaller grains. Bartolomé et al. [127] monitored physical and chemical changes during sealing and autosealing–ageing processes in anodic oxide films on aluminum using thermogravimetry and gas adsorption porosimetry techniques and reported approximate doubling of the pore volume (with pore diameter increasing from 140–150 to 200–220 Å) due to anhydrous alumina dissolution during the pore wall dissolution stage, coupled with about 50% increase in specific surface area (from ≈20 m2/g to 30 m2/g). In addition, a 2–3 orders of magnitude difference were reported [127] in the impedance of the sealed and unsealed anodic films over a wide frequency range which justifies the use of electrochemical impedance spectroscopy (EIS) a tool to for monitoring sealing quality or changes in the electrical responses of the constituent layers of the anodic oxide film on aluminum. This report demonstrates that impedance changes, mass changes and changes in specific surface area, and hence adsorption capacity accompany the sealing process.
Hu et al. [28] studied the structure evolution induced in anodized aluminum by sealing using different sealing processes (namely sealing in hot water, hot 5 g/L nickel acetate, cold 5 g/L nickel acetate, and cold saturated nickel acetate (180 g/L), respectively), and reported that whereas decrease in pore size was common in all the studied sealing methods, cold sealing and hot water sealing do not result in alterations in the cylindrical porous oxide framework of the anodic aluminum oxide layer, but hot nickel acetate sealing results in both filling of the pores (hence lowered porosity) and presence of deposits on aluminum oxide-air interface. With some sealing methods, other authors [45,127,175] have reported the presence of superficial (“hydrated oxide”) sealing layer on top of the anodic oxide film after sealing, of thickness in the range of 200–300 nm. The presence of such deposits on aluminum oxide-air interface is likely to result in reduced adhesive properties for hot nickel sealed anodized samples. According to Rocca et al. [87] during sealing there is a change in the local environment around aluminum cations from tetrahedral coordination to the more stable octahedral coordination coupled with the simultaneous uptake of water and ejection of some of the incorporated sulphate (or other incorporated electrolyte anion(s). Citing molecular theory calculations of the charge of Al atoms in different sites by Van Bokhoven et al. [176] which reported that whilst the calculated charge of Al atoms in octahedral sites is about +0.572, the charge of Al atoms in tetrahedral sites is about +0.737, they [87] highlighted the more electrophilic character of aluminum in tetrahedral sites. The apparent implication of this depletion of the more electrophilic tetrahedrally coordinated aluminum (and increase in the less electrophilic octahedrally coordinated Al sites) during sealing could be reduced reactivity of the pore surfaces to nucleophiles (anions such as OH− and SO42−) and thus can partially explain the reported ejection of sulphate during the sealing process. An undesirable physical change that can occur in sealed anodized samples, particularly hot-sealed samples is crazing. Crazing is thermal-shock induced cracking of the sealed anodic oxide layer due to very significant thermal expansion coefficient mismatch between the underlying metal and the sealed anodic oxide layer (ratio of mismatch ≈ 5:1 [177]). Systems and techniques of mitigating crazing in anodized metals is the subject of a recent patent [178]. From the review of the physical and chemical changes that occur due to sealing of anodic oxide layers and insights from literature, a flowchart demonstrating the changes that occur during the sealing step and its exploitation to monitor both the sealing process and seal quality is presented in Figure 6.
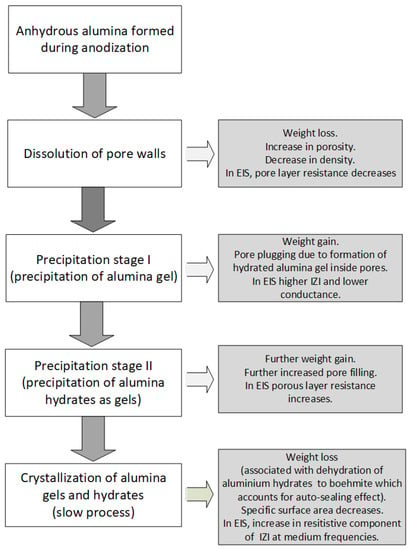
Figure 6.
Flowchart of changes that occur during the Sealing Step and Its Exploitation to Monitor Sealing Process and Seal Quality, based on deductions from Refs [170,179,180].
8. Monitoring Physical and Chemical Changes during the Sealing Step in Aluminum Anodizing
Theoretically each of the chemical and physical changes induced by the sealing process can be used to monitor the quality and/or progression of sealing. The physical and chemical changes that can be used to monitor sealing quality and or evolution of the sealing process are principally; impedance changes [25,57,181], weight changes [5,182], and changes in porosity [127]. However, ease of use, possibility of making measurements without interfering with the progression of the sealing process, and other factors limit the range of physical and chemical changes that are actually employed. In practice, the physical or chemical change actually employed in motoring sealing quality is diverse and dependent on application. In industrial practice the three common tests used to evaluate sealing quality are: (a) The dye spot test in which the qualification criterion is a spot strength below 2 [183,184]; (b) the phosphor-chromic acid dissolution test in which the allowed mass losses should be below the threshold value of 30 mg/dm2 of sample surface [185,186,187]; (c) and the 1 kHz admittance test at in which the pass criterion is an admittance value below 20 µS [188]. Table 4 presents the different techniques that have been employed to monitor changes in the anodic oxide layer due sealing, from which it is obvious that combinations of techniques are usually employed.
Table 4.
Table of techniques used to monitor changes in oxide layer due to sealing.
9. Electrochemical Impedance Spectroscopy (EIS) as a Tool for Monitoring the Sealing Process and the Efficiency of the Sealed Products in Aluminum Anodizing (Post-Sealing)
Electrochemical impedance spectroscopy has emerged as a very important tool for monitoring the sealing process both in-situ and post-sealing. González et al. [31] in their study on the use of EIS to monitor ageing and over-aging of sealed and unsealed aluminum oxide films, had reported that the most significant differences in the impedance spectra of unsealed and properly sealed anodized aluminum samples, were observable in the medium to high frequency regions, over a frequency range spanning 2–3 orders of magnitude in which higher impedances correlated with the degree of completeness of the sealing. According to Mansfeld and Kendig [190] the capacitive response of water-saturated porous anodized aluminum layer is so small that it is only detectable at frequencies higher than 107 Hz. Hence, they [190] concluded that at frequencies ≤ 105 Hz commonly employed in electrochemical impedance studies the major contribution to the impedance spectra of unsealed anodized aluminum films is from the barrier layer. Corroborating the position of Mansfeld and Kendig [190], Bartolomé et al. [127] in their studies on sealing and auto-sealing/ageing processes in anodized aluminum films had reported that the resistance of the pore walls (Rpw) is so low and contributes so little to the impedance response at all frequencies that it is not initially detected in the impedance spectra of unsealed anodized samples but become observable as the sealing or auto-sealing process progresses. Hence the presence and increase in the value of Rpw can be used as a measure of the quality and progress of the sealing process.
Zhao et al. [189] studied self-sealing process of un-sealed aluminum anodic films in neutral NaCl and Na2SO4 solutions using EIS in the frequency range 10−2 to 105 Hz, and reported only one capacitive arc (one time constant) for unsealed aluminum anodic films in neutral 0.1 M NaCl solution at short immersion times attributed to the response of the barrier layer [56,191,192], and the emergence of a second capacitive arc (second time constant) that increased with immersion time (up to a month) at high frequency which was attributed to increasing electrochemical impedance response of the pore walls with progression of the self-sealing process in the hitherto unsealed anodized aluminum samples. These reports demonstrate the efficacy of EIS as a tool to monitor evolution of the sealing process in unsealed anodized aluminum alloys. While using EIS to monitor the sealing process in anodized aluminum alloys, it is important to reinforce/highlight the caveat by Domingues et al. [193] that if the barrier layer is attacked by localized corrosion, the value of the resistance measured at low frequencies should not be attributed to the barrier layer, but instead to the charge transfer resistance associated with the corrosion process. The onset of localized corrosion attack of the barrier layer might be detected by decrease of the fitted barrier layer resistance with immersion time, instead of a fairly constant barrier layer resistance with time. In a recent report [34] a time constant observed at medium frequency range in anodized aluminum samples after sealing (in the frequency range ≈ 102–104 Hz) had been attributed to the precipitation of sealing products. In an earlier work [25] this medium frequency time constant had been attributed to precipitation and solidification of the sealing products as it only appeared in hot sealed samples after cooling by which time precipitates are deemed to significantly transform from gels to solids (crystalline) which having lower ionic mobilities (higher impedances) favors the detection of this time constant in the EIS spectra.
10. Perspective on Plausible Strategies for more Sustainable Sealing Procedures in Aluminum Anodizing
In spite of decades of research and search for alternative sealing methods for anodized aluminum alloys that does not involve the use of chromates, but meets the various requirements of the aerospace industry, no single replacement has been obtained [148,194,195,196]. These questions then arise: Why has it been difficult to find a chromium sealing alternative? What is unique about chromate sealing and its mechanism that makes it difficult to be replicated by other compounds in sealing anodized aluminum alloys? By a review of the relevant facts about chromate sealing, it is envisaged that these questions will be addressed herein and the important qualities expected from an ideal chromate alternative highlighted/delineated. Prior to delving into the intricacies of plausible strategies for development of more sustainable sealing procedures for anodized aluminum products, it will be important to take a closer look on the chromium sealing process which projected new methods will aspire to replace, in order to establish the desirable attributes of an ideal chromate alternative.
10.1. What Makes Chromium Sealing Unique and Accounts for Its Active Corrosion Inhibition Effects?
Limited knowledge on the mechanism of corrosion protection by chromium compounds is a significant constraint to development of non-chromate-based alternatives [197]. Active corrosion protection effect obtained by chromate sealing has been attributed [198,199,200] to the ability of chromates to form a hydrophobic barrier layer of chromium (III) oxyhydroxide, which contains residual hexavalent chromium (chromium in a higher oxidation state) that acts as a chromium reservoir [200,201], thus ensuring healing of discontinuities in the protective layer and improved corrosion resistance. Based on the Pourbaix diagram [202], Kulinich and Akhtara [196] had highlighted that whereas the solubility limit for aluminum oxides is pH ≈ 9, Cr(III) oxide (Cr2O3) uniquely has a much wider solubility limit at pH ≈ 15 compared to aluminum oxide and many other metal oxides such as CeO2 (pH ≈ 9.5), TiO2 (pH ≈ 12), SnO2 (pH ≈ 12.5), ZrO2 (pH ≈ 12.5), and Co2O3 (pH ≈ 13). Twite and Bierwagen [194] had attributed the use of chromates to their strong oxidizing powers, solubility in water, and the passive nature of their reduction products. Chromate sealing is reported [146,154] to start with adsorption of chromate anion onto the anodic oxide layer surface, which suggests that the ability of a candidate oxyanion surface to interact with the surface of the anodic oxide in the pores is vital. A recent report by Cui et al. [203] give interesting insights into the molecular level interaction of chromium/chromates (Cr(III)) with boehmite surface at very alkaline pH (pH ≈ 13). On the basis of their results they [203] concluded that Cr(III) adsorbed onto boehmite surface is surrounded by octahedral local structures, and coordinated to both oxy- and hydroxo- groups. This conclusion is consistent with observed significant incorporation of chromium into anodic oxides on aluminum sealed in chromate-containing sealing baths, in the light of reports [78,87] that hydrothermal sealing leads to increase in the proportion of octahedral sites in anodic oxides on aluminum. With regards to the progression of Cr(III) adsorption onto nanoboehmite surfaces, they [203] concluded that initial Cr(III) adsorption onto nanoboehmite under alkaline conditions occur by hydroxyl ligand exchange which leads to inner-sphere binding of Cr(OH)4−monomers, but at Cr(III) concentration in solution >20 ppm these monomers polymerize into clusters. Chromate ions are reported by several authors [204,205,206,207] to inhibit both hydration and deprotonation of anodic oxide films on aluminum.
Using an acoustic testing method based on the piezoelectric kinetic (PEK) effect [208,209], Kendig et al. [168] had reported that whereas reversible adsorption of positively charged Ce3+ cations onto the pore walls of anodized aluminum increases the zeta-potential within the pores of the anodic oxide layer, adsorption of anionic chromate species produces an opposite effect; decrease of the zeta-potential of the oxide in the pores of the porous anodized layer [209]. They [168] posited that a lowered zeta potential (due to adsorption of anionic chromate species) is quite likely to retard chloride transport into the pores anodic layer, and that Ce(III) cations adsorbs on some of the sites on which protons interact with the oxide surface. These observations and postulations are consistent with the arguments by Rocca et al. [78,87] with respect to mass transport into the pores of the anodic oxide layer and species selectivity on the basis of their charge(s).
10.2. Candidate Chromate Alternatives
From analysis of these reports, it is concluded that an ideal chromate alternative must be a compound/species that possesses multiple oxidation states, able to form a hydrophobic layer that is preferably less dense but harder than aluminum anodization products, contain relevant film forming element(s) in higher oxidation states, that the film forming specie must have sufficient solubility to be leached out of the film, sufficient mobility to present heterogeneities in the film, and on reduction form films that are stable over a very wide pH range. Analysis of the major sealing agent(s) in different sealing methods (Table 3) shows that the major sealing agent(s) (AlOHCrO4 and (AlO)2CrO4) in the industrial benchmark but environmentally unsustainable sealing procedure (dichromate sealing) are uniquely aluminum oxy and hydroxy compounds of the multivalent oxyanion (CrO4−). Since incorporation of the multivalent oxyanion (CrO4−) which serves as a sink for chromium and thus imparts active corrosion protection to chromate sealed anodized items, it might be possible to develop alternative and equally effective non-chromate sealing methods using environmentally benign oxyanions containing multivalent (transition) metals and able to form high specific volume insoluble compounds with aluminum oxides and hydroxides during sealing. Just like chromium with multiple oxidation states (common oxidation states: +2, +3, +6), manganese (common oxidation states: +2, +4, +7), tungsten (common oxidation states: +6 and +4), molybdenum (common oxidation states: +6 and +4), and vanadium (common oxidation states: +5, +4, +3, and +2) possess multiple oxidation states. Hence permanganate (MnO4−), tungstate (WO42−), molybdate (MoO42−), vanadate (VO43−) anions incorporated into the pores of the anodic oxide layers might act as reservoirs for release and transport of the corresponding multivalent metal species (Mn, W, Mo, and V) thus providing some active inhibition effects as with chromate sealing. Madden and Scully [210] had described in more detail how this might be possible with permanganate anion. This possibility is strengthened by the reported inhibitive effects of permanganates [210,211,212,213,214], tungstates [215,216,217], molybdates [67,152,211,212,213,214,215,216,217,218,219,220], and vanadates [221,222,223,224,225,226,227] on aluminum corrosion and enhanced corrosion resistance of the anodic films formed on aluminum alloys. These anions are known to form “alumino-oxy” compounds with aluminum like chromate anion such as; aluminum tungstate [228,229,230], aluminum molybdate [231,232,233,234], aluminum permanganate [235,236], and aluminum vanadate [237].
Due to the use of alumina as a catalyst support for molybdenum species in catalysis the mechanism of molybdate adsorption to alumina surface which is principally attributed to the influence of protonated surface hydroxyls has been well studied and reported [238,239,240,241,242]. Jones and Milne [243] had synthesized aluminum molybdate from mixtures of aluminum chloride and sodium molybdate by hydrothermal precipitation at temperatures ranging from 20 °C to 50 °C and reported that large amounts of molybdate were adsorbed onto aluminum minerals (boehmite and halloysite) exposed to solutions of sodium molybdate in the pH range 4.0–5.5 and at temperatures ranging from 20 °C to 90 °C.
Due to its varied interactions with copper in the presence and absence of sulphates [244,245,246,247,248], molybdenum/molybdates are employed in managing copper overdose and vice-versa [249,250], and thus might yield beneficial effects in sealing of copper-rich aluminum alloys. Its lower oxidizing power increases the chances of its compatibility with post-sealing applied organic coatings. Furthermore, on the basis of reports of the respective inhibitive effects of cerium [251,252,253,254,255,256,257], and molybdates [152,217,220,258,259,260,261,262,263,264] on corrosion of aluminum alloys, and active corrosion of aluminum by cerium molybdate [265,266,267], we postulate that simultaneous inclusion of both cerium and molybdate species in the sealing bath preferably as their respective sulphates or acetates or as cerium molybdate might be beneficial to the sealing of aluminum and impart some active corrosion protection to sealed materials. The preference for inclusion of these into the sealing baths as sulphates and/or acetate is premised on the known non-deleterious effects of these anions on the sealing process [37]. While cerium is believed to inhibit aluminum corrosion by formation of a protective oxide film [252,268,269], molybdates are postulated to inhibit by their adsorption and incorporation into the oxide film as hydrated aluminum molybdate [215,270]. Kulinich and Akhtara [196] had highlighted that although the pH ranges in which the reduced oxides of molybdenum, tungsten and vanadium are stable is significantly limited compared to chromium [202], their viability as chromium alternatives are not diminished, as they are known to form less soluble polyoxo species [200].
10.3. Important Factors to be Considered in Developing Sustainable and Efficient Sealing Alternatives
Beyond the need of an alternative to chromium sealing, current regulatory and environmental concerns necessitate the development of energy efficient and environmentally benign (i.e., sustainable) sealing procedures for anodic oxides on aluminum. Ideal sustainable sealing procedure(s) for anodized aluminum should employ environmentally friendly and inexpensive chemicals to ensure energy efficient low and mid temperature (preferably room temperature) alumina (Al2O3) hydration to form mono-hydrated alumina (AlOOH) with enhanced kinetics, so that the mono-hydrated alumina (AlOOH) having a volume greater than alumina (Al2O3) ensures fast filling and closure of the pores. Development of such procedure(s) will benefit immensely from a good knowledge of both the local environment (pH, ion composition, transport, and concentrations, etc.) inside the pores, and near the surface of the anodized layer, and its evolution at various stages of the sealing process. Currently, there appears to be a knowledge gap in this area.
10.3.1. pH inside the Pores and Its Effects
However, a recent publication by Etienne et al. [271] reports some information on the local pH evolution with time 10 to 20 µm above anodized aluminum surfaces in selected sealing solutions in which local pH measurements in the first 2 to 5 minutes manifested increase in pH by initial bulk pHs of 3.4 and 4.6 the measured pH maxima were ≈5.5 and >5.5 respectively but decreased to values tending towards the initial bulk values during 20 min. Runge [272] had highlighted the existence of a pH profile across the depth of anodic oxide layer with pH increasing with depth during anodization. The existence of a pH gradient inside the nano-confined space(s) of the nanopores of the anodic oxide layer during sealing is very likely to exert influences on the mechanism and/or kinetics of the sealing process.
Although information is scarce with respect to the pH profile inside the nano-confined space(s) of the pores and its evolution with the progress of the sealing process, some insight might be obtained by a review of reported pH values employed in the synthesis of boehmite and/or pseudo-boehmite. Lee et al. [161] studied the relationship between properties of pseudo-boehmite and its synthetic conditions and reported that pseudo-boehmite was formed in the pH range from 7.6 to 9.6 and that the pore size of the pseudo-boehmite increased with both increase in the ageing temperature, and increase in pH up to pH ≈ 9.6, while bayerite was formed at pH >10.6. Hence, we infer that irrespective of the bulk sealing solution pH, the pH of the sealing solution inside the pores most probably increases to the pH range ≈7.5 to ≥ 10 as sealing processes commence.
10.3.2. Effect of the Nano-Dimension of the Pores and the Surface Charge on the Pore Surfaces
Since a significant proportion of the sealing process take place in the nanopores of the anodic aluminum oxide layer of typical diameters in the range of 7–20 nm, the sealing process can be viewed as a process taking place in nanoconfined spaces (i.e., in a nanoporous electrode) [273,274,275,276], which are most likely to display peculiarities quite different from similar processes in/on micro- and macro-electrodes [277,278,279,280]. Taking the nano-dimensions of the pores of anodic aluminum oxide into consideration, Rocca et al. [78,87] had studied the possible effects of such dimensions on both mass transport into the pores and chemical reactivity of the pore walls and reported interesting results. On the basis of typical values of acidity constant for Al–OH groups (pKa1 = 6.5 for the Al–/Al–OH couple and pKa2 = 10.9 for the Al–OH/Al–O− couple), their electrokinetic (ζ potential) measurements which showed the PZC value of their unsealed anodic alumina samples to be around 10.5 [87], and the reported point of zero charge (PZC) of aluminum oxyhydroxide which is the range of pH 8.5–9 [281], Rocca et al. [87] had concluded that over a wide pH range (pH < PZC, i.e., pH < 10.5) which is within the pH range of most sealing solutions, the surface charge on anodic alumina oxide and inside the nanopores is positive. Hence it is established that the surface of the oxide layer inside the pores is positively charged under sealing conditions (Figure 7).
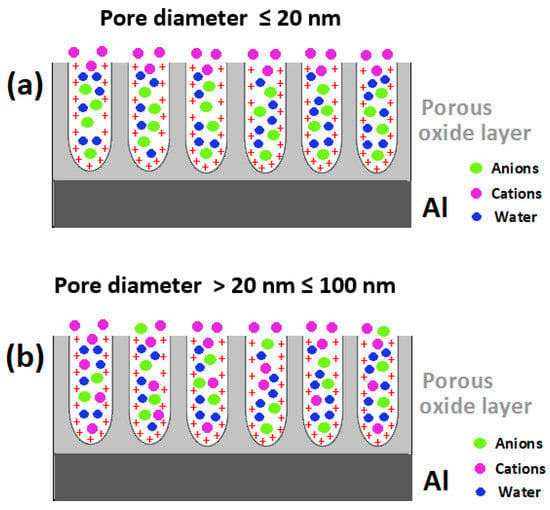
Figure 7.
Combined effect(s) of the positive surface charge and the nanometric dimensions of pores of anodic aluminum oxide films on the transport of species into the pores during sealing (The red “+” sign denotes the positive charge on the pore wall surface for simplicity the generated electric field is not presented).
With respect to the combined effect(s) of the positive surface charge and the nanometric dimensions of pores of anodic aluminum oxide films (10–20 nm diameter) on the transport of species into the pores during sealing (illustrated in Figure 7), they [87] had rightly highlighted the important roles of the combination of the nanometric dimensions of the nanopores in anodic alumina, the surface charge of the oxide layer inside the pores and its evolution as the sealing process progresses, on the transport and selectivity of species diffusing into the nanometric pores. They [87] mentioned that for very narrow pores (diameter < 20 nm) the electric field due to the charges on the pore surface can fill the entire volume of the nanopores, and demonstrated that whereas the transport of water and anions (CrO42− and/or Cr2O72−) were not hindered/affected, the transport of cations (Ce3+, Ni2+) more than 2 μm depth into the these nanometric diameter pores was apparently hindered [78,87]. These reports are supported by a recent publication by Cartigny et al. [29] that reported non-detection of nickel after a depth of 2 µm from the air-anodized layer interface in their nickel fluoride sealed anodized aluminum samples. In the light of these, it stands to reason that improvement of the kinetics of the sealing process and incorporation of inhibitive species might be achieved by manipulation of the surface charge density inside the nanoconfined space of the pores. Taking these factors into consideration, we postulate that incorporation of anionic inhibitive species such as manganate (VII) anion (MnO4−), molybdate anion (MoO42−), vanadate anion (VO43−), and tungstate anion (WO42−) into the pores of the anodic aluminum oxide film during the anodization step and/or during the sealing step might be feasible solution(s).
10.3.3. Improving Kinetics of Sealing Step at Lower Temperatures
Firstly, since bayerite (Al2O3·3H2O) is reported to be the stable aluminum hydroxide at room temperature [282,283,284,285,286], efforts at developing room and low temperature sealing procedures should be based on a good understanding of the mechanism of bayerite crystallization from alumina gel, under conditions consistent with those expected in the pores during the sealing step. This understanding is vital to developing less energy intensive procedures that promote the crystallization of boehmite (γ-AlO(OH)) at temperatures much lower than those employed in hydrothermal sealing (≥95 °C). Secondly, since pseudo-boehmite is another important intermediate product in the sealing process, a review of the abundant literature on pseudo-boehmite synthesis and crystallization [160,284,285,286,287,288,289,290,291,292,293,294,295,296,297,298,299,300,301,302,303,304,305] will be quite helpful in providing further insights on processes occurring during the sealing process and how to manipulate this process to achieve faster sealing kinetics at lower temperatures. Bayerite and pseudo-boehmite are known [287] to precipitate from alumina gels under certain conditions; a process that is exploited in the synthesis of pseudo-boehmite and boehmite for technological applications in catalysis [285,290,306,307,308,309,310]. Bye and Robinson [287] studied crystallization processes in aluminum hydroxide gels and reported that ethanol accelerated pseudo-boehmite formation but strongly inhibited bayerite formation, and attributed the strong differences in observed effects of ethanol on formation of the two phases to differences in the mechanism [311] of their respective formation, and concluded that while bayerite is formed by dissolution and recrystallization process, pseudo-boehmite formed by an inter- and intra-particle condensation that involved hydroxyl groups.
10.3.4. Effect Foreign Anions and Cations on the Crystallization of Aluminum Hydroxide
Since different substances have been known to exert influences on the kinetics of hydrothermal sealing of anodized aluminum [22,159,312] and on synthesis/crystallization of phases relevant to the sealing process [287,300,313,314], positive outcomes are plausible by identifying environmentally benign and inexpensive chemical agents that have accelerative effects on the kinetics of alumina gel transformation to hydrated alumina (AlOOH) and compatible with the chemistry of the sealing bath processes. In this regard, Bautista et al. [46,47,48] had reported that triethanolamine exerts catalytic effects on the sealing process thus altering the mechanism of alumina hydration, and concluded that for the organic molecules studied the concomitant presence of an alcohol group and an amine group in the same molecule tends to exert synergistic effects on the kinetics of the sealing process. In hot water sealing silicate and phosphate are known to be inhibitors of the sealing process while cupric, ferrous, fluoride, sulphate, and chloride ions are considered as harmful to the sealing process [22]. On the other hand, ammonium and acetate ions appear not to be harmful to hot water sealing of anodized aluminum as 1 g·L−1 ammonium acetate [159] is added to hot water sealing baths to combat rapid pH variations due to its buffering ability [22].
High concentrations of chromate ions are reported to inhibit the conversion of aluminum oxide to boehmite [22]. Silicate is reported to efficiently inhibit the conversion of aluminum oxide to boehmite [315]. Dabbs et al. [316] demonstrated that citric acid acts as a solubilizing agent that promotes aluminum oxyhydroxides solubility in aqueous solutions of high hydroxide-to-aluminum ratios. Phosphate ions are reported [164,317,318,319,320] to form stable oxide layers that inhibit hydration reactions in wet environments. Thus, the presence of phosphate ions in/on the unsealed anodic oxide layer or in the sealing bath electrolyte is likely to retard the kinetics of the sealing step by suppressing hydration processes [157,164,205,206,321,322,323]. This tendency has indeed been confirmed in various reports [37,324,325]. However, phosphates have been reported as components in multi-component sealing solutions [326]. Nitrate anion is reported to inhibit aluminum–water reaction [206] with the phenomenon attributed to general lowering of reaction kinetics associated with the surface oxide hydration in the presence of an oxidizing anion [270]. McCune et al. [270] had reported that the tendency of selected anions to inhibit film growth on aluminum followed the order; SiO32− > B4O72− > PO43− > MoO42− > WO42− > SO42− > NO3−. Moutarlier et al. [211] had proposed the inclusion of permanganate anions in the sulfuric acid anodizing bath on the strength of observed promotion of oxide film growth in presence of permanganate ions attributed to its oxidizing power and enhanced corrosion resistance of the anodic films formed in the presence of permanganate anion to values near to those obtained with anodic films from chromic acid anodization baths. Hence, the presence of permanganate anions in the sealing solution is likely to affect the mechanism and/or the kinetics of the sealing process.
10.3.5. Effect of anions having Enough Affinity for Al3+ to Displace Sulphate and other Electrolyte anions and promote Alumina Dissolution during Sealing
Working with alumina column and a pH 4 mobile phase Schmitt and Pietrzyk [327] had determined the order of anion retention on alumina to be: F− > SO42− > Cr2O72− > HCO2− > benzoate (C6H5COO−) > ClO2− > BrO3− > Cl− > NO2− > NO3− > Br− > ClO3− > SCN− > I− > ClO4− > C2H3OO− (acetate) which is in tandem with the reports of other workers [328,329,330,331,332,333]. This order is consistent with observations made by several authors with respect to sealing anodized aluminum; the non-deleterious influence of the acetate [31,159,327,334,335,336,337] and the efficacy of the fluoride ion in displacing sulphates and oxysulphates associated with alumina in the anodic oxide layer [327] which makes cold sealing with nickel fluoride feasible. The consistency of this ranking of anion retention on alumina makes it an important tool in the search for low temperature sealing solutions. According to the ranking of Schmitt and Pietrzyk [327], the only anion (F−) currently known to be capable of displacing sulphate incorporated in the alumina [26,327] in the anodic oxide layer and thus accelerate dissolution of some of the alumina in the pores necessary for sealing has already been employed in the Nickel fluoride sealing. Another set of questions arise; Can we get low temperature sealing solutions by making changes to the anions of the acids used in anodizing (i.e., using acids of anions with less affinity for alumina in anodizing) and then using salts of anions with higher affinities for alumina in the sealing? Can we search for and get other anions with affinities for alumina superior to that of fluoride ion so that salts of such an anion can be used for lower temperature sealing of anodized aluminum obtained from current anodizing bath chemistries?
Interestingly, Žutić and Stumm [338] in their studies on the influence of fluorides and organic acids on the dissolution kinetics of hydrous alumina had concluded that the affinity of the ligands studied for Al2O3 surface in aqueous media varied thus; formate (HCOO−) ≈ chloride (Cl−) ≈ carbonate (CO32−) < acetate (CH3COO−) < sulphate (SO42−) < salicylate (C7H5O3−) < fumarate (C4H2O42−) < maleate (C4H2O42−) < malonate (C3H2O42−) << oxalate (C2O42−) ≈ fluoride (F−) ≤ citrate (C6H5O73−). Pechenyuk et al. [339] compared data on sorption of anions (phosphate, carbonate, sulphate, chromate, oxalate, tartrate, and citrate) onto oxyhydroxides and ranked the adsorption affinity of the anions thus; PO43−, CO32− > C2O42−, C(OH)(CH2)2(COO)33−, (CHOH)2(COO)22− > CrO42− > SO42−. Missana et al. [340] analyzed effects of anion adsorption on stability of alumina nanoparticles and concluded that anion affinity to alumina surfaces and the consequent destabilization of colloidal alumina particles followed the order; SeO32− > SO42− > HCO3− > NO3− > ClO4−. Skeldon et al. [115] reported surface enrichment of anodic alumina films immersed in tungstate and molybdate solutions with tungsten and molybdenum atoms in concentrations (5–9 × 1014 atoms/cm2) which is suggestive of sub-monolayer presentation as they estimated monolayer concentration to be in the range of 2 × 1015 atom/cm2, but the effects of such concentrations of tungsten and molybdenum on anodic alumina surface on sealing was not reported.
10.3.6. Factors Relevant to Changes from Hot water sealing to Cold Sealing
In the search for efficient low temperature and preferably room temperature sealing solutions for anodized aluminum alloys it is important to highlight and appreciate plausible changes and scenarios in the mechanistic processes in the sealing bath. Whereas in the current high temperature sealing (temperatures ≥ 80 °C) the transformation of alumina to boehmite is favored [163], thermodynamic study by Deltombe and Pourbaix [341] suggest that on placing anodic coatings in water at lower temperatures (≤70 °C) the formation of hydrargillite (Al(OH)3) the most thermodynamically stable form of hydrated alumina will be favored instead of boehmite [12]. Since current hot water sealing technology is strongly reliant on the precipitation of boehmite into the pores of the anodized layer a lot of questions arise; can precipitation of hydrargillite in the pores at lower temperature yield good sealing results? Can processes be developed that ensure the precipitation of boehmite in the pores at significantly lower temperatures than (≥80 °C) at rates comparable to current practice (≈2 min/µm of anodized layer)?
The mechanism of current low temperature sealing processes like cold nickel acetate and cold nickel fluoride sealing processes are different from that of hot water sealing [342]. Future outlook for low temperature sealing based on nickel is bleak as the use of nickel comes under stringent regulations due to toxicity issues [343,344,345]. Table 5 presents solubility data of different aluminum compounds that are of relevance to the sealing process while Table 6 presents their respective specific volume data. These data are considered vital to successful manipulation of species precipitated inside the pores to obtain pore filling ratios better than that obtained in current practice.
Table 5.
Solubility data of aluminum compounds relevant to the sealing process.
Table 6.
Some relevant physical properties of aluminum oxides and (oxy)hydroxides.
10.3.7. One Pot Anodization and Sealing
A major potential benefit of low or intermediate temperature one-pot anodization and sealing strategy is probable reduced energy inputs. On the basis of the pH of the anodization/sealing bath, a one-pot strategy should be feasible as a survey of literature and patents indicate that sealing has been accomplished in sealing baths of pH ranging from acidic (as in anodizing baths) to alkaline. Development of a one-pot strategy for anodization and sealing of anodized aluminum will require information based and fundamental changes in the current anodizing bath chemistries, pH, cathode materials, and operating parameters such that the same bath chemistry supports both the formation of an anodic oxide layer and the sealing of the formed anodic oxide layer. A review of some relevant literature reports would be presented herein, in a bid to highlight plausible trajectories and considerations for development of a one-pot anodization and sealing procedure.
Recent reported modifications to the compositions of the sulfuric acid anodizing bath by the addition molybdates [219], tartaric acid in the tartaric-sulfuric acid (TSA) anodizing process [365], further addition of molybdenum/molybdates to the TSA bath in the MoTSA anodizing [366,367,368], and manganese oxyanion species [369] to the anodization bath are indicative of an emerging research towards achieving multi-functionalization in the anodization bath. Such a trend is obviously vital to development of a one-pot anodization/sealing strategies. Precipitation of rare-earths at alkaline pH (by NaOH) is reportedly inhibited completely in the presence of tartaric acid and tartarate solutions [370,371,372] which allows them to stay in solution at pH values higher than their respective precipitation pH. This suggests that introduction of tartrates or tartaric acid into the anodizing and/or sealing bath in the presence of rare-earth species is likely to exert significant effects on the mechanism and kinetics of the incorporation of the rare earths into the anodic oxide layer. Although tartaric acid currently features in some industrial scale anodization baths as reported in a patent on an adaptation of the sulfuric acid anodization process that incorporates tartaric acid [365] as the tartaric-sulfuric acid (TSA) anodization process, to the best of the authors’ knowledge tartaric acid has not been reported as a component of anodized aluminum sealing baths. In the light of the effects of tartaric acid and tartrates on the structure evolution of alumina [373] and as a peptizing agent [374,375,376], we posit the plausible emergence of tartaric acid and tartrates in future sealing bath compositions especially as research efforts move towards one-pot anodization and sealing strategies.
Purwani et al. had [377] reported oxidation of Ce3+ to Ce4+ by addition of (0.25–1.25 g) KMnO4 to (10 g of) RE hydroxide in 25 mL HNO3 at temperatures in the range of was 90–120 °C during reaction times that ranged from 15–75 min, and concluded the optimum processing parameters were 1.25 g: 10 g KMnO4 /REOH ratio, operating temperature of 120 °C and oxidation time of 75 min. Based on their results, they [377] also concluded that oxidation of Ce3+ to Ce4+ using KMnO4 was a first order reaction with the reaction rate constant of Ce ≈ 0.0291 min−1, and that the reaction is dependent on the ratio of KMnO4 /REOH, temperature and oxidation time. McNeice and Ghahreman [378] undertook a comparative study of selective oxidation abilities of four oxidants (hydrogen peroxide, sodium hypochlorite, potassium permanganate, and Caro’s acid (1:3 mixture of concentrated H2O2 and concentrated sulfuric acid (H2SO4) on cerium in rare-earth solutions and ranked their respective abilities to oxidize cerium thus; Caro’s acid > sodium hypochlorite > potassium permanganate > hydrogen peroxide. This ranking presents interesting implications on the possibilities of cerium incorporation into the anodic oxide layer in the presence of H2O2 and H2SO4. Itagaki et al. [379] achieved complete reduction of 4 ppm Mo(VI) in sulfuric acid solution to Mo(V) by reduction on glassy carbon electrode polarized to -400 mV (vs. saturated KCl/AgCl/Ag electrode) for 24 h and thus demonstrated that it is possible to manipulate the presentation of molybdenum species in solution. Such approach can be adapted to the anodization and/or sealing step to generate desirable chemical species in-situ and thus be very useful to the development of one-pot anodization/sealing systems. For the rare-earth, cerium; manipulation of its presentation in the anodization/sealing solution can be achieved via oxidation of trivalent cerium ion to tetravalent cerium ion by oxygen/air bubbling in alkaline solutions [380]. In acidic solutions (as in acidic anodizing solutions) conversion of introduced trivalent cerium ions to tetravalent cerium ions can be achieved either by the use of strong oxidants such as permanganate or persulphate, electrochemical oxidation and/or photochemical oxidation [381].
Yu et al. [382] introduced cerium nitrate and hydrogen peroxide (as an accelerant that supplied oxygen) into the sealing bath and reported that at commencement of the sealing the rare-earth laden solution was able to gain access into the pores of the anodized oxide layer and formed cerium oxide/hydroxides by precipitation, but as sealing progressed access of rare-earth solution into the pores is hindered by the formation of spherical deposits formed on the surface of the anodized oxide layer. According to their report [382] addition of H2O2 as an accelerator served as a source of O2 and simultaneously generated alkaline conditions that enhanced coating formation. Jiang et al. [383] had reviewed the incorporation of nanoparticles in coating systems for different metals and alloys, from which it can be easily inferred that the average sizes of most of the nanoparticles employed (mostly in the range of 15−300 nm) are not suitable for incorporation in typical anodized aluminum oxide layers with typical pore diameters in the range of 20 to 100 nm. One pot anodization and sealing strategy in which anodization and sealing with incorporation of corrosion inhibiting species is a strategy that can translate into much energy savings and thus demands investigation. However, a significant challenge to development of one pot anodization and sealing systems might be development of conditions/triggers that can initiate the sealing step instead of the anodic oxide dissolution and pore widening that occur when the anodizing power source is turned off and anodized samples are left immersed in anodization baths. A plausible solution to this challenge might be the introduction of an electrical impulse that is capable of exerting beneficial effects on the local environments inside the nanopores and thus enhance sealing kinetics during the sealing cycle of the one-pot anodization sealing process.
11. Minimizing the Energy Input of the Sealing Step in Aluminum Anodizing
Much of the energy consumption during the sealing step is associated with the heat input required to maintain the sealing bath at the operating temperature. In industrial practice, the volume of water in the sealing bath can be in the more than 5000 liters. As an illustration, assuming this volume of water (5000 liters) is at initial temperature of 20 °C but sealing bath operating temperature is 95 °C, ignoring heat losses due to conduction to the environment, heat transfer to immersed samples and evaporative losses, (and using specific heat capacity of water ≈ 4200 J/kg °C), at least 1575 MJ of energy will be needed to attain a bath operating temperature of 95 °C. However, if the sealing bath operating temperature is reduced to 50 °C, the energy input would be in the range of 630 MJ; resulting in about 945 MJ saving in energy consumption. Even a doubling of the sealing time at such lower bath operating (50 °C) compared to a sealing bath operating at 95 °C, is unlikely to totally offset such energy saving. Based on these assumptions, the energy input necessary to attain the different bath operating temperatures and the respective energy savings as the sealing bath operating temperature is reduced from 95 °C to 20 °C (ambient temperature) was calculated and presented in Figure 8.
Figure 8.
Energy considerations based on changing operating temperature of a 5000 liter sealing bath operating at 95 °C with an ambient temperature of 20 °C (specific heat capacity of water ≈ 4200 J/kg °C)).
Consequently, any efforts at improving the energy efficiency of the sealing process is best focused at either significant reduction of sealing bath operating temperatures or the sealing time, or both without compromising the quality of products. Application of insights highlighted earlier in Section 10.3.3, Section 10.3.4, Section 10.3.5, Section 10.3.6 and Section 10.3.7 is likely to result in both improvements in the kinetics of the sealing step and the lowering of sealing bath operating temperatures, and hence improvements in the energy efficiency of the sealing step. Based on these insights and the energy calculations (presented in Figure 8), we posit that reduction of the sealing bath operating temperature from ≥95 °C to ≤60 °C could be possible without a doubling of the sealing time.
12. Toxicity Concerns with Respect to Plausible Strategies for More Sustainable Sealing Procedures in Aluminum Anodizing
The current atmosphere of increased environmental impact awareness and accountability behooves scientists to factor in plausible environmental implications of new processes during the conceptualization and development stages. In this regard, it is important to highlight reports about vanadium toxicity which links both vanadate (VO3−, V5+) and vanadyl (VO2+, V4+) as reproductive and developmental toxicants in mammals [384,385,386,387], and neurobehavioral toxicity in workers occupationally exposed to vanadium [388]. Inhalation exposure to vanadium pentoxide has been linked to DNA damage [389] and vanadium pentoxide is reported to be a carcinogen [390,391,392,393,394]. These reports of vanadium toxicity diminish the viability of vanadate incorporating sealing solutions, as it is very likely to come under regulatory control. High concentrations (>15 μg/L) of vanadium in drinking water is already considered a potential health risk [395,396].
The corrosion inhibitive effects of tungstates have been much reported. However, most of these reports have been on its inhibitive effects on iron, carbon steels, and stainless steels (ferrous metals and alloys) [215,397,398,399,400,401,402,403,404], with only few reports on tunsgtate inhibition of aluminum corrosion [405,406,407,408]. Though the toxicity of tungsten/tungstate have hitherto been considered to be low [409,410,411,412], recent reports have brought it into scrutiny as an emergent toxicant [410,411,412,413,414,415,416,417,418,419,420,421,422,423,424,425,426,427,428,429,430,431,432], and thus the likelihood of tungsten/tungstate use coming under strict regulatory control in the near future is significant.
With respect to (potassium) permanganate, it is a strong oxidizing agent used as an oxidant in in the pre-oxidation phase of a drinking-water treatment/production [433,434,435,436,437,438,439,440], as an algicide [441,442,443,444], and as a bactericide in treatment of bacterial fish diseases in fish culture ponds [445,446,447,448,449,450,451]. Deichmann and Gerarde [452] reported that potassium permanganate has a probable lethal adult dose of about 10g. A recent EU substance evaluation document for Potassium permanganate [453] reported oral acute toxicity (LD50) value of 1090 to > 2000 mg/kg of body weight and dermal acute toxicity (LD50) value > 2000 mg/kg of body weight for potassium permanganate. Inadvertent poisoning from potassium permanganate is considered a rare occurrence [454,455,456,457,458]. In spite of its low toxicity, its very high oxidation power enables it to oxidize organic compounds [459,460,461] and eliminate taste and odor in water treatment by oxidizing both inorganic and organic materials that are responsible for taste and odor [462,463,464,465,466,467,468,469,470,471] might make permanganate unsuitable for sealing anodized aluminum products that need to be coated with organic primers and/or coatings after sealing.
Molybdates are generally classified as a low toxicity substance to humans [259,472,473,474,475,476,477,478] with its tolerable upper intake level (UL) of set at 2 mg/day for humans [479,480] and its content in drinking water limited to 70 ug/L [481,482,483,484]. Although molybdenum might be considered to be less toxic to humans, it is reported to be toxic to certain marine organisms [485,486,487,488,489] with its toxicity related to the solubility of the molybdenum compounds. Soluble molybdenum (VI) compounds (e.g., sodium molybdate dihydrate) are reported to be more toxic than insoluble ones [472,487]. Wang et al. [487] studied the effect of three common hexavalent molybdenum compounds (sodium molybdate (Na2MoO4·2H2O), ammonium molybdate ((NH4)6Mo7O24·4H2O) and molybdenum trioxide (MoO3) on an aquatic system and reported that the acute toxicities in solution follow the order: (NH4)6Mo7O24·4H2O > MoO3 > Na2MoO4·2H2O, and that the toxicity of molybdenum in the aquatic system is markedly dependent on the form of molybdenum salts used, and to some extent on the quality of the background water used.
Since toxicity data for humans for these plausible candidate alternative substances to chromates are not all available, some of the published data for these on a range of animals are presented in Table 7. To put these figures in perspective, published toxicity data for chromates are also included in Table 7. Auspiciously, a lot of comparative toxicity studies involving compounds of interest to the present work (chromate, molybdate, vanadate, and tungstate) had been carried out on a wide variety of animals [490,491,492,493,494,495]. On the basis of the results from their studies on the toxicity of these compounds on mice and rats, Pham and Cha [490] had concluded that sodium molybdate was the least toxic being 6 times less toxic than sodium chromate, 3 to 4 times less toxic than sodium-tungstate, while sodium metavanadate and sodium chromate were equally toxic to rats and mice, and more toxic than the molybdate (Table 7). Making a comparison of the toxicity of molybdates and chromates to fish using fish toxicity data presented by Armour and Robitaille [259] which showed no-observed-adverse-effect level (NOAEL) for tested fish in the range of 2400 to 7500 mg/L compared to 50 mg/L for chromates, it can be inferred that for these species molybdate is 48 to 150 times less toxic than chromates. In the light of available toxicity data, it is concluded that molybdates are highly favored for use in development of sealing systems and processes to replace chromates.
Table 7.
Toxicity data for chemistries of plausible sealing solutions.
13. Conclusions
Most current industrial technology solutions for sealing anodized aluminum are in need of improvements in their energy efficiencies and the environmental impacts. Hence there is a need to develop systems and processes that address these important issues from the process design stage. Such efforts must be based on in-depth consideration of many factors and at different size and process steps. The present work is an effort at an in-depth review that highlights some of the important factors relevant to development of sustainable anodized aluminum sealing solutions. Based on the review of the literature, available toxicity data, current regulatory position(s), and projected future regulatory trajectories, the development of energy efficient sealing solutions based on the use of molybdates, permanganates, and rare-earth metal compounds are deemed to be the most feasible trajectories. We posit that development of sustainable and energy efficient anodization and sealing strategies for aluminum alloys might have to involve significant and fundamental changes in the current anodizing bath chemical compositions, pH, cathode materials, and operating temperatures, which could permit in-situ and tailored incorporation of species that improve desired qualities of the anodic oxide layer such as corrosion resistance and abrasion resistance. Such significant changes in anodizing bath chemistry can ultimately lead to development of intermediate temperature (40–70 °C) and energy efficient, one-pot anodization and sealing systems. On the basis of both its much lower toxicity compared with chromates and the other oxyanions, and its (molybdate) severally reported ability to interact with copper, we postulate that the exploitation of these abilities by the use of molybdate incorporating sealing bath composition at temperatures ≤ 60 °C might be an environmentally benign and effective solution for sealing the problematic high copper content aluminum alloys that are much used in the aerospace industry.
Author Contributions
Conceptualization, Methodology, and Writing—Original Draft Preparation, S.U.O.; Writing—Review and Editing, S.U.O., F.A.O.F., and A.B.P; Supervision and Funding Acquisition, A.B.P.; Project Administration, F.A.O.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the projects: POCI-OT1-PI 1.2-TI 47-FEDER-033912—Competitiveness and Internationalization Operational Program, under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund; UIDB/00481/2020 and UIDP/00481/2020—FCT—Fundação para a Ciencia e a Tecnologia; and CENTRO-01-0145-FEDER-022083—Centro Portugal Regional Operational Programme (Centro2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund. The APC was funded by project POCI-OT1-PI 1.2-TI 47-FEDER-033912—Competitiveness and Internationalization Operational Program, under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund.
Acknowledgments
S.U.O. acknowledges post-doctoral grant (BPD/UI66/5308/2019) from the project: ANOD-IDT Copromotion (POCI-OT1-PI 1.2-TI 47-FEDER-033912). F.A.O.F. acknowledges Portuguese Science Foundation (FCT) for junior researcher grant (CEECIND/01192/2017).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grubbs, C.A. Anodizing of aluminum. Met. Finish. 1999, 97, 476–493. [Google Scholar] [CrossRef]
- Wernick, S.; Pinner, R.; Sheasby, P. The Surface Treatments of Aluminium and Its Alloys; ASM International and Finishing Publications Ltd.: Metals Park, OH, USA, 1987; p. 773. [Google Scholar]
- Brace, A.W. The Technology of Anodizing Aluminium; Technicopy Ltd.: Stonehouse, UK, 1979. [Google Scholar]
- Diggle, J.W.; Downie, T.C.; Goulding, C.W. Anodic oxide films on aluminum. Chem. Rev. 1969, 69, 365–405. [Google Scholar] [CrossRef]
- Kalantary, M.R.; Gabe, D.R.; Ross, D.H. Sealing of electrolytically formed porous films of aluminum by nickel fluoride process. Plat. Surf. Finish. 1993, 80, 52–56. [Google Scholar]
- Safrany, J.S. Anodisation de l’aluminium et de ses alliages. Techniques de l’ingénieur. Corrosion vieillissement 2008, COR2, M1630-1. [Google Scholar]
- Cieślak-Golonka, M. Toxic and mutagenic effects of chromium (VI). A review. Polyhedron 1996, 15, 3667–3689. [Google Scholar] [CrossRef]
- USEPA. Toxicological Review of Hexavalent Chromium; U.S. Environmental Protection Agency: Washington, DC, USA, 1998. Available online: https://cfpub.epa.gov/ncea/iris/iris_documents/documents/toxreviews/0144tr.pdf (accessed on 4 October 2019).
- Von Burg, R.; Liu, D. Chromium and hexavalent chromium. J. Appl. Toxicol. 1993, 13, 225–230. [Google Scholar] [CrossRef]
- Commission Regulation (EU). No 348/2013 of 17 April 2013. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32013R0348qid=%201487685993229from=EN (accessed on 7 October 2019).
- Berry, C.A. Guide to Hexavalent Chromium Cr (VI) for Industry. (Industry Guide 45); Occupational Safety and Health Division, N.C. Department of Labor: Raleigh, NC, USA, 2003. Available online: https://files.nc.gov/ncdol/oshApublications/ig45.pdf (accessed on 7 October 2019).
- EPA Chromium Compounds, Hazard Summary. 2000. Available online: https://www.epa.gov/sites/production/files/2016-09/documents/chromium-compounds.pdf (accessed on 7 October 2019).
- Are You Ready to Phase out Chromium 6? A Guide to Continue Production without Chromium 6. Version: 2017-03-06. Aquarden Technologies ApS White Paper. Available online: https://aquarden.com/wp-content/uploads/2017/03/White-paper-Chromium-6.pdf (accessed on 8 October 2019).
- Reisch, M.S. Confronting the looming hexavalent chromium ban. Chem. Eng. News 2017, 95, 28–29. [Google Scholar]
- Mason, R.B.; Clark, S.; Klingenberg, M.; Berman, E.; Voevodin, N. Alternatives to dichromate sealer in anodizing operations. Metal Finish. 2011, 109, 25–32. [Google Scholar] [CrossRef]
- Gharbi, O.; Thomas, S.; Smith, C.; Birbilis, N. Chromate replacement: What does the future hold? NPJ Mater. Degrad. 2018, 2, 1–8. [Google Scholar] [CrossRef]
- Koerner, T.; Kresse, J.; Lindener, J.; Roland, W.A. Henkel AG and Co KGaA, Method of Compacting Anodized Metals with Lithium and Fluoride-Containing Solutions without Using Heavy Metals. U.S. Patent 5,891,269, 6 April 1999. [Google Scholar]
- Papadopoulos, A.; Fatta, D.; Parperis, K.; Mentzis, A.; Haralambous, K.J.; Loizidou, M. Nickel uptake from a wastewater stream produced in a metal finishing industry by combination of ion-exchange and precipitation methods. Sep. Purif. Technol. 2004, 39, 181–188. [Google Scholar] [CrossRef]
- Wei, H.; Chen, D.; Hu, H.; Chang, M.; Ye, X.; Wang, M. Insights into energy-efficient and eco-friendly sealing of anodic aluminum oxide film holes with alkaline earth metal salts. RSC Adv. 2017, 7, 55653–55667. [Google Scholar] [CrossRef]
- Takaichi, H.; Moriguchi, T.; Horikawa, M.; Hongo, A.; Okuno Chemical Ind Co. Sealing Liquid for Anodic Oxide Coating Films of Aluminum Alloy, Concentrated Liquid and Sealing Method. U.S. Patent Application 16/088,595, 18 April 2019. [Google Scholar]
- Zuo, Y.; Zhao, P.H.; Zhao, J.M. The influences of sealing methods on corrosion behavior of anodized aluminum alloys in NaCl solutions. Surf. Coat. Technol. 2003, 166, 237–242. [Google Scholar] [CrossRef]
- Hao, L.; Cheng, B.R. Sealing processes of anodic coatings—Past, present, and future. Met. Finish. 2000, 98, 8–18. [Google Scholar] [CrossRef]
- Wood, G.C. Sealing Anodic Oxide Films on Aluminium. Trans. Inst. Met. Finish. 1959, 36, 220–229. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Jang, H.; Chung, W. Cr2O3 sealing of anodized aluminum alloy by heat treatment. Surf. Coat. Technol. 2014, 243, 34–38. [Google Scholar] [CrossRef]
- Carangelo, A.; Curioni, M.; Acquesta, A.; Monetta, T.; Bellucci, F. Application of EIS to in situ characterization of hydrothermal sealing of anodized aluminum alloys: Comparison between hexavalent chromium-based sealing, hot water sealing and cerium-based sealing. J. Electrochem. Soc. 2016, 163, C619–C626. [Google Scholar] [CrossRef]
- Kalantary, M.R.; Gabe, D.R.; Ross, D.H. A model for the mechanism of nickel fluoride cold sealing of anodized aluminium. J. Appl. Electrochem. 1992, 22, 268–276. [Google Scholar] [CrossRef]
- Tian, L.P.; Zhao, X.H.; Zhao, J.M.; Zhang, X.F.; Yu, Z.U.O. Electrochemical behaviors of anodic alumina sealed by Ce-Mo in NaCl solutions. Trans. Nonferrous Met. Soc. China 2006, 16, 1178–1183. [Google Scholar] [CrossRef]
- Hu, N.; Dong, X.; He, X.; Browning, J.F.; Schaefer, D.W. Effect of sealing on the morphology of anodized aluminum oxide. Corros. Sci. 2015, 97, 17–24. [Google Scholar] [CrossRef]
- Cartigny, V.; Veys-Renaux, D.; Desenne, P.; Rocca, E. Rapid sealing of an alumina nanoporous network grown by anodizing and dye-filled. Surf. Coat. Technol. 2019, 364, 369–376. [Google Scholar] [CrossRef]
- Herrera-Hernandez, H.; Vargas-Garcia, J.R.; Hallen-Lopez, J.M.; Mansfeld, F. Evaluation of different sealing methods for anodized aluminum-silicon carbide (Al/SiC) composites using EIS and SEM techniques. Mater. Corros. 2007, 58, 825–832. [Google Scholar] [CrossRef]
- González, J.A.; López, V.; Otero, E.; Bautista, A.; Lizarbe, R.; Barba, C.; Baldonedo, J.L. Overaging of sealed and unsealed aluminium oxide films. Corros. Sci. 1997, 39, 1109–1118. [Google Scholar] [CrossRef]
- Zhao, J.M.; Chen, S.L.; Huo, P. Comparative studies on corrosion behavior of sealed aluminium with cerium salt under bidirectional pulse electric field. Corros. Eng. Sci. Technol. 2012, 47, 203–208. [Google Scholar] [CrossRef]
- Zhao, J.M.; Liu, H.X.; Chen, S.L.; Zhao, X.H. EIS evolution of anodised aluminium by cerium salt sealing in NaCl solution with exposure time. Corros. Eng. Sci. Technol. 2013, 48, 44–47. [Google Scholar] [CrossRef]
- Carangelo, A.; Curioni, M.; Acquesta, A.; Monetta, T.; Bellucci, F. Cerium-based sealing of anodic films on AA2024T3: Effect of pore morphology on anticorrosion performance. J. Electrochem. Soc. 2016, 163, C907–C916. [Google Scholar] [CrossRef]
- Andreeva, R.A.; Stoyanova, E.A.; Tsanev, A.S.; Stoychev, D.S. Corrosion behavior of anodically formed oxide films on aluminum, sealed in cerium-ions containing solutions. Bulg. Chem. Commun. 2016, 48, 96–102. [Google Scholar]
- Cheng, B.R.; Hao, L. Comparative study of the effects of sealing processes on the wear resistance and the sealing quality of hard anodic coatings. Met. Finish. 2000, 98, 48–55. [Google Scholar] [CrossRef]
- Mansfeld, F.; Chen, C.; Breslin, C.B.; Dull, D. Sealing of anodized aluminum alloys with rare earth metal salt solutions. J. Electrochem. Soc. 1998, 145, 2792–2798. [Google Scholar] [CrossRef]
- Whelan, M.; Cassidy, J.; Duffy, B. Sol–gel sealing characteristics for corrosion resistance of anodised aluminium. Surf. Coat. Technol. 2013, 235, 86–96. [Google Scholar] [CrossRef]
- Costenaro, H.; Lanzutti, A.; Fedrizzi, L.; Terada, M.; de Melo, H.G.; Olivier, M.G. Corrosion resistance of 2524 Al alloy anodized in tartaric-sulphuric acid at different voltages and protected with a TEOS-GPTMS hybrid sol-gel coating. Surf. Coat. Technol. 2017, 324, 438–450. [Google Scholar] [CrossRef]
- Zemanova, M.; Chovancova, M. Sol-gel method for sealing anodized aluminum. Met. Finish. 2003, 101, 14–16. [Google Scholar] [CrossRef]
- Xue, B.; Yu, M.; Liu, J.; Liu, J.; Li, S.; Xiong, L. Corrosion protection of AA2024-T3 by sol-gel film modified with graphene oxide. J. Alloys Compd. 2017, 725, 84–95. [Google Scholar] [CrossRef]
- Yu, M.; Dong, H.; Shi, H.; Xiong, L.; He, C.; Liu, J.; Li, S. Effects of graphene oxide-filled sol-gel sealing on the corrosion resistance and paint adhesion of anodized aluminum. Appl. Surf. Sci. 2019, 479, 105–113. [Google Scholar] [CrossRef]
- Kim, M.; Yoo, H.; Choi, J. Non-nickel-based sealing of anodic porous aluminum oxide in NaAlO2. Surf. Coat. Technol. 2017, 310, 106–112. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, J.; Liu, S.; Wang, H.; Dong, P. Effect of NaAlO2 sealing on corrosion resistance of 2024 aluminum alloy anodized film. Mater. Corros. 2019, 70, 120–127. [Google Scholar] [CrossRef]
- Ono, S.; Okura, M.; Asoh, H.; Tanaka, H.; Yamamoto, T. Sealing Mechanism of Anodic Porous Oxide Films Formed on Aluminum in Lithium Hydroxide Solution. In Proceedings of the 12th International Conference on Aluminium Alloys, Yokohama, Japan, 5–9 September 2010; pp. 1463–1468. [Google Scholar]
- Bautista, A.; González, J.A.; Lopez, V. Influence of triethanolamine additions on the sealing mechanism of anodised aluminium. Surf. Coat. Technol. 2002, 154, 49–54. [Google Scholar] [CrossRef]
- Gonzalez, J.A.; Otero, E.; Bautista, A.; Lopez, V. Effect of triethanolamine addition on sealing time. Plat. Surf. Finish. 1997, 84, 59–63. [Google Scholar]
- Bautista, A.; González, J.A.; López, V. Sealing Mechanism of Anodized Aluminum in Triethanolamine Solutions. In Corrosion and Corrosion Prevention of Low-Density Metals and Alloys: Proceedings of the International Symposium; The Electrochemical Society: Pennington, NJ, USA, 2001; Volume 2000, pp. 146–155. [Google Scholar]
- Mata, D.; Serdechnova, M.; Mohedano, M.; Mendis, C.L.; Lamaka, S.V.; Tedim, J.; Hack, T.; Nixon, S.; Zheludkevich, M.L. Hierarchically organized Li–Al-LDH nano-flakes: A low-temperature approach to seal porous anodic oxide on aluminum alloys. RSC Adv. 2017, 7, 35357–35367. [Google Scholar] [CrossRef]
- Kuznetsov, B.; Serdechnova, M.; Tedim, J.; Starykevich, M.; Kallip, S.; Oliveira, M.P.; Hack, T.; Nixon, S.; Ferreira, M.G.S.; Zheludkevich, M.L. Sealing of tartaric sulfuric (TSA) anodized AA2024 with nanostructured LDH layers. RSC Adv. 2016, 6, 13942–13952. [Google Scholar] [CrossRef]
- Yang, J.; Yang, Y.; Balaskas, A.; Curioni, M. Development of a Chromium-Free Post-Anodizing Treatment Based on 2-Mercaptobenzothiazole for Corrosion Protection of AA2024T3. J. Electrochem. Soc. 2017, 164, C376–C382. [Google Scholar] [CrossRef]
- Wang, S.; Peng, H.; Shao, Z.; Zhao, Q.; Du, N. Sealing of anodized aluminum with phytic acid solution. Surf. Coat. Technol. 2016, 286, 155–164. [Google Scholar] [CrossRef]
- Shang, Y.; Wang, L.; Liu, Z.; Niu, D.; Wang, Y.; Liu, C. The Effects of Different Sealing Techniques for Anodic Film of Al-12.7 Si 0.7 Mg Alloys. Int. J. Electrochem. Sci. 2016, 11, 5234–5244. [Google Scholar] [CrossRef]
- Pearlstein, F.; Agarwala, V.S. Trivalent chromium solutions for applying chemical conversion coatings to aluminum alloys or for sealing anodized aluminum. Plat. Surf. Finish. 1994, 81, 50–55. [Google Scholar]
- Pearlstein, F.; Agarwala, V.S.; US Secretary of Navy. Trivalent Chromium Solutions for Sealing Anodized Aluminum. U.S. Patent 5,374,347, 20 December 1994. [Google Scholar]
- Hoar, T.P.; Wood, G.C. The sealing of porous anodic oxide films on aluminium. Electrochim. Acta 1962, 7, 333–353. [Google Scholar] [CrossRef]
- Wood, G.C.; Marron, V.J.J. Studies of the Sealing of Anodized Aluminium using A C Impedance Techniques and Electron Probe Microanalysis: Part I—Sealing in Typical Salt Solutions. Trans. Inst. Met. Finish. 1967, 45, 17–26. [Google Scholar] [CrossRef]
- Murphy, O.J.; Wainright, J.S.; Lenczewski, J.J.; Gibson, J.H.; Santana, M.W. Spectroscopic Investigations of Porous and Sealed Anodic Alumina Films. J. Electrochem. Soc. 1989, 136, 3518–3525. [Google Scholar] [CrossRef]
- Booker, C.J.L.; Wood, J.L.; Walsh, A. Electron micrographs from thick oxide layers on aluminium. Nature 1955, 176, 222–223. [Google Scholar] [CrossRef]
- Booker, C.J.L.; Wood, J.L.; Walsh, A. Electron micrographs from thick oxide layers on aluminium. Br. J. Appl. Phys. 1957, 8, 347. [Google Scholar] [CrossRef]
- Brace, A.W. The Technology of Anodized Aluminum; Robert Droper: Teddington, UK, 1968; pp. 1–11. [Google Scholar]
- Keller, F.; Hunter, M.S.; Robinson, D.L. Structural Features of Oxide Coatings on Aluminum. J. Electrochem. Soc. 1953, 100, 411. [Google Scholar] [CrossRef]
- Hunter, M.S.; Fowle, P. Factors Affecting the Formation of Anodic Oxide Coatings. J. Electrochem. Soc. 1954, 101, 514–519. [Google Scholar] [CrossRef]
- Edwards, J.D.; Keller, F. The structure of anodic oxide coatings. Trans. Am. Inst. Min. Metall. Pet. Eng. 1944, 156, 288–299. [Google Scholar]
- Feliu, S.; Bartolomé, M.J.; González, J.A. XPS characterization of porous and sealed anodic films on aluminum alloys. J. Electrochem. Soc. 2007, 154, C241–C248. [Google Scholar] [CrossRef]
- Feliu, S.; González, J.A.; López, V.; Bartolome, M.J.; Escudero, E.; Otero, E. Characterisation of porous and barrier layers of anodic oxides on different aluminium alloys. J. Appl. Electrochem. 2007, 37, 1027–1037. [Google Scholar] [CrossRef][Green Version]
- Moutarlier, V.; Gigandet, M.P.; Pagetti, J.; Linget, S. Influence of molybdate species added to sulphuric acid on composition and morphology of the anodic layers formed on 2024 aluminium alloy. Thin Solid Films 2005, 483, 197–204. [Google Scholar] [CrossRef]
- Le Coz, F.; Arurault, L.; Fontorbes, S.; Vilar, V.; Datas, L.; Winterton, P. Chemical composition and structural changes of porous templates obtained by anodising aluminium in phosphoric acid electrolyte. Surf. Interface Anal. 2010, 42, 227–233. [Google Scholar] [CrossRef]
- Thompson, G.E. Porous anodic alumina: Fabrication, characterization and applications. Thin Solid Films 1997, 297, 192–201. [Google Scholar] [CrossRef]
- Xu, Y.; Thompson, G.E.; Wood, G.C.; Bethune, B. Anion incorporation and migration during barrier film formation on aluminium. Corros. Sci. 1987, 27, 83–102. [Google Scholar] [CrossRef]
- Han, S. Anodic Film Formation on Aluminium (II) Anodic Film Composition. J. Met. Finish. Soc. Korea 1988, 21, 183–192. [Google Scholar]
- Takahashi, H.; Nagayama, M. Distribution of Phosphate Ions in the Porous Anodic Oxide Film Formed on Aluminium in Phosphoric Acid Solution. Nippon Kagaku Kaishi 1974, 1974, 453–458. [Google Scholar] [CrossRef]
- Zhou, X.; Thompson, G.E.; Skeldon, P.; Wood, G.C.; Shimizu, K.; Habazaki, H. Film formation and detachment during anodizing of Al–Mg alloys. Corros. Sci. 1999, 41, 1599–1613. [Google Scholar] [CrossRef]
- Bocchetta, P.; Sunseri, C.; Chiavarotti, G.; Di Quarto, F. Microporous alumina membranes electrochemically grown. Electrochim. Acta 2003, 48, 3175–3183. [Google Scholar] [CrossRef]
- Parkhutik, V.P.; Makushak, Y.E.; Kudryavtsev, V.I.; Sokol, V. X-Ray Electronic Study of the Formation of Anodic Oxide Films on Aluminum in Nitric Acid. (Translation). Sov. Electrochem. 1987, 23, 1439–1444. [Google Scholar]
- Alexander, M.R.; Beamson, G.; Bailey, P.; Noakes, T.C.Q.; Skeldon, P.; Thompson, G.E. The distribution of hydroxyl ions at the surface of anodic alumina. Surf. Interface Anal. 2003, 35, 649–657. [Google Scholar] [CrossRef]
- El-Mashri, S.M.; Jones, R.G.; Forty, A.J. An electron-yield EXAFS study of anodic-oxide and hydrated-oxide films on pure aluminium. Philos. Mag. A 1983, 48, 665–683. [Google Scholar] [CrossRef]
- Rocca, E.; Vantelon, D.; Reguer, S.; Mirambet, F. Structural evolution in nanoporous anodic aluminium oxide. Mater. Chem. Phys. 2012, 134, 905–911. [Google Scholar] [CrossRef]
- Oka, Y.; Takahashi, T.; Okada, K.; Iwai, S.I. Structural analysis of anodic alumina films. J. Non-Cryst. Solids 1979, 30, 349–357. [Google Scholar] [CrossRef]
- Farnan, I.; Dupree, R.; Forty, A.J.; Jeong, Y.S.; Thompson, G.E.; Wood, G.C. Structural information about amorphous anodic alumina from 27Al MAS NMR. Philos. Mag. Lett. 1989, 59, 189–195. [Google Scholar] [CrossRef]
- Farnan, I.; Dupree, R.; Jeong, Y.; Thompson, G.E.; Wood, G.C.; Forty, A.J. Structural chemistry of anodic alumina. Thin Solid Films 1989, 173, 209–215. [Google Scholar] [CrossRef]
- Hashimoto, H.; Yazawa, K.; Asoh, H.; Ono, S. NMR Spectroscopic Analysis of the Local Structure of Porous-Type Amorphous Alumina Prepared by Anodization. J. Phys. Chem. C 2017, 121, 12300–12307. [Google Scholar] [CrossRef]
- Stumm, W.; Furrer, G.; Kunz, B. The role of surface coordination in precipitation and dissolution of mineral phases. Croat. Chem. Acta 1983, 56, 593–611. [Google Scholar]
- Stumm, W.; Wollast, R. Coordination chemistry of weathering: Kinetics of the surface-controlled dissolution of oxide minerals. Rev. Geophys. 1990, 28, 53–69. [Google Scholar] [CrossRef]
- Furrer, G.; Stumm, W. The coordination chemistry of weathering: I. Dissolution kinetics of δ-Al2O3 and BeO. Geochim. Cosmochim. Acta 1986, 50, 1847–1860. [Google Scholar] [CrossRef]
- Dove, P.M.; Han, N.; Wallace, A.F.; De Yoreo, J.J. Kinetics of amorphous silica dissolution and the paradox of the silica polymorphs. Proc. Natl. Acad. Sci. USA 2008, 105, 9903–9908. [Google Scholar] [CrossRef] [PubMed]
- Rocca, E.; Vantelon, D.; Gehin, A.; Augros, M.; Viola, A. Chemical reactivity of self-organized alumina nanopores in aqueous medium. Acta Mater. 2011, 59, 962–970. [Google Scholar] [CrossRef]
- Baker, B.R.; Pearson, R.M. Sealing Studies of Anodic Oxides by Wide-Line NMR Spectroscopy. J. Electrochem. Soc. 1972, 119, 160–165. [Google Scholar] [CrossRef]
- Murphy, J.F. Chemical and electrochemical factors in the mechanism of formation and poroperties of anodic coatings. In Proceedings of the International Symposium on Anodizing. (Aluminium Federation, UK), Birmingham, UK, April 1967; p. 3. [Google Scholar]
- Shimizu, K.; Brown, G.M.; Habazaki, H.; Kobayashi, K.; Skeldon, P.; Thompson, G.E.; Wood, G.C. Impurity distributions in barrier anodic films on aluminium: A GDOES depth profiling study. Electrochim. Acta 1999, 44, 2297–2306. [Google Scholar] [CrossRef]
- Morlidge, J.R.; Skeldon, P.; Thompson, G.E.; Habazaki, H.; Shimizu, K.; Wood, G.C. Gel formation and the efficiency of anodic film growth on aluminium. Electrochim. Acta 1999, 44, 2423–2435. [Google Scholar] [CrossRef]
- Xu, Y. The Growth Mechanisms of Anodic Films on Aluminium. Ph.D. Thesis, University of Manchester Institute of Science and Technology (UMIST), Manchester, UK, 1983. [Google Scholar]
- Wernick, S.; Pinner, R.; Sheasby, P. The Surface Treatments of Aluminium and Its Alloys; ASM International and Finishing Publications Ltd.: Metals Park, OH, USA, 1987; pp. 292–298, 407–430. [Google Scholar]
- Lee, W.; Schwirn, K.; Steinhart, M.; Pippel, E.; Scholz, R.; Gösele, U. Structural engineering of nanoporous anodic aluminium oxide by pulse anodization of aluminium. Nat. Nanotechnol. 2008, 3, 234–239. [Google Scholar] [CrossRef]
- Venables, J.D.; McNamara, D.K.; Chen, J.M.; Sun, T.S.; Hopping, R.L. Oxide morphologies on aluminum prepared for adhesive bonding. Appl. Surf. Sci. 1979, 3, 88–98. [Google Scholar] [CrossRef]
- Mata-Zamora, M.E.; Saniger, J.M. Thermal evolution of porous anodic aluminas: A comparative study. Rev. Mex. Fis. 2005, 51, 502–509. [Google Scholar]
- Fukuda, Y. Role of oxalate ion in the formation of oxide film of aluminum in oxalic acid electrolyte. Nippon Kagaku Kaishi 1974, 1974, 1868–1875. [Google Scholar] [CrossRef]
- Fukuda, Y. Concentration and/or pH Effects of Oxalic Acid Bath on the Formation or Dissolution of Aluminum Oxide Films. Nippon Kagaku Kaishi 1975, 1975, 1299–1304. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Baba, N. Nature of the carboxylate species incorporated in anodic alumina films formed in oxalic acid solution. Thin Solid Films 1983, 101, 329–338. [Google Scholar] [CrossRef]
- Wood, G.C.; Skeldon, P.; Thompson, G.E.; Shimizu, K. A model for the incorporation of electrolyte species into anodic alumina. J. Electrochem. Soc. 1996, 143, 74–83. [Google Scholar] [CrossRef]
- Surganov, V.; Jansson, C.; Nielsen, J.G.; Morgen, P.; Gorokh, G.; Larsen, A.N. Different stages of aluminium anodization studied with depth profiling techniques. Electrochim. Acta 1988, 33, 517–519. [Google Scholar] [CrossRef]
- Thanh, P.T.; Akiyama, A.; Saji, T. Behavior of Phosphoric Acid Anodized Oxide on Aluminum in Phosphate Buffer Solutions. Denki Kagaku Oyobi Kogyo Butsuri Kagaku 1982, 50, 149–155. [Google Scholar] [CrossRef]
- Fukuda, Y.; Fukushima, T. Anodic oxidation of aluminium in sulphuric acid containing aluminium sulphate or magnesium sulphate. Electrochim. Acta 1983, 28, 47–56. [Google Scholar] [CrossRef]
- Fukuda, Y.; Fukushima, T. Behavior of Sulfate Ions daring Formation of Anodic Oxide Film on Aluminium. Bull. Chem. Soc. Jpn. 1980, 53, 3125–3130. [Google Scholar] [CrossRef]
- Runge, J.; Pomis, A. Anodic oxide film formation: Relating mechanism to composition and structure. In Proceedings of the AESF SUR/FIN Technical Conference, Chicago, IL, USA, 25–29 June 2000. [Google Scholar]
- Spooner, R.C. The anodic treatment of aluminum in sulfuric acid solutions. J. Electrochem. Soc. 1955, 102, 156–162. [Google Scholar] [CrossRef]
- Pullen, N.D. Oxide Films on Aluminum. Some of Their Physical Characteristics. Metal Ind. Lond. 1939, 54, 327–329. [Google Scholar]
- Mason, R.B. Factors affecting the formation of anodic oxide coatings in sulfuric acid electrolytes. J. Electrochem. Soc. 1955, 102, 671–675. [Google Scholar] [CrossRef]
- Brace, A.W.; Baker, H. The Incorporation of Sulphate into Anodic Coatings using Radioactive Sulphur as Tracer. Trans. Inst. Met. Finish. 1963, 40, 31–34. [Google Scholar] [CrossRef]
- Thompson, G.E.; Furneaux, R.C.; Wood, G.C. Electron microscopy of ion beam thinned porous anodic films formed on aluminium. Corros. Sci. 1978, 18, 481–498. [Google Scholar] [CrossRef]
- Shimizu, K.; Thompson, G.E.; Wood, G.C.; Xu, Y. Direct observations of ion-implanted xenon marker layers in anodic barrier films on aluminium. Thin Solid Films 1982, 88, 255–262. [Google Scholar] [CrossRef]
- Shimizu, B.K.; Kobayashi, K.; Thompson, G.E.; Wood, G.C. A novel marker for the determination of transport numbers during anodic barrier oxide growth on aluminium. Philos. Mag. B 1991, 64, 345–353. [Google Scholar] [CrossRef]
- Skeldon, P.; Shimizu, K.; Thompson, G.E.; Wood, G.C. Mobile tracers: Their use in understanding key features of anodic alumina film formation. Philos. Trans. Phys. Sci. Eng. 1994, 295–314. Available online: https://www.jstor.org/stable/54245 (accessed on 10 October 2019).
- Cherki, C.; Siejka, J. Study by nuclear microanalysis and O18 tracer techniques of the oxygen transport processes and the growth laws for porous anodic oxide layers on aluminum. J. Electrochem. Soc. 1973, 120, 784–791. [Google Scholar] [CrossRef]
- Skeldon, P.; Skeldon, M.; Thompson, G.E.; Wood, G.C. Incorporation of tungsten and molybdenum into anodic alumina films. Philos. Mag. B 1989, 60, 513–521. [Google Scholar] [CrossRef]
- Xu, Y.; Li, H.; Shen, Y.; Liu, S.; Wang, W.; Tao, J. Improvement of adhesion performance between aluminum alloy sheet and epoxy based on anodizing technique. Int. J. Adhes. Adhes. 2016, 70, 74–80. [Google Scholar] [CrossRef]
- Bjørgum, A.; Lapique, F.; Walmsley, J.; Redford, K. Anodising as pre-treatment for structural bonding. Int. J. Adhes. Adhes. 2003, 23, 401–412. [Google Scholar] [CrossRef]
- Descamps, P.; Iker, J.; Wolf, A.T. Effects of anodized aluminium surface parameters on the long-term adhesion of silicone structural glazing sealants. Constr. Build. Mater. 1996, 10, 527–538. [Google Scholar] [CrossRef]
- Johnsen, B.B.; Lapique, F.; Bjørgum, A. The durability of bonded aluminium joints: A comparison of AC and DC anodising pretreatments. Int. J. Adhes. Adhes. 2004, 24, 153–161. [Google Scholar] [CrossRef]
- Park, S.Y.; Choi, W.J.; Choi, H.S.; Kwon, H.; Kim, S.H. Recent trends in surface treatment technologies for airframe adhesive bonding processing: A review (1995–2008). J. Adhes. 2010, 86, 192–221. [Google Scholar] [CrossRef]
- Li, Y.D.; Zhao, P.Z.; Feng, Y.J.; Cao, H.L. Influence of anodic oxide film structure on adhesive bonding performance of 5754 aluminum alloy. Trans. Nonferrous Met. Soc. 2019, 29, 1836–1841. [Google Scholar] [CrossRef]
- Runge, J.M. Enhancing anodic aluminum oxide for bonding applications. In Proceedings of the 24th Annual AAC Conference, San Diego, CA, USA, 2–4 October 2015; Available online: https://pdfs.semanticscholar.org/2fb5/d59019d0e989c1b842def0c512356b84685e.pdf (accessed on 15 November 2019).
- Abrahami, S.T.; Hauffman, T.; de Kok, J.M.; Mol, J.M.; Terryn, H. Effect of anodic aluminum oxide chemistry on adhesive bonding of epoxy. J. Phys. Chem. C 2016, 120, 19670–19677. [Google Scholar] [CrossRef]
- Abrahami, S.T.; de Kok, J.M.; Gudla, V.C.; Ambat, R.; Terryn, H.; Mol, J.M. Interface strength and degradation of adhesively bonded porous aluminum oxides. NPJ Mater. Degrad. 2017, 1, 8. [Google Scholar] [CrossRef]
- Carre, A.; Schultz, J. Polymer-aluminium adhesion. I. The surface energy of aluminium in relation to its surface treatment. J Adhes. 1983, 15, 151–161. [Google Scholar] [CrossRef]
- Lefebvre, D.R.; Ahn, B.K.; Dillard, D.A.; Dillard, J.G. The effect of surface treatments on interfacial fatigue crack initiation in aluminum/epoxy bonds. Int. J. Frac. 2002, 114, 191–202. [Google Scholar] [CrossRef]
- Bartolomé, M.J.; López, V.; Escudero, E.; Caruana, G.; González, J.A. Changes in the specific surface area of porous aluminium oxide films during sealing. Surf. Coat. Technol. 2006, 200, 4530–4537. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, P.; Feng, Y.; Cao, H. Effects of anodizing pore structure on the adhesive bonding performance of AA5754 automotive sheets. In Proceedings of the 16th International Aluminum Alloys Conference (ICAA16), Montreal, QC, Canada, 17–21 June 2018. [Google Scholar]
- Underhill, P.R.; Rider, A.N. Hydrated oxide film growth on aluminium alloys immersed in warm water. Surf. Coat. Technol. 2005, 192, 199–207. [Google Scholar] [CrossRef]
- Underhill, P.R.; Rider, A.N.; DuQuesnay, D.L. The effect of warm water surface treatments on the fatigue life in shear of aluminum joints. Int. J. Adhes. Adhes. 2006, 26, 199–205. [Google Scholar] [CrossRef]
- Rider, A.N.; Arnott, D.R. Boiling water and silane pre-treatment of aluminium alloys for durable adhesive bonding. Int. J. Adhes. Adhes. 2000, 20, 209–220. [Google Scholar] [CrossRef]
- Strålin, A.; Hjertbergc, T. Improved adhesion strength between aluminum and ethylene copolymers by hydration of the aluminum surface. J. Appl. Polym. Sci. 1993, 49, 511–521. [Google Scholar] [CrossRef]
- Rider, A.N. The influence of porosity and morphology of hydrated oxide films on epoxy-aluminium bond durability. J. Adhes. Sci. Technol. 2001, 15, 395–422. [Google Scholar] [CrossRef]
- Oh, Y.H.; Rhee, C.K.; Kim, D.H.; Lee, G.H.; Kim, W.W. Formation characteristics of an aluminum hydroxide fiber by a hydrolysis of aluminum nano powder. J. Mater. Sci. 2006, 41, 4191–4195. [Google Scholar] [CrossRef]
- Allington, R.D.; Porritt, N.; Shaw, S.J. Proceedings of the 24th Annual Meeting of Adhesive Society, Williamsburg, VA, USA, 25–28 February 2001; Adhesion Society: Blacksburg, VA, USA, 2001; pp. 386–388. [Google Scholar]
- Semoto, T.; Tsuji, Y.; Yoshizawa, K. Molecular understanding of the adhesive force between a metal oxide surface and an epoxy resin. J. Phys. Chem. C 2011, 115, 11701–11708. [Google Scholar] [CrossRef]
- Luo, Y.R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Kalantary2007, M.R.; Gabe, D.R.; Ross, D.H. Processes for Sealing of Anodic Films on Aluminium. Alum. Finish. 1993, 13, 33–37. [Google Scholar]
- Spooner, R.C. Water Sealing of Detached Aluminium Oxide Anodic Film. Nature 1956, 178, 1113–1114. [Google Scholar] [CrossRef]
- Spooner, R.C. Sealing of Anodic Films on Aluminum and Its Alloys. Part 1. Met. Finish. 1968, 66, 44–49. [Google Scholar]
- Wefers, K. The Mechanism of Sealing of Anodic Oxide Coatings on Aluminum. Aluminium 1973, 49 Pt 1, 553–561. [Google Scholar]
- Wefers, K. The Mechanism of Sealing of Anodic Oxide Coatings on Aluminum (II). Aluminium 1973, 49, 622–625. [Google Scholar]
- Yaffe, B. Sealing of Anodized Aluminum—A Review. Mater. Australas. 1990, 22, 20–23. [Google Scholar]
- Baker, B.R.; Balser, J.D.; Pearson, R.M.; Strahl, E.O. Sealing of Anodic Oxide Films on Aluminum. Aluminium 1976, 52, 241–245. [Google Scholar]
- Li, L.; Zhang, Y.; Lei, J.; He, J.; Lv, R.; Li, N.; Pan, F. Water-only hydrothermal method: A generalized route for environmentally-benign and cost-effective construction of superhydrophilic surfaces with biomimetic micronanostructures on metals and alloys. Chem. Commun. 2014, 50, 7416–7419. [Google Scholar] [CrossRef]
- Tomashov, N.; Tyukina, M. Chromate Sealing of Anodic Films. Light Met. 1946, 9, 22–35. [Google Scholar]
- Steele, L.S.; Brandewie, B. Goodrich Corp, Treated Aluminum Article and Method for Making Same. U.S. Patent 7,527,872, 5 May 2009. [Google Scholar]
- Shulman, G.P.; Bauman, A.J. Organic acid sealants for anodized aluminum—A new method for corrosion protection. Met. Finish. 1995, 93, 16–19. [Google Scholar] [CrossRef]
- Shulman, G.P.; Bauman, A.J. Corrosion Protection with Organic Acid Sealants for Anodized Aluminum. In Organic Coatings for Corrosion Control; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1996; Volume 689, Chapter 34; pp. 420–422. [Google Scholar] [CrossRef]
- Kramer, I.R.; Burrows, C.F. Method of completely impregnating a medium hard anodized surface with molten straight-chain saturated aliphatic compounds and the product thereof. U.S. Patent 3,510,411, 5 May 1970. [Google Scholar]
- Kramer, I.R.; Burrows, C.F. Dual-seal anodized aluminum. U.S. Patent 3,440,150, 22 April 1969. [Google Scholar]
- Moutarlier, V.; Pelletier, S.; Lallemand, F.; Gigandet, M.P.; Mekhalif, Z. Characterisation of the anodic layers formed on 2024 aluminium alloy, in tetraborate electrolyte containing molybdate ions. Appl. Surf. Sci. 2005, 252, 1739–1746. [Google Scholar] [CrossRef]
- Hart, R.K. A study of boehmite formation on aluminium surfaces by electron diffraction. Trans. Faraday Soc. 1954, 50, 269–273. [Google Scholar] [CrossRef]
- Tomashov, N.D.; Tyukina, M.N. Mechanism of Chromate Filling of Anodic Films on Aluminum. In Bulletin of the Academy of Sciences of the USSR, Division of Chemical Sciences; Consultants Bureau: New York, NY, USA, 1944; pp. 325–336. [Google Scholar]
- Murphy, J.F. Practical implications of research on anodic coatings on aluminum. Plating 1967, 54, 1241–1245. [Google Scholar]
- Spooner, R.C. Proceedings of the 48th Annual Technical Proceedings; American Electroplaters’ Society: Boston, MA, USA, 1961; pp. 69–81. [Google Scholar]
- Richaud, H.; Franklin, R.W. The Influence of Sealing Water Impurities on the Quality of the Oxide Films. In Proceedings of the Conference on Anodising Aluminium, Nottingham, UK, 12–14 September 1961; pp. 181–185. [Google Scholar]
- Wernick, S.; Pinner, R. The Surface Treatment and Finishing of Aluminum; Robert Draper Ltd.: Teddington, UK, 1964; p. 497. [Google Scholar]
- Sheasby, P.G.; Bancroft, G. The Use of an Ammonium Acetate Buffer Solution for Sealing Anodized Aluminium. Trans. Inst. Met. Finish. 1970, 48, 140–144. [Google Scholar] [CrossRef]
- Okada, K.; Nagashima, T.; Kameshima, Y.; Yasumori, A.; Tsukada, T. Relationship between formation conditions, properties, and crystallite size of boehmite. J. Colloid Interface Sci. 2002, 253, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Park, B.K.; Tian, M.Y.; Lee, J.M. Relationship between properties of pseudo-boehmite and its synthetic conditions. J. Ind. Eng. Chem. 2006, 12, 295–300. [Google Scholar] [CrossRef]
- Alwitt, R.S. The growth of hydrous oxide films on aluminum. J. Electrochem. Soc. 1974, 121, 1322–1328. [Google Scholar] [CrossRef]
- Ito, G.; Sawayanagi, F.; Shimizu, Y. Rikagaku Kenkyusho Hohoku, 1959; 35, 409/18 (CA 54,19418a).
- Troutner, V.H. Observations on the Mechanisms and Kinetics of Aqueous Aluminum Corrosion (Part 1—Role of the Corrosion Product Film in the Uniform Aqueous Corrosion of Aluminum). Corrosion 1959, 15, 25–28. [Google Scholar] [CrossRef]
- Draley, J.E.; Ruther, W.E. Aqueous Corrosion of Aluminum (Part 2-Methods of Protection Above 200 °C). Corrosion 1956, 12, 20–30, 480t–490t. [Google Scholar] [CrossRef]
- Kondo, T.; Akutsu, C.; Kawasaki, M. Corrosion of Aluminum in High Pressure Steam at Temperatures above and below 320 °C. Trans. Jpn. Inst. Met. 1962, 3, 110–117. [Google Scholar] [CrossRef][Green Version]
- Griess, J.C.; Savage, H.C.; Mauney, T.H.; English, J.L.; Rainwater, J.G. Effect of Heat Flux on the Corrosion of Aluminum by Water. Part II. Influence of Water Temperature, Velocity, and pH on Corrosion-Product Formation (No. ORNL-3056); Oak Ridge National Lab. Tenn.: Oak Ridge, TN, USA, 1961. [Google Scholar]
- Kendig, M.; Addison, R.; Jeanjaquet, S. Adsorption of Ce (III) on anodized aluminum. Electrochem. Solid-State Lett. 2000, 3, 266–267. [Google Scholar] [CrossRef]
- Burwell, R.L.; May, T.P. The Measurement of Permeability Characteristics of Anodic Films on Aluminum. J. Electrochem. Soc. 1948, 94, 195–213. [Google Scholar] [CrossRef]
- González, J.A.; López, V.; Otero, E.; Bautista, A. Postsealing Changes in Porous Aluminum Oxide Films Obtained in Sulfuric Acid Solutions. J. Electrochem. Soc. 2000, 147, 984–990. [Google Scholar] [CrossRef]
- Dorsey, G.A. Structural Features of Anodic Aluminas Adjusted to Influence the “Degree-of-Seal”. J. Electrochem. Soc. 1970, 117, 1181–1183. [Google Scholar] [CrossRef]
- Thompson, G.E.; Furneaux, R.C.; Wood, G.C. The Morphology of Sealed Anodic Films Formed on Aluminium in Phosphoric Acid. Trans. Inst. Met. Finish. 1975, 53, 97–102. [Google Scholar] [CrossRef]
- Patermarakis, G.; Papandreadis, N. Effect of the structure of porous anodic Al2O3 films on the mechanism of their hydration and pore closure during hydrothermal treatment. Electrochim. Acta 1993, 38, 1413–1420. [Google Scholar] [CrossRef]
- Gonzalez, J.A.; Feliu, S.; Bautista, A.; Otero, E. Changes in cold sealed aluminium oxide films during ageing. J. Appl. Electrochem. 1999, 29, 843–852. [Google Scholar] [CrossRef]
- Chahboun, N.; Rocca, E.; Veys-Renaux, D.; Augros, M.; Boutoba, M.; Caldeira, N. Sealing of Anodized Multiphase Aluminum Alloys with Cr(+III)/Zr(+IV) Salts: Characterization and Corrosion Behavior. J. Electrochem. Soc. 2015, 163, C69–C75. [Google Scholar] [CrossRef]
- van Bokhoven, J.A.; Sambe, H.; Ramaker, D.E.; Koningsberger, D.C. Al K-edge near-edge X-ray absorption fine structure (NEXAFS) study on the coordination structure of aluminum in minerals and Y zeolites. J. Phys. Chem. B 1999, 103, 7557–7564. [Google Scholar] [CrossRef]
- Schenk, M. Werkstoffe Aluminium und Seine Anodische Oxydation; A. Francke: Bern, Switzerland, 1948. [Google Scholar]
- Maloney, M.A.; Sung, D.J. Elimination of Crazing in Anodized Layers. U.S. Patent No. 9,702,053, 11 July 2017. [Google Scholar]
- Boisier, G.; Pébère, N.; Druez, C.; Villatte, M.; Suel, S. FESEM and EIS study of sealed AA2024 T3 anodized in sulfuric acid electrolytes: Influence of tartaric acid. J. Electrochem. Soc. 2008, 155, C521–C529. [Google Scholar] [CrossRef]
- Suay, J.J.; Gimenez, E.; Rodrıguez, T.; Habbib, K.; Saura, J.J. Characterization of anodized and sealed aluminium by EIS. Corros. Sci. 2003, 45, 611–624. [Google Scholar] [CrossRef]
- Survila, E.; Andrews, D.P. Correlation of Non-Destructive Sealing Tests with Destructive Tests for Anodic Oxidation Coatings on Aluminium. Trans. Inst. Met. Finish. 1976, 54, 163–173. [Google Scholar] [CrossRef]
- Spooner, R.C. The Sealing of Sulfuric Acid Anodic Films on Aluminum. In Proceedings of the 44th Annual Technology Proceeding Amercian Electroplaters’ Society, Montreal, QC, Canada, 17–20 June 1957; pp. 132–142. [Google Scholar]
- ISO 2143:2017. Anodizing of Aluminium and Its Alloys—Estimation of Loss of Absorptive Power of Anodic Oxidation Coatings after Sealing—Dye-Spot Test with Prior Acid Treatment; International Organization for Standardization: Geneva, Switzerland, 2017. [Google Scholar]
- ASTM B136-84(2018). Standard Method for Measurement of Stain Resistance of Anodic Coatings on Aluminium ASTM International; ASTM International: West Conshohocken, PA, USA, 2018. [Google Scholar]
- Manhart, J.H.; Cochran, W. Acid dissolution tests for seal quality of anodized aluminium. Plating 1971, 58, 219–224. [Google Scholar]
- ASTM B680-80(2019). Standard Test Method for Seal Quality of Anodic Coatings on Aluminum by Acid Dissolution; ASTM International: West Conshohocken, PA, USA, 2019. [Google Scholar]
- ISO 3210:2017 Anodizing of Aluminium and Its Alloys—Assessment of Quality of Sealed Anodic Oxidation Coatings by Measurement of the Loss of Mass after Immersion in Acid Solution(s); International Organization for Standardization: Geneva, Switzerland, 2017.
- ISO 2931:2017 Anodizing of Aluminium and Its Alloys—Assessment of Quality of Sealed Anodic Oxidation Coatings by Measurement of Admittance; International Organization for Standardization: Geneva, Switzerland, 2017.
- Zhao, X.H.; Zuo, Y.; Zhao, J.M.; Xiong, J.P.; Tang, Y.M. A study on the self-sealing process of anodic films on aluminum by EIS. Surf. Coat. Technol. 2006, 200, 6846–6853. [Google Scholar] [CrossRef]
- Mansfeld, F.; Kendig, M.W. Evaluation of anodized aluminum surfaces with electrochemical impedance spectroscopy. J. Electrochem. Soc. 1988, 135, 828–833. [Google Scholar] [CrossRef]
- Hitzig, J.; Jüttner, K.; Lorenz, W.J.; Paatsch, W. AC-impedance measurements on porous aluminium oxide films. Corros. Sci. 1984, 24, 945–952. [Google Scholar] [CrossRef]
- Hitzig, J.; Jüttner, K.; Lorenz, W.J.; Paatsch, W. AC-Impedance measurements on corroded porous aluminum oxide films. J. Electrochem. Soc. 1986, 133, 887–892. [Google Scholar] [CrossRef]
- Domingues, L.; Fernandes, J.C.S.; Belo, M.D.C.; Ferreira, M.G.S.; Guerra-Rosa, L. Anodising of Al 2024-T3 in a modified sulphuric acid/boric acid bath for aeronautical applications. Corros. Sci. 2003, 45, 149–160. [Google Scholar] [CrossRef]
- Twite, R.L.; Bierwagen, G.P. Review of alternatives to chromate for corrosion protection of aluminum aerospace alloys. Prog. Org. Coat. 1998, 33, 91–100. [Google Scholar] [CrossRef]
- Analysis of Alternatives for Chromium Sealing in the Aerospace Industry—GCCA_AoA_Potassium Dichromate—2007. Available online: https://echa.europa.eu/documents/10162/d6c88cd1-ef18-460b-8493-e1d59211a888 (accessed on 5 November 2019).
- Kulinich, S.A.; Akhtara, A.S. On Conversion Coating Treatments to Replace Chromating for Al Alloys: Recent Developments and Possible Future Directions. Russ. J. Non-Ferr. Met. 2012, 53, 176–203. [Google Scholar] [CrossRef]
- Cohen, S.M. Replacements for chromium pretreatments on aluminum. Corrosion 1995, 51, 71–78. [Google Scholar] [CrossRef]
- Katzman, H.A.; Malouf, G.M.; Bauer, R.; Stupian, G.W. Corrosion-protective chromate coatings on aluminum. Appl. Surf. Sci. 1979, 2, 416–432. [Google Scholar] [CrossRef]
- Kendig, M.W.; Davenport, A.J.; Isaacs, H.S. The mechanism of corrosion inhibition by chromate conversion coatings from X-ray absorption near edge spectroscopy (XANES). Corros. Sci. 1993, 34, 41–49. [Google Scholar] [CrossRef]
- Kendig, M.W.; Buchheit, R.G. Corrosion inhibition of aluminum and aluminum alloys by soluble chromates, chromate coatings, and chromate-free coatings. Corrosion 2003, 59, 379–400. [Google Scholar] [CrossRef]
- Kendig, M.; Bucheit, R. Corrosion Inhibition of Al and Al Alloys by Hexavalent Cr compounds, A Mechanistic Overview,” Research Topical Symposium on Surface Conversion for Aluminum and Ferrous Alloys. In Proceedings of the Corrosion 2000, NACE/2000, Orlando, FL, USA, 26–31 March 2000. [Google Scholar]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions; National Association of Corrosion Engineers: Houston, TX, USA, 1974. [Google Scholar]
- Cui1974, W.; Zhang, X.; Pearce, C.I.; Chen, Y.; Zhang, S.; Liu, W.; Engelhard, M.H.; Kovarik, L.; Zong, M.; Zhang, H.; et al. Cr (III) adsorption by cluster formation on boehmite nanoplates in highly alkaline solution. Environ. Sci. Technol. 2019, 53, 11043–11055. [Google Scholar] [CrossRef]
- Chidambaram, D.; Clayton, C.R.; Halada, G.P. A duplex mechanism-based model for the interaction between chromate ions and the hydrated oxide film on aluminum alloys. J. Electrochem. Soc. 2003, 150, B224–B237. [Google Scholar] [CrossRef]
- Konno, H.; Kobayashi, S.; Takahashi, H.; Nagayama, M. The hydration of barrier oxide films on aluminium and its inhibition by chromate and phosphate ions. Corros. Sci. 1982, 22, 913–923. [Google Scholar] [CrossRef]
- Vermilyea, D.A.; Vedder, W. Inhibition of the aluminum + water reaction. Trans. Faraday Soc. 1970, 66, 2644–2654. [Google Scholar] [CrossRef]
- Gancs, L.; Besing, A.S.; Bujak, R.; Kolics, A.; Nemeth, Z.; Wieckowski, A. Interaction of chromate with aluminum in NaCl solutions. Electrochem. Solid-State Lett. 2002, 5, B16–B19. [Google Scholar] [CrossRef]
- Bernstein, J.J.; White, R.M. Piezo-Electrocapillary Effect: A New Effect Observed in Porous Anodic Oxide Films. J. Electrochem. Soc. 1984, 131, 1050–1053. [Google Scholar] [CrossRef]
- Kendig, M.; Addison, R.; Jeanjaquet, S. The Influence of Adsorbed Oxo-Cr (VI) Species on the Zeta Potential in the Porous Oxide of Anodized Aluminum. J. Electrochem. Soc. 1999, 146, 4419–4423. [Google Scholar] [CrossRef]
- Madden, S.B.; Scully, J.R. Inhibition of AA2024-T351 corrosion using permanganate. J. Electrochem. Soc. 2014, 161, C162–C175. [Google Scholar] [CrossRef]
- Moutarlier, V.; Gigandet, M.P.; Ricq, L.; Pagetti, J. Electrochemical characterisation of anodic oxidation films formed in presence of corrosion inhibitors. Appl. Surf. Sci. 2001, 183, 1–9. [Google Scholar] [CrossRef]
- Moutarlier, V.; Gigandet, M.P.; Normand, B.; Pagetti, J. EIS characterisation of anodic films formed on 2024 aluminium alloy, in sulphuric acid containing molybdate or permanganate species. Corros. Sci. 2005, 47, 937–951. [Google Scholar] [CrossRef]
- Mohammadi, M.; Yazdani, A.; Bahrololoom, M.E.; Alfantazi, A. Corrosion behavior of 2024 aluminum alloy anodized in presence of permanganate and phosphate ions. J. Coat. Technol. Res. 2013, 10, 219–229. [Google Scholar] [CrossRef]
- Crouse, M.; Miller, A.E.; Pujar, M.G.; Vasanth, K.L. Evaluation of Potassium Permanganate (KMnO4) as a Green Corrosion Inhibitor/Sealant for Anodized Al 2024 and Al 6061 at Different pH Values. In Proceedings of the Corrosion 2002, Denver, CO, USA, 7–11 April 2002. [Google Scholar]
- Robertson, W.D. Molybdate and tungstate as corrosion inhibitors and the mechanism of inhibition. J. Electrochem. Soc. 1951, 98, 94–100. [Google Scholar] [CrossRef]
- El Abedin, S.Z. Role of chromate, molybdate and tungstate anions on the inhibition of aluminium in chloride solutions. J. Appl. Electrochem. 2001, 31, 711–718. [Google Scholar] [CrossRef]
- Vukasovich, M.S.; Farr, J.P.G. Molybdate in corrosion inhibition—A review. Polyhedron 1986, 5, 551–559. [Google Scholar] [CrossRef]
- Yoganandan, G.; Balaraju, J.N. Synergistic effect of V and Mn oxyanions for the corrosion protection of anodized aerospace aluminum alloy. Surf. Coat. Technol. 2014, 252, 35–47. [Google Scholar] [CrossRef]
- Moutarlier, V.; Gigandet, M.P.; Pagetti, J.; Ricq, L. Molybdate/sulfuric acid anodising of 2024-aluminium alloy: Influence of inhibitor concentration on film growth and on corrosion resistance. Surf. Coat. Technol. 2003, 173, 87–95. [Google Scholar] [CrossRef]
- Lopez-Garrity, O.F.G.S.; Frankel, G.S. Corrosion inhibition of aluminum alloy 2024-T3 by sodium molybdate. J. Electrochem. Soc. 2014, 161, C95–C106. [Google Scholar] [CrossRef]
- Guan, H.; Buchheit, R.G. Corrosion protection of aluminum alloy 2024-T3 by vanadate conversion coatings. Corrosion 2004, 60, 284–296. [Google Scholar] [CrossRef]
- Iannuzzi, M.; Young, T.; Frankel, G.S. Aluminum alloy corrosion inhibition by vanadates. J. Electrochem. Soc. 2006, 153, B533–B541. [Google Scholar] [CrossRef]
- Iannuzzi, M.; Frankel, G.S. Inhibition of aluminum alloy 2024 corrosion by vanadates: An in situ atomic force microscopy scratching investigation. Corrosion 2007, 63, 672–688. [Google Scholar] [CrossRef]
- Iannuzzi, M.; Frankel, G.S. Mechanisms of corrosion inhibition of AA2024-T3 by vanadates. Corros. Sci. 2007, 49, 2371–2391. [Google Scholar] [CrossRef]
- Ralston, K.D.; Chrisanti, S.; Young, T.L.; Buchheit, R.G. Corrosion inhibition of aluminum alloy 2024-T3 by aqueous vanadium species. J. Electrochem. Soc. 2008, 155, C350–C359. [Google Scholar] [CrossRef]
- Kharitonov, D.S.; Örnek, C.; Claesson, P.M.; Sommertune, J.; Zharskii, I.M.; Kurilo, I.I.; Pan, J. Corrosion inhibition of aluminum alloy AA6063-T5 by vanadates: Microstructure characterization and corrosion analysis. J. Electrochem. Soc. 2018, 165, C116–C126. [Google Scholar] [CrossRef]
- Iannuzzi, M.; Kovac, J.; Frankel, G.S. A study of the mechanisms of corrosion inhibition of AA2024-T3 by vanadates using the split cell technique. Electrochim. Acta 2007, 52, 4032–4042. [Google Scholar] [CrossRef]
- Thomas, R.; Kerkhof, F.P.J.M.; Moulijn, J.A.; Medema, J.; De Beer, V.H.J. On the formation of aluminum tungstate and its presence in tungsten oxide on γ-alumina catalysts. J. Catal. 1980, 61, 559–561. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Egawa, T.; Tamura, S.; Imanaka, N.; Adachi, G.Y. Trivalent Al3+ ion conduction in aluminum tungstate solid. Chem. Mater. 1997, 9, 1649–1654. [Google Scholar] [CrossRef]
- Dąbkowski, A.; Dąbkowska, H.A.; Greedan, J.E.; Adachi, G.; Kobayashi, Y.; Tamura, S.; Hiaraiwa, M.; Imanaka, N. Crystal growth of aluminum tungstate Al2(WO4)3 by the Czochralski method from nonstoichiometric melt. J. Cryst. Growth 1999, 197, 879–882. [Google Scholar] [CrossRef]
- Mushran, S.P. The preparation of transparent jellies of aluminium molybdate. Curr. Sci. 1949, 18, 48. [Google Scholar]
- Kassem, M. Phase relations in the Al2O3-MoO3 and Al-MoO3 systems, investigated by X-ray powder diffraction, FTIR, and DTA techniques. Inorg. Mater. 2006, 42, 165–170. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Shimanouchi, R. Synthesis of Al2(MoO4)3 by Two Distinct Processes, Hydrothermal Reaction and Solid-State Reaction. Procedia Eng. 2016, 148, 158–162. [Google Scholar] [CrossRef]
- László, K. Synthesis and Properties of Simple and Complex Salts of Permanganic Acid. Ph.D. Thesis, Hungarian Academy of Science, Budapest, Hungary, 2006. [Google Scholar]
- Theodore, J. Carus Chemical Co, Process of Making Alkaline Earth Metal Permanganates. U.S. Patent 2,504,130, 18 April 1950. [Google Scholar]
- Kótai, L.; Banerji, K.K. An Improved Method for the Preparation of High-Purity Permanganate Salts. Synth. React. Inorg. Met. Org. Chem. 2001, 31, 491–495. [Google Scholar] [CrossRef]
- Pecquenard, B.; Zavalij, P.Y.; Whittingham, M.S. Hydrothermal synthesis and characterization of a new aluminium vanadium oxide hydroxide Al2(OH)3(VO4). J. Mater. Chem. 1998, 8, 1255–1258. [Google Scholar] [CrossRef]
- Ferreiro, E.A.; Helmy, A.K.; De Bussetti, S.G. Molybdate sorption by oxides of aluminium and iron. Zeitschrift für Pflanzenernährung und Bodenkunde 1985, 148, 559–566. [Google Scholar] [CrossRef]
- Spanos, N.; Vordonis, L.; Kordulis, C.; Lycourghiotis, A. Molybdenum-oxo species deposited on alumina by adsorption: I. Mechanism of the Adsorption. J. Catal. 1990, 124, 301–314. [Google Scholar] [CrossRef]
- Spanos, N.; Vordonis, L.; Kordulis, C.; Koutsoukos, P.G.; Lycourghiotis, A. Molybdenum-oxo species deposited on alumina by adsorption: II. Regulation of the surface MoVI concentration by control of the protonated surface hydroxyls. J. Catal. 1990, 124, 315–323. [Google Scholar] [CrossRef]
- Spanos, N.; Lycourghiotis, A. Molybdenum-oxo species deposited on alumina by adsorption: III. Advances in the mechanism of Mo (VI) deposition. J. Catal. 1994, 147, 57–71. [Google Scholar] [CrossRef]
- Bourikas, K.; Goula, M.A.; Lycourghiotis, A. Kinetics of Deposition of the Mo−Oxo Species on the Surface of γ-Alumina. Langmuir 1998, 14, 4819–4826. [Google Scholar] [CrossRef]
- Jones, L.H.P.; Milne, A.A. Aluminium Molybdate and its Formation in Clay Minerals. Nature 1956, 178, 1115. [Google Scholar] [CrossRef]
- Deosthale, Y.G.; Gopalan, C. The effect of molybdenum levels in sorghum (Sorghum vulgare Pers.) on uric acid and copper excretion in man. Br. J. Nutr. 1974, 31, 351–355. [Google Scholar] [CrossRef]
- Compere, R.; Burny, A.; Riga, A.; Francois, E.; Vanuytrecht, S. Copper in the treatment of molybdenosis in the rat: Determination of the dose of the antidote. J. Nutr. 1965, 87, 412–418. [Google Scholar] [CrossRef]
- Nederbragt, H.; van den Hamer, C.J. Changes in the binding of copper in the plasma of molybdenum supplemented rats. J. Inorg. Biochem. 1981, 15, 293–306. [Google Scholar] [CrossRef]
- Nederbragt, H. Changes in the distribution of copper and molybdenum after Mo administration and subsequent additional oral or intraperitoneal Cu administration to rats. Br. J. Nutr. 1982, 48, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Quagraine, E.K.; Reid, R.S. UV/visible spectrophotometric studies of the interactions of thiomolybdates, copper (II) and other ligands. J. Inorg. Biochem. 2001, 85, 53–60. [Google Scholar] [CrossRef]
- Morgan, P.L.; Grace, N.D.; Lilley, D.P. Using sodium molybdate to treat chronic copper toxicity in dairy cows: A practical approach. N. Z. Vet. J. 2014, 62, 167–170. [Google Scholar] [CrossRef]
- Suzuki, K.T.; Ogura, Y. Biological regulation of copper and selective removal of copper: Therapy for Wilson disease and its molecular mechanism. Yakugaku Zasshi J. Pharm. Soc. Jpn. 2000, 120, 899–908. [Google Scholar] [CrossRef][Green Version]
- Hinton, B.R.W. The inhibition of aluminum alloy corrosion by cerous cations. Met. Forum 1984, 7, 221. [Google Scholar]
- Hinton, B.R.W. Corrosion inhibition with rare earth metal salts. J. Alloys Compd. 1992, 180, 15–25. [Google Scholar] [CrossRef]
- Aldykiewicz, A.J.; Davenport, A.J.; Isaacs, H.S. Studies of the Formation of Cerium-Rich Protective Films Using X-Ray Absorption Near-Edge Spectroscopy and Rotating Disk Electrode Methods. J. Electrochem. Soc. 1996, 143, 147–154. [Google Scholar] [CrossRef]
- Shi, H.; Han, E.H.; Liu, F. Corrosion protection of aluminium alloy 2024-T3 in 0.05 M NaCl by cerium cinnamate. Corros. Sci. 2011, 53, 2374–2384. [Google Scholar] [CrossRef]
- Arnott, D.R.; Ryan, N.E.; Hinton, B.R.W.; Sexton, B.A.; Hughes, A.E. Auger and XPS studies of cerium corrosion inhibition on 7075 aluminum alloy. Appl. Surface Sci. 1985, 22, 236–251. [Google Scholar] [CrossRef]
- Hu, T.H.; Shi, H.W.; Wei, T.; Fan, S.H.; Liu, F.C.; Han, E.H. Corrosion protection of AA2024-T3 by cerium malate and cerium malate-doped sol–gel coatings. Acta Metall. Sin.-Engl. 2019. [Google Scholar] [CrossRef]
- Ho, D.; Brack, N.; Scully, J.; Markley, T.; Forsyth, M.; Hinton, B. Cerium dibutylphosphate as a corrosion inhibitor for AA2024-T3 aluminum alloys. J. Electrochem. Soc. 2006, 153, B392–B401. [Google Scholar] [CrossRef]
- Emregül, K.C.; Aksüt, A.A. The effect of sodium molybdate on the pitting corrosion of aluminum. Corros. Sci. 2003, 45, 2415–2433. [Google Scholar] [CrossRef]
- Armour, A.W.; Robitaille, D.R. Corrosion inhibition by sodium molybdate. J. Chem. Technol. Biotechnol. 1979, 29, 619–628. [Google Scholar] [CrossRef]
- Kwolek, P.; Kamiński, A.; Dychtoń, K.; Drajewicz, M.; Sieniawski, J. The corrosion rate of aluminium in the orthophosphoric acid solutions in the presence of sodium molybdate. Corros. Sci. 2016, 106, 208–216. [Google Scholar] [CrossRef]
- Kwolek, P.; Dychtoń, K.; Szydełko, J.; Gradzik, A.; Drajewicz, M.; Sieniawski, J. The influence of sodium molybdate on the rate of corrosion of aluminum in phosphoric (V) acid. In Materials Science Forum; Trans Tech Publications: Zürich, Switzerland, 2016; Volume 844, pp. 31–37. [Google Scholar] [CrossRef]
- Moshier, W.C.; Davis, G.D. Interaction of molybdate anions with the passive film on aluminum. Corrosion 1990, 46, 43–50. [Google Scholar] [CrossRef]
- Kartsonakis, I.A.; Balaskas, A.C.; Kordas, G.C. Influence of cerium molybdate containers on the corrosion performance of epoxy coated aluminium alloys 2024-T3. Corros. Sci. 2011, 53, 3771–3779. [Google Scholar] [CrossRef]
- Taylor, S.R.; Chambers, B.D. The discovery of non-chromate corrosion inhibitors for aerospace alloys using high-throughput screening methods. Corros. Rev. 2007, 25, 571–590. [Google Scholar] [CrossRef]
- Yasakau, K.A.; Tedim, J.; Zheludkevich, M.L.; Drumm, R.; Shem, M.; Wittmar, M.; Veith, M.; Ferreira, M.G.S. Cerium molybdate nanowires for active corrosion protection of aluminium alloys. Corros. Sci. 2012, 58, 41–51. [Google Scholar] [CrossRef]
- Yasakau, K.A.; Kallip, S.; Zheludkevich, M.L.; Ferreira, M.G.S. Active corrosion protection of AA2024 by sol–gel coatings with cerium molybdate nanowires. Electrochim. Acta 2013, 112, 236–246. [Google Scholar] [CrossRef]
- Yasakau, K.A.; Tedim, J.; Montemor, M.F.; Salak, A.N.; Zheludkevich, M.L.; Ferreira, M.G. Mechanisms of localized corrosion inhibition of AA2024 by cerium molybdate nanowires. J. Phys. Chem. C 2013, 117, 5811–5823. [Google Scholar] [CrossRef]
- Yasakau, K.A.; Zheludkevich, M.L.; Lamaka, S.V.; Ferreira, M.G. Mechanism of corrosion inhibition of AA2024 by rare-earth compounds. J. Phys. Chem. B 2006, 110, 5515–5528. [Google Scholar] [CrossRef]
- Rodič, P.; Milošev, I. Corrosion inhibition of pure aluminium and alloys AA2024-T3 and AA7075-T6 by cerium (III) and cerium (IV) salts. J. Electrochem. Soc. 2015, 163, C85. [Google Scholar] [CrossRef]
- McCune, R.C.; Shilts, R.L.; Ferguson, S.M. A study of film formation on aluminum in aqueous solutions using Rutherford backscattering spectroscopy. Corros. Sci. 1982, 22, 1049–1065. [Google Scholar] [CrossRef]
- Etienne, M.; Rocca, E.; Chahboun, N.; Veys-Renaux, D. Local Evolution of pH with Time Determined by Shear Force-based Scanning Electrochemical Microscopy: Surface Reactivity of Anodized Aluminium. Electroanalysis 2016, 28, 2466–2471. [Google Scholar] [CrossRef]
- Runge, J.M. The Metallurgy of Anodizing Aluminum; Connecting Science to Practice; Springer International Publishing AG: Cham, Switzerland, 2018; Volume 305. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Y.; White, H.S. The nanopore electrode. Anal. Chem. 2004, 76, 6229–6238. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Y.; White, H.S. Steady-state voltammetric response of the nanopore electrode. Anal. Chem. 2006, 78, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, B.; White, H.S. Electrochemistry of nanopore electrodes in low ionic strength solutions. J. Phys. Chem. B 2006, 110, 1768–1774. [Google Scholar] [CrossRef]
- Watkins, J.J.; Zhang, B.; White, H.S. Electrochemistry at nanometer-scaled electrodes. J. Chem. Educ. 2005, 82, 712–719. [Google Scholar] [CrossRef]
- Hummer, G.; Rasaiah, J.; Noworyta, J. Water conduction through the hydrophobic channel of a carbon nanotube. Nature 2001, 414, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Tian, Y.W.; Shao, W.T.; Yuan, C.G.; Wang, K.; Xia, X.H. Solution pH regulating mass transport in highly ordered nanopore array electrode. Electrochem Commun. 2014, 42, 1–5. [Google Scholar] [CrossRef]
- Wang, C.; Xu, J.; Chen, H.; Xia, X. Mass transport in nanofluidic devices. Sci. China Chem. 2012, 55, 453–468. [Google Scholar] [CrossRef]
- Ma, C.; Xu, W.; Wichert, W.R.; Bohn, P.W. Ion accumulation and migration effects on redox cycling in nanopore electrode arrays at low ionic strength. ACS Nano 2016, 10, 3658–3664. [Google Scholar] [CrossRef]
- Ermakova, L.; Sidorova, M.; Bogdanova, N.; Klebanov, A. Electrokinetic and adsorption characteristics of (hydr) oxides and oxide nanostructures in 1: 1 electrolytes. Colloids Surf. A Physicochem. Eng. Asp. 2001, 192, 337–348. [Google Scholar] [CrossRef]
- Barkman, E.F. Sealing and Post Anodic Treatments, in Aodized Aluminum. ASTM STP 1965, 388, 85–120. [Google Scholar]
- Panias, D.; Paspaliaris, I. Thermodynamic determination of the stability area of boehmite in Al2O3-Na2O-H2O and Al2O3-H2O systems. World Metall.—Erzmetalls 1999, 52, 585–595. [Google Scholar]
- Wang, J.Q.; Liu, J.L.; Liu, X.Y.; Qiao, M.H.; Pei, Y.; Fan, K.N. Hydrothermal transformation of bayerite to boehmite. Sci. Adv. Mater. 2009, 1, 77–85. [Google Scholar] [CrossRef]
- Digne, M.; Sautet, P.; Raybaud, P.; Toulhoat, H.; Artacho, E. Structure and stability of aluminum hydroxides: A theoretical study. J. Phys. Chem. B 2002, 106, 5155–5162. [Google Scholar] [CrossRef]
- Sato, T. The thermal transformation of alumina monohydrate, boehmite. J. Appl. Chem. 2007, 12, 9–12. [Google Scholar] [CrossRef]
- Bye, G.C.; Robinson, J.G. Crystallization processes in aluminium hydroxide gels. Kolloid-Zeitschrift und Zeitschrift für Polymere 1964, 198, 53–60. [Google Scholar] [CrossRef]
- Sato, T. The transformation of bayerite into hydrargillite. J. Appl. Chem. 1961, 11, 207–209. [Google Scholar] [CrossRef]
- Sato, T. Hydration Phenomena of Aluminum Oxides. J. Jpn. Inst. Light Met. 1987, 37, 729–735. [Google Scholar] [CrossRef]
- Rocek, J.; Heeg, K.H.; Steinike, U.; Jirátová, K. Porous structure of aluminium hydroxide and its content of pseudoboehmite. Appl. Catal. 1991, 74, 29–36. [Google Scholar] [CrossRef]
- Xianqi, Z.; Ting’an, Z.; Wei, F.; Shaoyong, Y.; Junmin, S.; Guozhi, L.; Huibin, Y. Preparation of pseudo-boehmite by using high-alumina coal fly ash. In Light Metals; Springer: Cham, Switzerland, 2015; pp. 147–151. [Google Scholar] [CrossRef]
- Derakhshan, A.A.; Rajabi, L.; Ghorabi, S. Synthesis of boehmite-like nanorods through inverse phase transformation. J. Nanoeng. Nanomanuf. 2011, 1, 182–187. [Google Scholar] [CrossRef]
- Rajabi, L.; Derakhshan, A.A. Room temperature synthesis of boehmite and crystallization of nanoparticles: Effect of concentration and ultrasound. Sci. Adv. Mater. 2010, 2, 163–172. [Google Scholar] [CrossRef]
- Aldcroft, D.; Bye, G.C.; Hughes, C.A. Crystallisation processes in aluminium hydroxide gels. IV. Factors influencing the formation of the crystalline trihydroxides. J. Appl. Chem. 1969, 19, 167–172. [Google Scholar] [CrossRef]
- Violante, A.; Huang, P.M. Formation mechanism of aluminum hydroxide polymorphs. Clays Clay Miner. 1993, 41, 590–597. [Google Scholar] [CrossRef]
- Jun, J.H.; Kim, H.J.; Choi, D.J. Effect of the hydration on the properties of an aluminum oxide film. J. Ceram. Process Res. 2008, 9, 75–78. [Google Scholar]
- Shirai, T.; Watanabe, H.; Fuji, M.; Takahashi, M. Structural Properties and Surface Characteristics on Aluminum Oxide Powders. In Annual Report of the Ceramics Research Laboratory; Nagoya Institute of Technology: Aichi, Japan, 2010; Volume 9, pp. 23–31. Available online: http://id.nii.ac.jp/1476/00002232 (accessed on 6 October 2019).
- Munhoz, A.H., Jr.; Martins, M.V.S.; Ussui, V.; Cruz, K.; Zandonadi, A.R.; de Miranda, L.F. The influence of ageing in pseudoboehmites synthesis. In Materials Science Forum; Trans Tech Publications: Zürich, Switzerland, 2012; Volume 727, pp. 1795–1801. [Google Scholar] [CrossRef]
- Santos, P.D.S.; Coelho, A.C.V.; Santos, H.D.S.; Kiyohara, P.K. Hydrothermal synthesis of well-crystallised boehmite crystals of various shapes. Mater. Res. 2009, 12, 437–445. [Google Scholar] [CrossRef]
- Abdollahifar, M.; Hidaryan, M.; Jafari, P. The role anions on the synthesis of AlOOH nanoparticles using simple solvothermal method. Bol. Soc. Esp. Ceram. V 2018, 57, 66–72. [Google Scholar] [CrossRef]
- Bellotto, M.; Rebours, B.; Euzen, P. Mechanism of pseudo-boehmite dehydration: Influence of reagent structure and reaction kinetics on the transformation sequence. In Materials Science Forum; Trans Tech Publications: Zürich, Switzerland, 1998; Volume 278, pp. 572–577. [Google Scholar] [CrossRef]
- Conroy, M.; Soltis, J.A.; Wittman, R.S.; Smith, F.N.; Chatterjee, S.; Zhang, X.; Ilton, E.S.; Buck, E.C. Importance of interlayer H bonding structure to the stability of layered minerals. Sci. Rep. 2017, 7, 13274. [Google Scholar] [CrossRef] [PubMed]
- Gayer, K.H.; Thompson, L.C.; Zajicek, O.T. The solubility of aluminum hydroxide in acidic and basic media at 25 °C. Can. J. Chem. 1958, 36, 1268–1271. [Google Scholar] [CrossRef]
- May, H.M.; Helmke, P.A.; Jackson, M.L. Gibbsite solubility and thermodynamic properties of hydroxy-aluminum ions in aqueous solution at 25 °C. Geochim. Cosmochim. Acta 1979, 43, 861–868. [Google Scholar] [CrossRef]
- Dimitrios, P.; Athina, K. Alumina hydrate Precipitates in the system NaAl(OH)4 (supersaturated)/HNO3. In Proceedings of the EMC, Chicago, IL, USA, 8–12 August 2005; pp. 1–15. [Google Scholar]
- Bai2005, B.; Bai, H.; Zhang, L.; Huang, W. Catalytic activity of γ-AlOOH (0 0 1) surface in syngas conversion: Probing into the mechanism of carbon chain growth. Appl. Surf. Sci. 2018, 455, 123–131. [Google Scholar] [CrossRef]
- Karami, C.; Abdollahifar, M.; Jahani, F.; Farrokhi, A.; Taher, M.A. The preparation and characterization of flower-like boehmite nanoparticles-SA: A new and reusable nanocatalyst for the synthesis of 2-aryl-1H-benzimidazoles. Inorg. Nano-Met. Chem. 2017, 47, 626–631. [Google Scholar] [CrossRef]
- Park, B.K.; Lee, Y.S.; Koo, K.K. Preparation of highly porous aluminum hydroxide gels by hydrolysis of an aluminum sulfate and mineralizer. J. Ceram Process Res. 2010, 11, 64–68. [Google Scholar]
- Sumeng, Z.; Qinghe, Y.; Shuangqin, Z.; Hong, N. The form of sulfate in pseudo-boehmite and its effect on properties of pseudo-boehmite. China Pet. Process Petrochem. Technol. 2014, 16, 1–6. [Google Scholar]
- Guzman-Castillo, M.L.; Bokhimi, X.; Toledo-Antonio, A.; Salmones-Blasquez, J.; Hernandez-Beltran, F. Effect of boehmite crystallite size and steaming on alumina properties. J. Phys. Chem. B 2001, 105, 2099–2106. [Google Scholar] [CrossRef]
- Feitknecht, W. Ordnungsvorgänge bei kolloiddispersen Hydroxyden und Hydroxysalzen. Colloid Polym. Sci. 1954, 136, 52–66. [Google Scholar]
- Arenas, M.A.; Conde, A.; De Damborenea, J.J. Effect of acid traces on hydrothermal sealing of anodising layers on 2024 aluminium alloy. Electrochim. Acta 2010, 55, 8704–8708. [Google Scholar] [CrossRef]
- Bentur, S.; Marboe, E.C. Atomistic Interpretation of the Aging of Aluminium Hydroxide; Technical Report. 74; U.S. Office of Naval Research on Contract N6ONR-249(08): Washington, DC, USA, 1957. [Google Scholar]
- Gastuche, M.C.; Herbillon, A. Etude des gels dalumine-cristallisation en milieu desionise. Bull. Soc. Chim. Fr. 1962, 1962, 1404–1412. [Google Scholar]
- Wernick, S.; Pinner, R.; Sheasby, P.G. The Surface Treatment and Finishing of Aluminum and Its Alloys, 5th ed.; Finishing Publications Ltd.: Teddington, UK, 1996; Volume 2, Chapters 6 and 11. [Google Scholar]
- Dabbs, D.M.; Ramachandran, U.; Lu, S.; Liu, J.; Wang, L.Q.; Aksay, I.A. Inhibition of aluminum oxyhydroxide precipitation with citric acid. Langmuir 2005, 21, 11690–11695. [Google Scholar] [CrossRef]
- Rider, A.N.; Arnott, D.R. Durability of bonds formed between epoxy adhesive and aluminium alloy treated with phosphonate inhibitors. Surf. Interface Anal. 1996, 24, 583–590. [Google Scholar] [CrossRef]
- De Wilde, W.P.; Van Vinckenroy, G.; Tirry, L.; Cardon, A.H. Effects of the environment and curing on the strength of adhesive joints. J. Adhes. Sci. Technol. 1995, 9, 149–158. [Google Scholar] [CrossRef]
- Arrowsmith, D.J.; Moth, D.A.; Rose, S.P. The enhancement of adhesive joint strength by extending the surface of anodized aluminium. Int. J. Adhes. Adhes. 1992, 12, 67–72. [Google Scholar] [CrossRef]
- Wilson, B.A. Identification of inhibitor films on aluminium in silicate solutions. Corros. Sci. 1971, 11, 527–531. [Google Scholar] [CrossRef]
- Hunter, H.S.; Turner, P.F.; Robinson, D.L. Technical Proceedings of the Golden Jubilee Convention; Amercian Electroplaters’ Society: Detroit, MI, USA, 1959; Volume 40, pp. 220–225. [Google Scholar]
- Bogoyavlenskii, A.F.; Belov, V.T. Zh. Prikl. Khim. 1966; 39, 2368–2371.
- Ammar, I.A.; Darwish, S.; Ammar, E.A. Stability of the anodic film on aluminium in relation to the nature of some anions. Mater. Corros. 1973, 24, 200–206. [Google Scholar] [CrossRef]
- Carter, B.G. Enhanced Hydrothermal Sealing of Anodized Aluminum. U.S. Patent No. 4,288,299, 8 September 1981. [Google Scholar]
- Raj, V.; Kanagaraj, D.; Mohan, S. Effect of sealing agents on the corrosion behavior of sulphamic acid anodized alumina films. Trans. Inst. Met. Finish. 2003, 81, 83–88. [Google Scholar] [CrossRef]
- Fabregas, J.B. Process for Sealing Anodic Oxidation Layers on Aluminium Surfaces and Its Alloys. U.S. Patent 4,084,014, 11 April 1978. [Google Scholar]
- Schmitt, G.L.; Pietrzyk, D.J. Liquid chromatographic separation of inorganic anions on an alumina column. Anal. Chem. 1985, 57, 2247–2253. [Google Scholar] [CrossRef] [PubMed]
- Lederer, M.; Polcaro, C. Thin-layer chromatography of inorganic anions on aluminium oxide. J. Chromatogr. A 1973, 84, 379–386. [Google Scholar] [CrossRef]
- Schwab, G.M.; Ghosh, A.N. Inorganic Chromatography V. Announcement. As an aid of micro analysis. Angew. Chem. 1940, 53, 39–40. [Google Scholar] [CrossRef]
- Amphlett, C.B. Inorganic Ion Exchangers; Elsevier: Amsterdam, The Netherlands, 1964; p. 84. [Google Scholar]
- Clearfield, A. (Ed.) Inorganic ion Exchange Materials; CRC Press: Boca Raton, FL, USA, 1982; Volume 16. [Google Scholar]
- Paterson, R. An Introduction to Ion Exchange; Heyden: London, UK, 1970; pp. 15, 31, 90. [Google Scholar]
- Michal, J. Inorganic Chromatographic Analysis; Van Nostrand Reinhold Company: New York, NY, USA, 1973; p. 75. [Google Scholar]
- Remaley, E.G.; Robert, W.B.; Raymond, J.M. Low Temperature Sealing of Anodized Aluminum. U.S. Patent 4,045,599, 30 August 1977. [Google Scholar]
- Treiber, R.L. Method and Composition for Sealing Anodized Aluminum Surfaces. U.S. Patent 3,647,649, 7 March 1972. [Google Scholar]
- Basaly, M.A.; Suhail, K.M.; Luis, F.A. Process and Composition for Sealing Anodized Aluminum Surfaces. U.S. Patent 5,411,607, 2 May 1995. [Google Scholar]
- Dull, D.L.; Florian, B.M. Use of Rare Earth Metal Salt Solutions for Sealing or Anodized Aluminum for Corosion Protection and Paint Adhesion. U.S. Patent 6,248,184, 19 June 2001. [Google Scholar]
- Žutić, V.; Stumm, W. Effect of organic acids and fluoride on the dissolution kinetics of hydrous alumina. A model study using the rotating disc electrode. Geochim. Cosmochim. Acta 1984, 48, 1493–1503. [Google Scholar] [CrossRef]
- Pechenyuk, S.I.; Semushina, Y.P.; Kuz’mich, L.F. Adsorption affinity of anions on metal oxyhydroxides. Russ. J. Phys. Chem. A 2013, 87, 490–496. [Google Scholar] [CrossRef]
- Missana, T.; Benedicto, A.; Mayordomo, N.; Alonso, U. Analysis of anion adsorption effects on alumina nanoparticles stability. Appl. Geochem. 2014, 49, 68–76. [Google Scholar] [CrossRef]
- Deltombe, E.; Pourbaix, M. The electrochemical behavior of aluminum—Potential pH diagram of the system AI-H2O at 25 °C. Corrosion 1958, 14, 16–20. [Google Scholar] [CrossRef]
- Kalantary, M.R. Development of Cold Sealing Processes for Anodized Aluminium. Ph.D. Thesis, Loughborough University, Loughborough, UK, February 1990. Available online: https://dspace.lboro.ac.uk/2134/13910 (accessed on 20 September 2019).
- Forgacs, Z.; Massányi, P.; Lukac, N.; Somosy, Z. Reproductive toxicology of nickel–review. J. Environ. Sci. Health A Tox. Hazard. Subst. Environ. Eng. 2012, 47, 1249–1260. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, X.; Castranova, V.; Ding, M. Occupational toxicology of nickel and nickel compounds. J. Environ. Pathol. Toxicol. Oncol. 2009, 28, 177–208. [Google Scholar] [CrossRef]
- Davidson, T.; Ke, Q.; Max Costa, M. Selected Molecular Mechanisms of Metal Toxicity and Carcinogenicity. In Handbook on the Toxicology of Metals, 3rd ed.; Nordberg, G.F., Fowler, B.A., Nordberg, M., Friberg, L.T., Eds.; Academic Press, Inc.: Cambridge, MA, USA, 2007; pp. 79–95. [Google Scholar]
- Rubin, A.J.; Hayden, P.L. Studies on the Hydrolysis and Precipitation of Aluminum (III). 1973. Available online: https://kb.osu.edu/bitstream/handle/1811/36328/3/OH_WRC_364X%5B1%5D.pdf (accessed on 20 October 2019).
- Russell, A.S.; Edwards, J.D.; Taylor, C.S. Solubility and density of hydrated aluminas in NaOH solutions. JOM-US 1955, 7, 1123–1128. [Google Scholar] [CrossRef]
- Apps, J.A.; Neil, J.M.; Jun, C.H. Thermochemical Properties of Gibbsite, Bayerite, Boehmite, Diaspore, and the Aluminate Ion between 0 and 350 °C; Lawrence Berkeley Laboratory Report; Nuclear Regulatory Commission: Washington, DC, USA, 1989; p. 21482.
- Verdes, G.; Gout, R.; Castet, S. Thermodynamic properties of the aluminate ion and of bayerite, boehmite, diaspore and gibbsite. Eur. J. Mineral. 1992, 767–792. [Google Scholar] [CrossRef]
- Castet, S.; Dandurand, J.L.; Schott, J.; Gout, R. Boehmite solubility and aqueous aluminum speciation in hydrothermal solutions (90–350 °C): Experimental study and modeling. Geochim. Cosmochim. Acta 1993, 57, 4869–4884. [Google Scholar] [CrossRef]
- Bénézeth, P.; Palmer, D.A.; Wesolowski, D.J. Aqueous high-temperature solubility studies. II. The solubility of boehmite at 0.03 m ionic strength as a function of temperature and pH as determined by in situ measurements. Geochim. Cosmochim. Acta 2001, 65, 2097–2111. [Google Scholar] [CrossRef]
- Hem, J.D.; Roberson, C.E. Form and Stability of Aluminum Hydroxide Complexes in Dilute Solution; Water-Supply Paper 1827-A; United States Geological Survey: Washington, DC, USA, 1967. Available online: https://pubs.usgs.gov/wsp/1827a/report.pdf (accessed on 11 November 2019).
- Hwang, C.S.; Lin, W.H. Preparation and sinterability of zirconia-toughened-alumina composite powder. J. Ceram. Soc. Jpn. 1991, 99, 271–275. [Google Scholar] [CrossRef][Green Version]
- Okada, K.; Nagashima, T.; Kameshima, Y.; Yasumori, A. Effect of crystallite size of boehmite on sinterability of alumina ceramics. Ceram. Int. 2003, 29, 533–537. [Google Scholar] [CrossRef]
- Kaur, P.; Singh, G.P.; Kaur, S.; Singh, D.P. Modifier role of cerium in lithium aluminium borate glasses. J. Mol. Struct. 2012, 1020, 83–87. [Google Scholar] [CrossRef]
- Gutierrez, G. Atomistic simulation of densified amorphous alumina. Rev. Mex. Fís. 2002, 48 (Suppl. 3), 60–62. [Google Scholar]
- Lizárraga, R.; Holmström, E.; Parker, S.C.; Arrouvel, C. Structural characterization of amorphous alumina and its polymorphs from first-principles XPS and NMR calculations. Phys. Rev. B 2011, 83. [Google Scholar] [CrossRef]
- Koski, K.; Hölsä, J.; Juliet, P. Properties of aluminium oxide thin films deposited by reactive magnetron sputtering. Thin Solid Films 1999, 339, 240–248. [Google Scholar] [CrossRef]
- Kaledin, L.A.; Tepper, F.; Kaledin, T.G. Pristine point of zero charge (ppzc) and zeta potentials of boehmite’s nanolayer and nanofiber surfaces. Int. J. Smart Nano Mater. 2016, 7, 1–21. [Google Scholar] [CrossRef]
- Bohnstedt-Kupletskaya, E.M.; Veodavetz, N.I. Boehmite from the nepheline pegmatites of the Vishnevye Mountains in the central Urals. Comptes Rendus L’académie Sciences l’URSS 1945, 49, 587–589. [Google Scholar]
- Dana, J.D.; Dana, E.S.; Palache, C.; Berman, H.M.; Frondel, C. The System of Mineralogy of James Dwight Dana and Edward Salisbury Dana, Yale University, (No. 549); John Wiley and Sons, Inc.: Hoboken, NJ, USA, 1944; pp. 1837–1892. [Google Scholar]
- Montero, V. Kristallstruktur des Bayerit. Ricerca Sci. 1942, 13, 565–571. [Google Scholar]
- Clark, G.R.; Rodgers, K.A.; Henderson, G.S. The crystal chemistry of doyleite, Al(OH)3. Z Kristallogr Cryst Mater. 1998, 213, 96–100. [Google Scholar] [CrossRef]
- Cardarelli, F. Materials Handbook: A Concise Desktop Reference; Springer Science Business Media: Berlin/Heidelberg, Germany, 2008; p. 897. [Google Scholar]
- Aeronautica, S.A. Anodizing Process, with Low Environmental Impact, for a Workpiece of alUminium or Aluminium Alloys. Euopean Patent AP1233084A2, 21 August 2002. [Google Scholar]
- Ocon Esteban, P.; Garcia Rubio, M.; Garcia Diego, I.; Lavia Gonzalez-escalada, M.A. Procedure for Anodising Aluminium or Aluminium Alloys. U.S. Patent Application 12/0732009,632, 30 April 2009. [Google Scholar]
- García-Rubio, M.; Ocón, P.; Climent-Font, A.; Smith, R.W.; Curioni, M.; Thompson, G.E.; Skeldon, P.; Lavía, A.; García, I. Influence of molybdate species on the tartaric acid/sulphuric acid anodic films grown on AA2024 T3 aerospace alloy. Corros. Sci. 2009, 51, 2034–2042. [Google Scholar] [CrossRef]
- García Rubio, M. Optimisation of a Non-Chromium-Containing Tartaric Acid/Sulphuric Acid Anodising Bath for Aluminium Alloys for Aerospace Industry Application. Ph.D. Thesis, Universidad Autónoma de Madrid, Madrid, Spain, October 2009. Available online: http://hdl.handle.net/10486/14205 (accessed on 4 December 2019).
- Nagabushan, B.J.; Yoganandan, G. A Process for the Preparation of Corrosion Resistance Sealed Anodized Coatings on Aluminum Alloy. U.S. Patent Application 15/543,153, 4 January 2018. [Google Scholar]
- Deshmukh, G.S.; Nayak, K.V. On the solubility of rare earth hydroxides in aqueous sodium potassium tartrate. Proc. Indian Acad. Sci.-Sect. A 1951, 34, 316. [Google Scholar] [CrossRef]
- Mellor, J.W. A Comprehensive Treatise on Inorganic and Theoretical Chemistry; Longmans Green and Company: New York, NY, USA, 1924; Volume 5, p. 570. [Google Scholar]
- Thomson, T. XLVIII. Experiments on allanite a new mineral from greenland. Philos. Mag. 1811, 37, 278–288. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, A.; Wang, X.; Zhang, T. Mesoporous γ-alumina synthesized by hydro-carboxylic acid as structure-directing agent. Microporous Mesoporous Mater. 2006, 92, 10–21. [Google Scholar] [CrossRef]
- Thomas, A.W.; Vartanian, R.D. The Action of Acids upon Hydrous Alumina. J. Am. Chem. Soc. 1935, 57, 4–7. [Google Scholar] [CrossRef]
- Sen, K.C. On the theory of peptization. J. Phys. Chem. 1925, 29, 1533–1547. [Google Scholar] [CrossRef]
- Tregubenko, V.Y.; Udras, I.E.; Belyi, A.S. Preparation of Mesoporous γ-Al2O3 from Aluminum Hydroxide Peptized with Organic Acids. Russ. J. Appl. Chem. 2017, 90, 1961–1968. [Google Scholar] [CrossRef]
- Purwani, M.V.; Trinopiawan, K.; Poernomo, H.; Pusporini, N.D.; Amiliana, R.A. Separation of Ce, La and Nd in Rare Earth Hydroxide (REOH) by Oxidation with Potassium Permanganate and Precipitation. J. Phys. Conf. Ser. 2019, 11988, 032003. [Google Scholar] [CrossRef]
- McNeice, J.; Ghahreman, A. Selective oxidation of cerium in rare earth solutions, a comparison of four oxidants. In TMS Annual Meeting Exhibition; Springer: Cham, Switzerland, 2018; pp. 15–25. [Google Scholar] [CrossRef]
- Itagaki, M.; Suzuki, T.; Watanabe, K. Channel flow double electrode study on anodic dissolution of molybdenum in sulfuric acid solution. Electrochim. Acta 1997, 42, 1081–1086. [Google Scholar] [CrossRef]
- Gupta, C.K.; Krishnamurthy, N. Extractive Metallurgy of Rare Earths; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Kedari, C.S.; Pandit, S.S.; Ramanujam, A. Studies on the in situ electrooxidation and selective permeation of cerium (IV) across a bulk liquid membrane containing tributyl phosphate as the ion transporter. Sep. Sci. Technol. 1999, 34, 1907–1923. [Google Scholar] [CrossRef]
- Yu, X.; Yan, C.; Cao, C. Study on the rare earth sealing procedure of the porous film of anodized Al6061/SiCp. Mater. Chem. Phys. 2002, 76, 228–235. [Google Scholar] [CrossRef]
- Jiang, C.C.; Cao, Y.K.; Xiao, G.Y.; Zhu, R.F.; Lu, Y.P. A review on the application of inorganic nanoparticles in chemical surface coatings on metallic substrates. RSC Adv. 2017, 7, 7531–7539. [Google Scholar] [CrossRef]
- Domingo, J.L.; Gomez, M.; Sanchez, D.J.; Llobet, J.M.; Keen, C.L. Toxicology of vanadium compounds in diabetic rats: The action of chelating agents on vanadium accumulation. Mol. Cell. Biochem. 1995, 153, 233–240. [Google Scholar] [CrossRef]
- Domingo, J.L. Vanadium: A review of the reproductive and developmental toxicity. Reprod. Toxicol. 1996, 10, 175–182. [Google Scholar] [CrossRef]
- Aureliano, M.; Ohlin, C.A. Decavanadate in vitro and in vivo effects: Facts and opinions. J. Inorg. Biochem. 2014, 137, 123–130. [Google Scholar] [CrossRef]
- Assem, F.L.; Levy, L.S. A review of current toxicological concerns on vanadium pentoxide and other vanadium compounds: Gaps in knowledge and directions for future research. J. Toxicol. Environ. Health B Crit. Rev. 2009, 12, 289–306. [Google Scholar] [CrossRef]
- Li, H.; Zhou, D.; Zhang, Q.; Feng, C.; Zheng, W.; He, K.; Lan, Y. Vanadium exposure-induced neurobehavioral alterations among Chinese workers. Neurotoxicology 2013, 36, 49–54. [Google Scholar] [CrossRef]
- Ehrlich, V.A.; Nersesyan, A.K.; Hoelzl, C.; Ferk, F.; Bichler, J.; Valic, E.; Schaffer, A.; Schulte-Hermann, R.; Fenech, M.; Wagner, K.; et al. Inhalative exposure to vanadium pentoxide causes DNA damage in workers: Results of a multiple end point study. Environ. Health Perspect. 2008, 116, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Ress, N.B.; Chou, B.J.; Renne, R.A.; Dill, J.A.; Miller, R.A.; Roycroft, J.H.; Hailey, J.R.; Haseman, J.K.; Bucher, J.R. Carcinogenicity of inhaled vanadium pentoxide in F344/N rats and B6C3F1 mice. Toxicol. Sci. 2003, 74, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Duffus, J.H. Carcinogenicity classification of vanadium pentoxide and inorganic vanadium compounds, the NTP study of carcinogenicity of inhaled vanadium pentoxide, and vanadium chemistry. Regul. Toxicol. Pharmacol. 2007, 47, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, M.C.; Straif, K.; Siemiatycki, J. IARC carcinogen update. Environ. Health Perspect. 2005, 113, A580–A581. [Google Scholar] [CrossRef] [PubMed]
- Galanis, A.; Karapetsas, A.; Sandaltzopoulos, R. Metal-induced carcinogenesis, oxidative stress and hypoxia signalling. Mutat Res. Genet. Toxicol Environ. Mutagen. 2009, 674, 31–35. [Google Scholar] [CrossRef]
- Fortoul, T.I.; Rodriguez-Lara, V.; González-Villalva, A.; Rojas-Lemus, M.; Cano-Gutiérrez, G.; Ustarroz-Cano, M.; Jimenez-Martinez, R.S. Inhalation of vanadium pentoxide and its toxic effects in a mouse model. Inorg. Chim. Acta 2014, 420, 8–15. [Google Scholar] [CrossRef]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Vanadium. (Draft for Public Comment); U.S. Department of Health and Human Services, Public Health Service: Atlanta, GA, USA, 2009.
- Howd, R.A. Proposed Notification Level for Vanadium; California Office of Environmental Health Hazard Assessment: Sacramento, CA, USA, 2008.
- Uhlig, H.H.; King, P.F. The flade potential of iron passivated by various inorganic corrosion inhibitors. J. Electrochem. Soc. 1959, 106, 1–7. [Google Scholar] [CrossRef]
- Devasenapathi, A.; Raja, V.S. Effect of externally added molybdate on repassivation and stress corrosion cracking of type 304 stainless steel in hydrochloric acid. Corrosion 1996, 52, 243–249. [Google Scholar] [CrossRef]
- Mu, G.; Li, X.; Qu, Q.; Zhou, J. Molybdate and tungstate as corrosion inhibitors for cold rolling steel in hydrochloric acid solution. Corros. Sci. 2006, 48, 445–459. [Google Scholar] [CrossRef]
- Qu, Q.; Li, L.; Bai, W.; Jiang, S.; Ding, Z. Sodium tungstate as a corrosion inhibitor of cold rolled steel in peracetic acid solution. Corros. Sci. 2009, 51, 2423–2428. [Google Scholar] [CrossRef]
- Sastri, V.S.; Tjan, C.; Roberge, P.R. Corrosion inhibition by tungstate in aqueous solutions and its application to coal–water slurries. Br. Corros. J. 1991, 26, 251–254. [Google Scholar] [CrossRef]
- Fujioka, E.; Nishihara, H.; Aramaki, K. The inhibition of pit nucleation and growth on the passive surface of iron in a borate buffer solution containing Cl− by oxidizing inhibitors. Corros. Sci. 1996, 38, 1915–1933. [Google Scholar] [CrossRef]
- Jabeera, B.; Shibli, S.M.A.; Anirudhan, T.S. Synergistic inhibitive effect of tartarate and tungstate in preventing steel corrosion in aqueous media. Appl. Surf. Sci. 2006, 252, 3520–3524. [Google Scholar] [CrossRef]
- Hu, J.; Wang, T.; Wang, Z.; Wei, L.; Zhu, J.; Zheng, M.; Chen, Z. Corrosion protection of N80 steel in hydrochloric acid medium using mixed C15H15NO and Na2WO4 inhibitors. Coatings 2018, 8, 315. [Google Scholar] [CrossRef]
- Monticelli, C.; Zucchi, F.; Brunoro, G.; Trabanelli, G. Corrosion and corrosion inhibition of alumina particulate/aluminium alloys metal matrix composites in neutral chloride solutions. J. Appl. Electrochem. 1997, 27, 325–334. [Google Scholar] [CrossRef]
- Sarangapani, K.B.; Balaramachandran, V.; Kapali, V.; Iyer, S.V.; Potdar, M.G.; Rajagopalan, K.S. Aluminium as anode in primary alkaline batteries. Influence of additives on the corrosion and anodic behavior of 2S aluminium in alkaline citrate solution. J. Appl. Electrochem. 1984, 14, 475–480. [Google Scholar] [CrossRef]
- Lomakina, S.V.; Shatova, T.S.; Kazansky, L.P. Heteropoly anions as corrosion inhibitors for aluminium in high temperature water. Corros. Sci. 1994, 36, 1645–1651. [Google Scholar] [CrossRef]
- Silva, J.W.J.; Codaro, E.N.; Nakazato, R.Z.; Hein, L.R.O. Influence of chromate, molybdate and tungstate on pit formation in chloride medium. Appl. Surf. Sci. 2005, 252, 1117–1122. [Google Scholar] [CrossRef]
- Haneke, K.E.; Carson, B.L.; Gregorio, C.A.; Emanuel, A.; Wood, M.R. Tungsten and Selected Tungsten Compounds—Review of Toxicological Literature. National Institute of Environmental Health Sciences. 2003. Available online: https://ntp.niehs.nih.gov/ntp/htdocs/chem_background/exsumpdf/tungsten_508.pdf (accessed on 5 November 2019).
- Wasel, O.; Freeman, J.L. Comparative Assessment of Tungsten Toxicity in the Absence or Presence of Other Metals. Toxics 2018, 6, 66. [Google Scholar] [CrossRef]
- Van der Voet, G.B.; Todorov, T.I.; Centeno, J.A.; Jonas, W.; Ives, J.; Mullick, F.G. Metals and health: A clinical toxicological perspective on tungsten and review of the literature. Mil. Med. 2007, 172, 1002–1005. [Google Scholar] [CrossRef]
- Keith, S. Toxicological Profile for Tungsten; Agency for Toxic Substances and Disease Registry: Atlanta, CA, USA, 2005.
- Cheraghi, G.; Hajiabedi, E.; Niaghi, B.; Nazari, F.; Naserzadeh, P.; Hosseini, M.J. High doses of sodium tungstate can promote mitochondrial dysfunction and oxidative stress in isolated mitochondria. J. Biochem. Mol. Toxicol. 2019, 33, e22266. [Google Scholar] [CrossRef] [PubMed]
- Zoroddu, M.A.; Medici, S.; Peana, M.; Nurchi, V.M.; Lachowicz, J.I.; Laulicht-Glickc, F.; Costa, M. Tungsten or Wolfram: Friend or foe? Curr. Med. Chem. 2018, 25, 65–74. [Google Scholar] [CrossRef]
- Bolt, A.M.; Mann, K.K. Tungsten: An emerging toxicant, alone or in combination. Curr. Environ. Health Rep. 2016, 3, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Lemus, R.; Venezia, C.F. An update to the toxicological profile for water-soluble and sparingly soluble tungsten substances. Crit. Rev. Toxicol. 2015, 45, 388–411. [Google Scholar] [CrossRef] [PubMed]
- USEPA Provisional Peer-Reviewed Toxicity Values for Soluble Tungsten Compounds (Various CASRNs) United States Environmental Protection Agency (EPA/690/R-15/015F) Final 9-29-2015. Available online: https://cfpub.epa.gov/ncea/pprtv/documents/Tungsten.pdf (accessed on 4 November 2019).
- Keith, S.; Moffett, D.; Rosemond, Z.A.; Wohlers, D. Addendum to the Toxicological Profile for Tungsten; ATSDR: Atlanta, GA, USA, 2015. [Google Scholar]
- Tyrrell2015, J.; Galloway, T.S.; Abo-Zaid, G.; Melzer, D.; Depledge, M.H.; Osborne, N.J. High urinary tungsten concentration is associated with stroke in the National Health and Nutrition Examination Survey 1999–2010. PLoS ONE 2013, 8, e77546. [Google Scholar] [CrossRef]
- Kennedy, A.J.; Johnson, D.R.; Seiter, J.M.; Lindsay, J.H.; Boyd, R.E.; Bednar, A.J.; Allison, P.G. Tungsten toxicity, bioaccumulation, and compartmentalization into organisms representing two trophic levels. Environ. Sci. Technol. 2012, 46, 9646–9652. [Google Scholar] [CrossRef] [PubMed]
- Thomas, V.G.; Roberts, M.J.; Harrison, P.T. Assessment of the environmental toxicity and carcinogenicity of tungsten-based shot. Ecotox. Environ. Saf. 2009, 72, 1031–1037. [Google Scholar] [CrossRef]
- Schell, J.D.; Pardus, M.J. Preliminary risk-based concentrations for tungsten in soil and drinking water. Land Contam. Reclamat. 2009, 17, 179–188. [Google Scholar] [CrossRef]
- Pardus, M.J.; Lemus-Olalde, R.; Hepler, D.R. Tungsten human toxicity: A compendium of research on metallic tungsten and tungsten compounds. Land Contam. Reclamat. 2009, 17, 217–222. [Google Scholar] [CrossRef]
- Clausen, J.L.; Korte, N. Environmental fate of tungsten from military use. Sci. Total Environ. 2009, 407, 2887–2893. [Google Scholar] [CrossRef]
- Lavin, R.P. Histologic Changes as Indicators of Carcinogenicity of Tungsten Alloy in Rodents; Uniformed Services University of The Health Sciences: Bethesda, MD, USA, 2005. [Google Scholar]
- Keith2008, L.S.; Wohlers, D.W.; Moffett, D.B.; Rosemond, Z.A. ATSDR evaluation of potential for human exposure to tungsten. Toxicol. Ind. Health 2007, 23, 309–345. [Google Scholar] [CrossRef] [PubMed]
- Koutsospyros, A.; Braida, W.; Christodoulatos, C.; Dermatas, D.; Strigul, N. A review of tungsten: From environmental obscurity to scrutiny. J. Hazard. Mater. 2006, 136, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Strigul, N.; Koutsospyros, A.; Arienti, P.; Christodoulatos, C.; Dermatas, D.; Braida, W. Effects of tungsten on environmental systems. Chemosphere 2005, 61, 248–258. [Google Scholar] [CrossRef]
- Strigul, N.; Koutsospyros, A.; Christodoulatos, C. Tungsten speciation and toxicity: Acute toxicity of mono-and poly-tungstates to fish. Ecotox. Environ. Saf. 2010, 73, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Osterburg, A.R.; Robinson, C.T.; Schwemberger, S.; Mokashi, V.; Stockelman, M.; Babcock, G.F. Sodium tungstate (Na2WO4) exposure increases apoptosis in human peripheral blood lymphocytes. J. Immunotoxicol. 2010, 7, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Marquet, P.; Francois, B.; Vignon, P.; Lachâtre, G. A soldier who had seizures after drinking quarter of a litre of wine. Lancet 1996, 348, 1070. [Google Scholar] [CrossRef]
- Lison, D.; Bucket, J.P.; Hoet, P. Toxicity of tungsten. Lancet 1997, 349, 58. [Google Scholar] [CrossRef]
- Zhao, F.W.; Li, X.; Graham, N. The application of potassium manganate in water treatment. Water Sci Technol. Water Supply 2011, 11, 612–620. [Google Scholar] [CrossRef]
- Szlag, D.C.; Spies, B.; Szlag, R.G.; Westrick, J.A. Permanganate Oxidation of Microcystin-LA: Kinetics, Quantification, and Implications for Effective Drinking Water Treatment. J. Toxicol. 2019, 2019. [Google Scholar] [CrossRef]
- He, X.; Liu, Y.L.; Conklin, A.; Westrick, J.; Weavers, L.K.; Dionysiou, D.D.; Lenhart, J.J.; Mouser, P.J.; Szlag, D.; Walker, H.W. Toxic cyanobacteria and drinking water: Impacts, detection, and treatment. Harmful Algae 2016, 54, 174–193. [Google Scholar] [CrossRef]
- Xie, P.; Ma, J.; Fang, J.; Guan, Y.; Yue, S.; Li, X.; Chen, L. Comparison of permanganate preoxidation and preozonation on algae containing water: Cell integrity, characteristics, and chlorinated disinfection byproduct formation. Environ. Sci. Technol. 2013, 47, 14051–14061. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, G.B.; Chen, Z.L.; Xu, G.R.; Cai, G.Q. Enhanced coagulation of surface waters with high organic content by permangante preoxidation. Water Sci. Technol. Water Supply 2001, 1, 51–61. [Google Scholar] [CrossRef]
- Hidayah, E.N.; Yeh, H.H. Effect of Permanganate Preoxidation to Natural Organic Matter and Disinfection by-Products Formation Potential Removal. J. Phys. Conf. Ser. 2018, 953, 012218. [Google Scholar] [CrossRef]
- Welch, W.A. Potassium permanganate in water treatment. J. Am. Water Works Assoc. 1963, 55, 735–741. [Google Scholar] [CrossRef]
- Ficek, K.J. Raw Water Reservoir Treatment with Potassium Permanganate. In Proceedings of the 74th Annual Meeting Illinois Section American Water Works Association, Chicago, IL, USA, 24 March 1983. [Google Scholar]
- Kemp, H.T.; Fuller, R.G.; Davidson, R.S. Potassium permanganate as an algicide. J. Am. Water Works Assoc. 1966, 58, 255–263. [Google Scholar] [CrossRef]
- Fitzgerald, G.P. Evaluation of potassium permanganate as an algicide for water cooling towers. Ind. Eng. Chem. Prod. Res. Dev. 1964, 3, 82–85. [Google Scholar] [CrossRef]
- Chen, J.J.; Yeh, H.H. The mechanisms of potassium permanganate on algae removal. Water Res. 2005, 39, 4420–4428. [Google Scholar] [CrossRef]
- Wilde, E.W.; McLaughlin, B.D. Selecting an algicide for use with aluminum alloys. Water Res. 1981, 15, 1117–1124. [Google Scholar] [CrossRef]
- Yahya, M.T.; Landeen, L.K.; Forsthoefel, N.R.; Kujawa, K.; Gerba, C.P. Evaluation of potassium permanganate for inactivation of bacteriophage MS-2 in water systems. J. Environ. Sci. Health Part A Tox. Hazard. Subst. Environ. Eng. 1989, 24, 979–989. [Google Scholar] [CrossRef]
- Yahya, M.T.; Landeen, L.K.; Gerba, C.P. Inactivation of Legionella pneumophila by potassium permanganate. Environ. Technol. 1990, 11, 657–662. [Google Scholar] [CrossRef]
- Darwish, A.M.; Mitchell, A.J.; Hobbs, M.S. In vitro and in vivo evaluation of potassium permanganate treatment efficacy for the control of acute experimental infection by Flavobacterium columnare in channel catfish. N. Am. J. Aquac. 2008, 70, 314–322. [Google Scholar] [CrossRef]
- Darwish, A.M.; Mitchell, A.J.; Straus, D.L. Evaluation of potassium permanganate against an experimental subacute infection of Flavobacterium columnare in channel catfish, Ictalurus punctatus (Rafinesque). J. Fish. Dis. 2009, 32, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Tucker, C.S.; Boyd, C.E. Relationships between potassium permanganate treatment and water quality. Trans. Am. Fish. Soc. 1977, 106, 481–488. [Google Scholar] [CrossRef]
- Tucker, C.S. Potassium permanganate demand of pond waters. Prog. Fish.-Cult. 1984, 46, 24–28. [Google Scholar] [CrossRef]
- Jee, L.K.; Plumb, J.A. Effects of organic load on potassium permanganate as a treatment for Flexibacter columnaris. Trans. Am. Fish. Soc. 1981, 110, 86–89. [Google Scholar] [CrossRef]
- Deichmann, W.B.; Gerarde, H.W. Toxicology of Drugs and Chemicals; Academic Press: New York, NY, USA, 1969. [Google Scholar]
- Evaluation Report for Potassium Permanganate (CAS No 7722-64-7) EC No: 231-760-3. 30 November 2018. Available online: https://echa.europa.eu/documents/10162/f91eb21d-12bb-7a7a-9708-9534f87c3440 (accessed on 7 November 2019).
- Ong, K.L.; Tan, T.H.; Cheung, W.L. Potassium permanganate poisoning—A rare cause of fatal self-poisoning. Emerg. Med. J. 1997, 14, 43–45. [Google Scholar] [CrossRef]
- Young, R.J.; Critchley, J.A.J.H.; Young, K.K.; Freebairn, R.C.; Reynolds, A.P.; Lolin, Y.I. Fatal acute hepatorenal failure following potassium permanganate ingestion. Hum. Exp. Toxicol. 1996, 15, 259–261. [Google Scholar] [CrossRef]
- Karthik, R.; Veerendranath, H.P.K.; Wali, S.; Mohan, M.N.; Kumar, P.A.; Trimurty, G. Suicidal ingestion of potassium permanganate crystals: A rare encounter. Toxicol. Int. 2014, 21, 331. [Google Scholar] [CrossRef][Green Version]
- Agrawal, V.K.; Bansal, A.; Kumar, R.; Kumawat, B.L.; Mahajan, P. Potassium permanganate toxicity: A rare case with difficult airway management and hepatic damage. Indian J. Crit. Care Med. 2014, 18, 819–821. [Google Scholar] [CrossRef]
- Nadikuda, S.K.; Venkategowda, P.M.; Rao, S.M.; Kondala, R. Effective Approach to Potassium Permanganate Poisoning: Case Report and Review of Literature. Asia Pac. J. Med. Toxicol. 2015, 4, 127–130. [Google Scholar]
- Shaabani, A.; Tavasoli-Rad, F.; Lee, D.G. Potassium permanganate oxidation of organic compounds. Synth. Commun. 2005, 35, 571–580. [Google Scholar] [CrossRef]
- Spicher, R.G.; Skrinde, R.T. Potassium permanganate oxidation of organic contaminants in water supplies. J. Am. Water Works Assoc. 1963, 55, 1174–1194. [Google Scholar] [CrossRef]
- Ma, X.; Hu, S.; Wang, H.; Li, J.; Huang, J.; Zhang, Y.; Lu, W.; Li, Q. Kinetics of oxidation of dimethyl trisulfide by potassium permanganate in drinking water. Front. Environ. Sci. Eng. 2012, 6, 171–176. [Google Scholar] [CrossRef]
- Pal, P. Chemical Treatment Technology. In Industrial Water Treatment Process Technology, Industrial Water Treatment Process Technology; Butterworth-Heinemann: Oxford, UK, 2017; pp. 21–63. [Google Scholar] [CrossRef]
- Montiel, A.J. Municipal drinking water treatment procedures for taste and odour abatement—A review. Water Sci. Technol. 1983, 15, 279–289. [Google Scholar] [CrossRef]
- Li, W.G.; Gao, Y.N.; Huang, X.D.; Sun, S.Q.; Zhang, J.S. Study on Taste and Odor Removal in Drinking Water by Potassium Permanganate Combined with Powdered Activated Carbon. China Water Wastewater 2007, 23, 18. [Google Scholar]
- Dietrich, A.M.; Hoehn, R.C.; Dufresne, L.C.; Buffin, L.W.; Rashash, D.M.C.; Parker, B.C. Oxidation of odorous and non-odorous algal metabolites by permanganate, chlorine, and chlorine dioxide. Water Sci. Technol. 1995, 31, 223–228. [Google Scholar] [CrossRef]
- Cherry, A.K. Use of potassium permanganate in water treatment. J. Am. Water Works Assoc. 1962, 54, 417–424. [Google Scholar] [CrossRef]
- Jung, S.W.; Baek, K.H.; Yu, M.J. Treatment of taste and odor material by oxidation and adsorption. Water Sci. Technol. 2004, 49, 289–295. [Google Scholar] [CrossRef]
- McGuire, M.J. Advances in treatment processes to solve off-flavor problems in drinking water. Water Sci. Technol. 1999, 40, 153–163. [Google Scholar] [CrossRef]
- Bruchet, A.; Duguet, J.P. Role of oxidants and disinfectants on the removal, masking and generation of tastes and odours. Water Sci. Technol. 2004, 49, 297–306. [Google Scholar] [CrossRef]
- Boland, J.J.; DeArment, W.E. Potassium permanganate removal of tastes and odors from paper mill wastes. J. Am. Water Works Assoc. 1965, 57, 1451–1455. [Google Scholar] [CrossRef]
- He, Q.; Wang, H.; Zhong, L.; Yang, K.; Zou, Z. The use of potassium permanganate, ozone and associated coupled processes for odor removal in drinking water: Bench and pilot scale tests. J. Water Supply Res. Technol.—Aqua 2017, 66, 249–256. [Google Scholar] [CrossRef]
- Vyskočil, A.; Viau, C. Assessment of molybdenum toxicity in humans. J. Appl. Toxicol. 1999, 19, 185–192. [Google Scholar] [CrossRef]
- Hodge, A.; Sterner, B. Toxicity Classes. Canadian Center for Occupational Health and Safety. 2005. Available online: http://www.ccohs.ca/oshanswers/chemicals/id50.htm (accessed on 5 October 2019).
- Williams, R.J.P. The Biological Role of Molybdenum; Climax Molybdenum Co. Ltd.: London, UK, 1978. [Google Scholar]
- Farnsworth, C.G. Study of molybdenum in public water supply. Water Sewage Works 1970, 117, 418–421. [Google Scholar]
- Lehman, A.J.; Patterson, W.I.; Davidow, B.; Hagan, E.C. Procedures for the appraisal of the toxicity of chemicals in foods, drugs and cosmetics. Food Drug Cosmet. Law J. 1955, 10, 679–748. [Google Scholar]
- Fairhall, L.T.; Dunn, R.C.; Sharpless, N.E.; Pritchard, E.A. The Toxicity of Molybdenum; U.S. Public Health Service. Public Health Bulletin. U.S. Government Printing Office: Washington, DC, USA, 1945; Volume 293, pp. 5–7.
- United States Environmental Protection Agency (USEPA) Inert Reassessment Document for Sodium Molybdate—CAS No. 7631-95-0. Available online: https://www.epa.gov/ingredients-used-pesticide-products/inert-reassessment-document-sodium-molybdate-cas-no-7631-95-0 (accessed on 12 November 2019).
- IOM/NAS; Food and Nutation Board (FNB); Institute of Medicine (IOM). Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; National Academies Press: Washington, DC, USA, 2000; Available online: http://books.napedu/catalog/100026.html (accessed on 20 November 2019).
- Trumbo, P.; Yates, A.A.; Schlicker, S.; Poos, M. Dietary reference intakes: Vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J. Acad. Nutr. Diet. 2001, 101, 294–301. [Google Scholar]
- World Health Organization (WHO). Molybdenum in Drinking Water. Background Document for Development of WHO Guidelines for Drinking Water Quality. (WHO/SDE/WSH/03.04/11/Rev/1). 2011. Available online: https://www.who.int/water_sanitation_health/dwq/chemicals/molybdenum.pdf (accessed on 12 November 2019).
- World Health Organization. Molybdenum in Drinking-Water: Background Document for Development of WHO Guidelines for Drinking-wAter Quality (No. WHO/SDE/WSH/03.04/11). World Health Organization. 2003. Available online: https://apps.who.int/iris/bitstream/handle/10665/75372/WHO_SDE_WSH_03.04_11_eng.pdf (accessed on 10 November 2019).
- Frisbie, S.H.; Mitchell, E.J.; Sarkar, B. Urgent need to reevaluate the latest World Health Organization guidelines for toxic inorganic substances in drinking water. Environ. Health 2015, 14, 63. [Google Scholar] [CrossRef]
- Smedley, P.L.; Kinniburgh, D.G. Molybdenum in natural waters: A review of occurrence, distributions and controls. Appl. Geochem. 2017, 84, 387–432. [Google Scholar] [CrossRef]
- Heijerick, D.G.; Regoli, L.; Stubblefield, W. The chronic toxicity of molybdate to marine organisms. I. Generating reliable effects data. Sci. Total Environ. 2012, 430, 260–269. [Google Scholar] [CrossRef]
- Heijerick, D.G.; Carey, S. The toxicity of molybdate to freshwater and marine organisms. III. Generating additional chronic toxicity data for the refinement of safe environmental exposure concentrations in the US and Europe. Sci. Total Environ. 2017, 609, 420–428. [Google Scholar] [CrossRef]
- Wang, C.W.; Liang, C.; Yeh, H.J. Aquatic acute toxicity assessments of molybdenum (+VI) to Daphnia magna. Chemosphere 2016, 147, 82–87. [Google Scholar] [CrossRef] [PubMed]
- 17 CAS No.: 7439-98-7 (Molybdenum) Substance Substance: Molybdenum and Its Compounds. Chemical Substances Control Law Reference No.: PRTR Law Cabinet Order No.*: 1–453 (Molybdenum and Its Compounds) Revised Cabinet Order Enacted on 1 October 2009. Available online: https://www.env.go.jp/en/chemi/chemicals/profile_erac/profile10/pf1-17.pdf (accessed on 12 November 2019).
- Rey, S.P. Molybdate and Non-Molybdate Options for Closed Systems. Molybdate and Non-Molybdate Options for Closed Systems Association of Water Technologies (AWT). 2005. Available online: http://www.natcoll.com/wp-content/uploads/Molybdate-and-Non-Molybdate-Options-for-Closed-Systems.pdf (accessed on 6 October 2019).
- Pham, H.U.; Cha, H. The Comparative Toxicity of Sodium Chromate, Molybdate, Tungstate and Metavanadate. I. Experiments on Mice and Rats. Arch. Int. Pharmacodyn. Ther. 1965, 154, 243–249. [Google Scholar]
- Chanh, P.H. The comparative toxicity of sodium chromate, molybdate, tungstate and metavanadate. II. Experiments on rabbits. Arch. Int. Pharmacodyn. Ther. 1965, 157, 109–114. [Google Scholar]
- Pham, H.C.; Azum-Gelade, M.C.; Som-Chanyattey, M.C. Comparative toxicity of sodium chromate, molybdate, tungstate and metaanadate. 3. Tests Cats Agressologie: Revue Internationale Physio-Biologie Pharmacologie Appliquées Aux Effets L’agression 1967, 8, 51–60. [Google Scholar]
- Caujolle, F.; Pham, H.C. Comparative toxicity of sodium chromate, molybdate, tungstate and metavandadate. IV. Tests on dogs. Agressologie: Revue internationale Physio-Biologie Pharmacologie Appliquées Aux Effets L’agression 1967, 8, 265–273. [Google Scholar]
- Pham, H.C.; Chanvattey, S. Comparative study of sodium chromate, molybdate, tungstate and metavandate. V. Experiments on pigeons, chickens and rats Pham Huu Chanh; Chanvattey, S. Agressologie: Revue Internationale Physio-Biologie Pharmacologie Appliquees Aux Effets l’Agression 1967, 8, 433–439. [Google Scholar]
- Caujolle, F.; Godfrain, J.; Meynier, D. Comparative toxicities of sodium chromate, sodium molybdats and sodium tungstate in rats and mice after intraperitoneal administration. Comptes Rendus Hebdomadaires Des Seances l’Academie Sci. 1959, 248, 2667–2669. [Google Scholar]
- Stokinger, H.E. The Metals. In Patty’s Industrial Hygiene and Toxicology; Clayton, G.D., Clayton, F.E., Eds.; John and Wiley and Sons: Hoboken, NJ, USA, 1981; Volume 2A, pp. 1749–1769. [Google Scholar]
- Saganuwan, S.A.; Ahur, V.M.; Yohanna, C.A. Acute toxicity studies of potassium permanganate in Swiss albino mice. Niger. J. Physiol. Sci. 2008, 23, 31–35. [Google Scholar] [CrossRef][Green Version]
- Francis, A.A.; Forsyth, C. Toxicity Summary for Manganese. Oak Ridge Reservation Environmental Restoration Program, préparé pour le US Department of Energy. 1995. Available online: http://hydratechnm.org/documents/manganese/toxicity_summary_for_manganese.pdf (accessed on 16 November 2019).
- Lewis, R.J.; Sweet, D.V. (Eds.) Registry of Toxic Effects of Chemical Substances; U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control, National Institute for Occupational Safety and Health: Cincinnati, OH, USA, 1984; Volume 1.
- Bruce, D.W.; Hietbrink, B.E.; DuBois, K.P. The acute mammalian toxicity of rare earth nitrates and oxides. Toxicol. Appl. Pharmacol. 1963, 5, 750–759. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).