Adsorption Kinetics of a Cationic Surfactant Bearing a Two-Charged Head at the Air-Water Interface



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Equilibrium Surface Tension
2.2.2. Dynamic Surface Tension
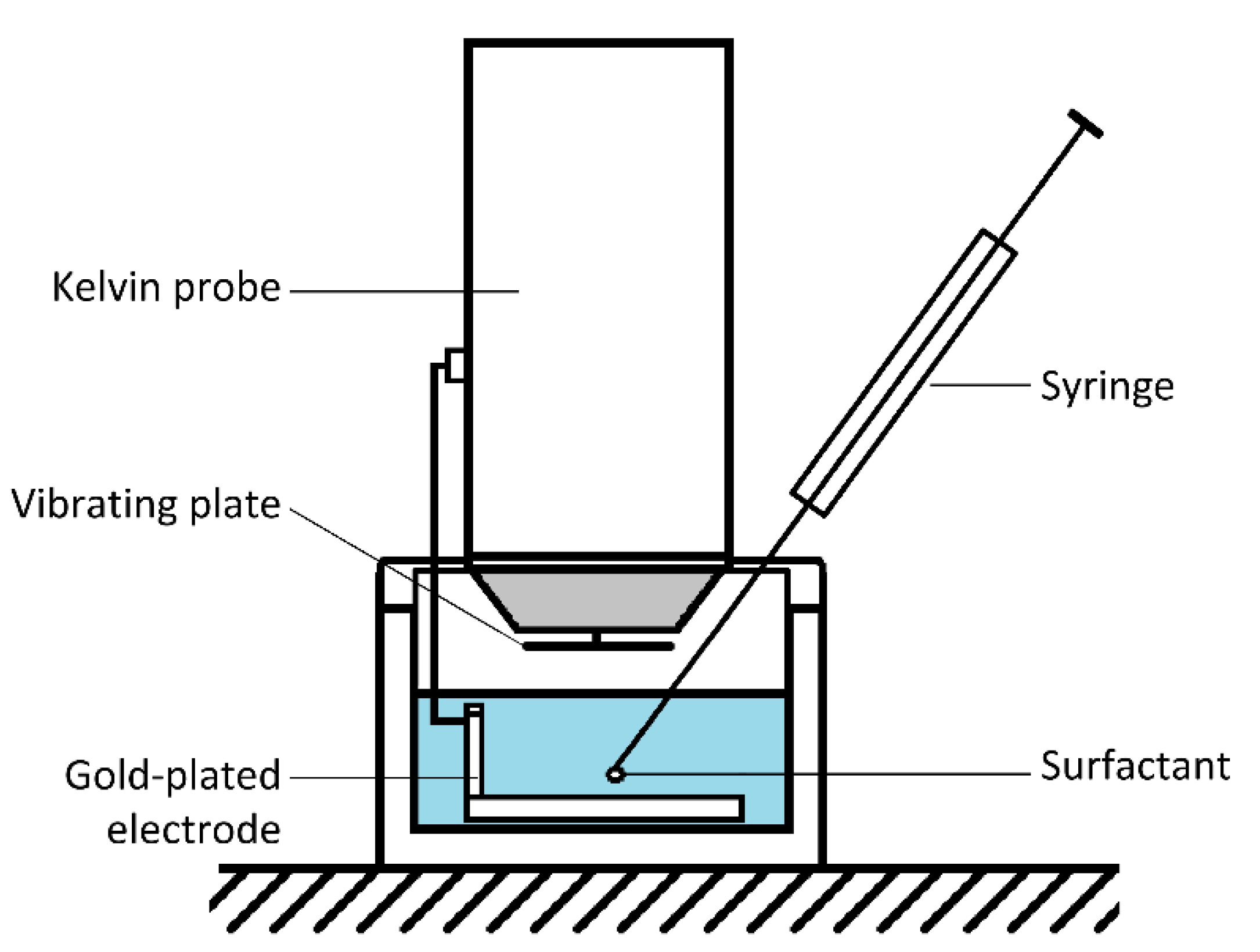
2.2.3. Time-Resolved Surface Potential Experiments
3. Results
3.1. Equilibrium Surface Tension Isotherm
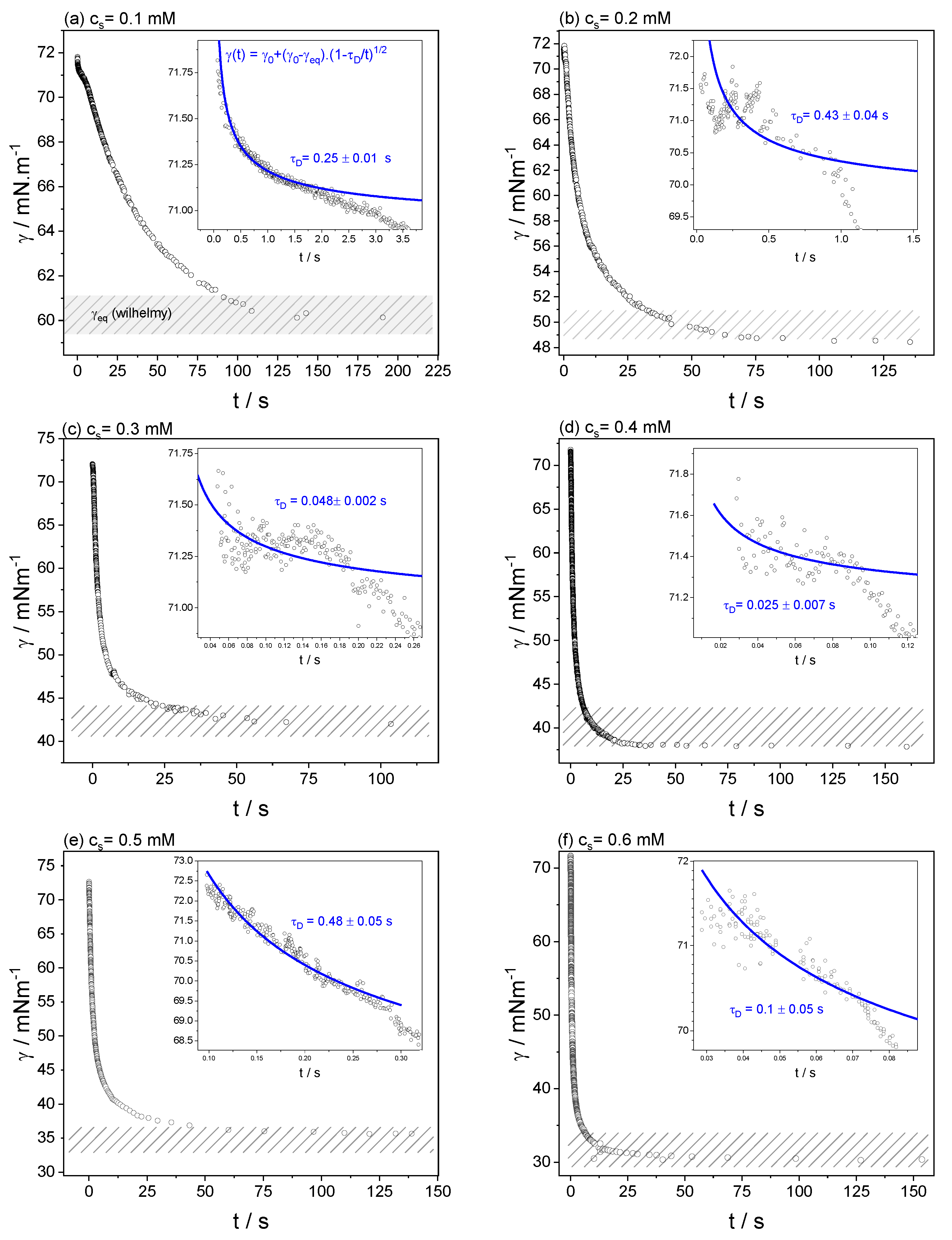
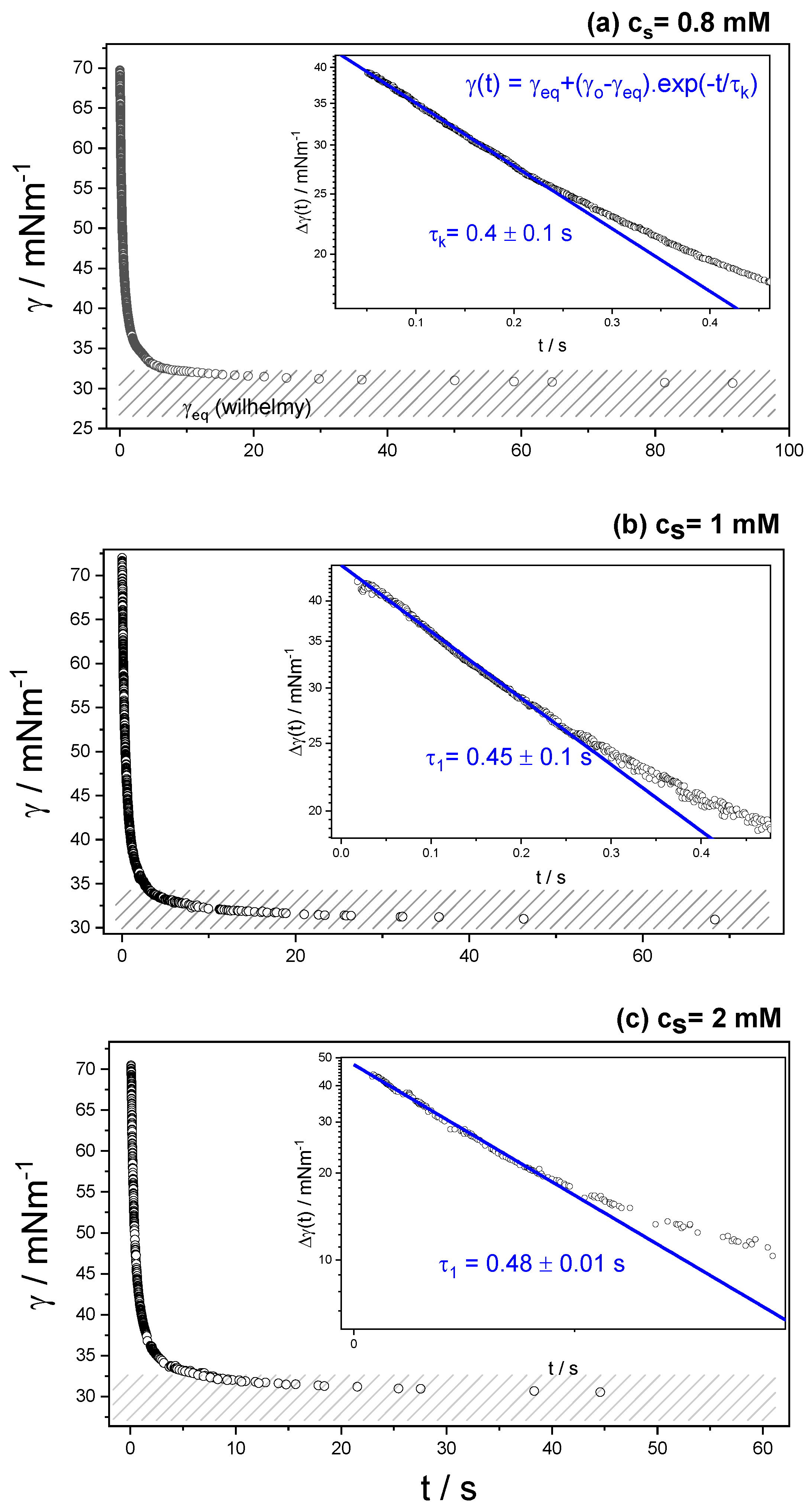
3.2. Dynamic Surface Tension
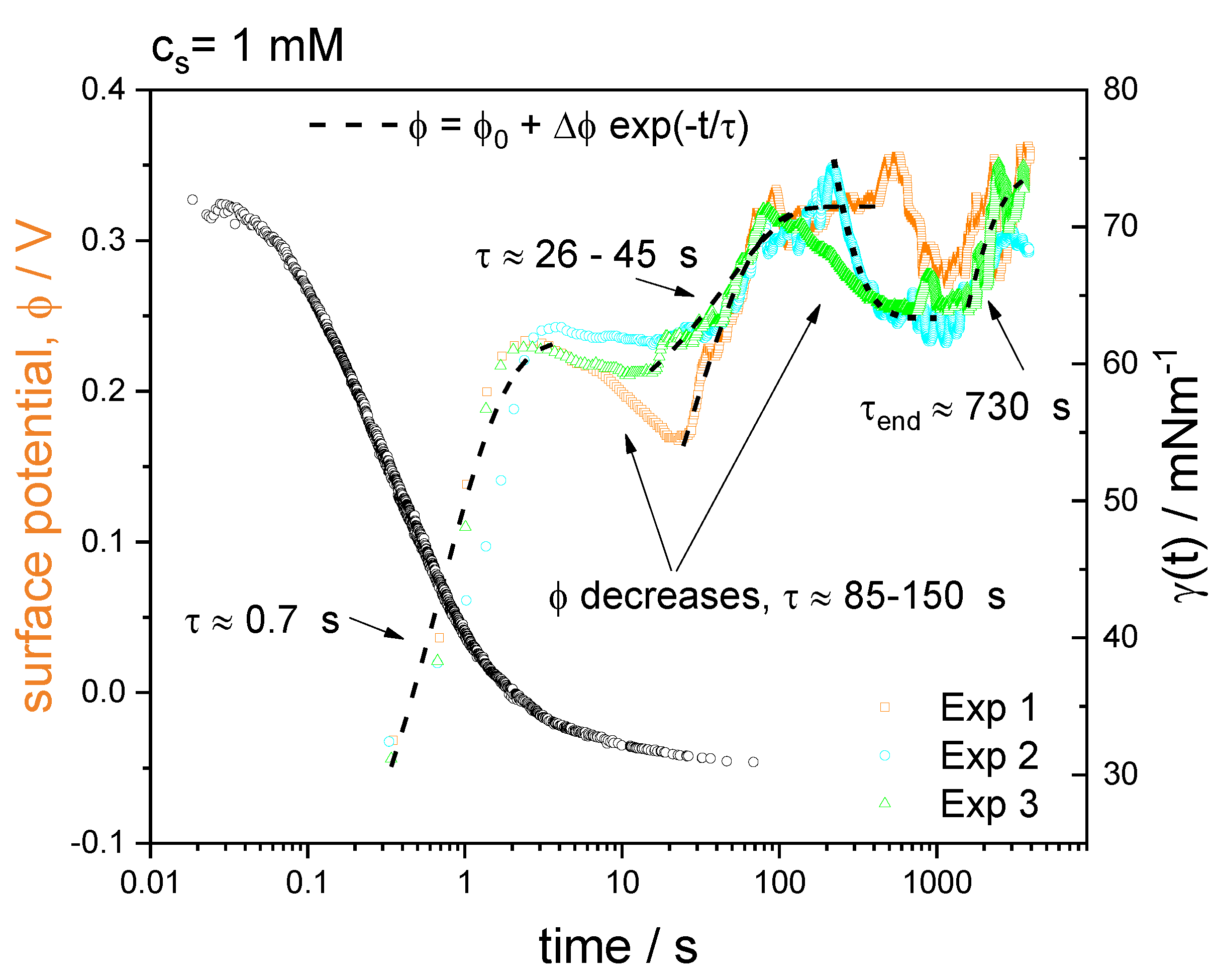
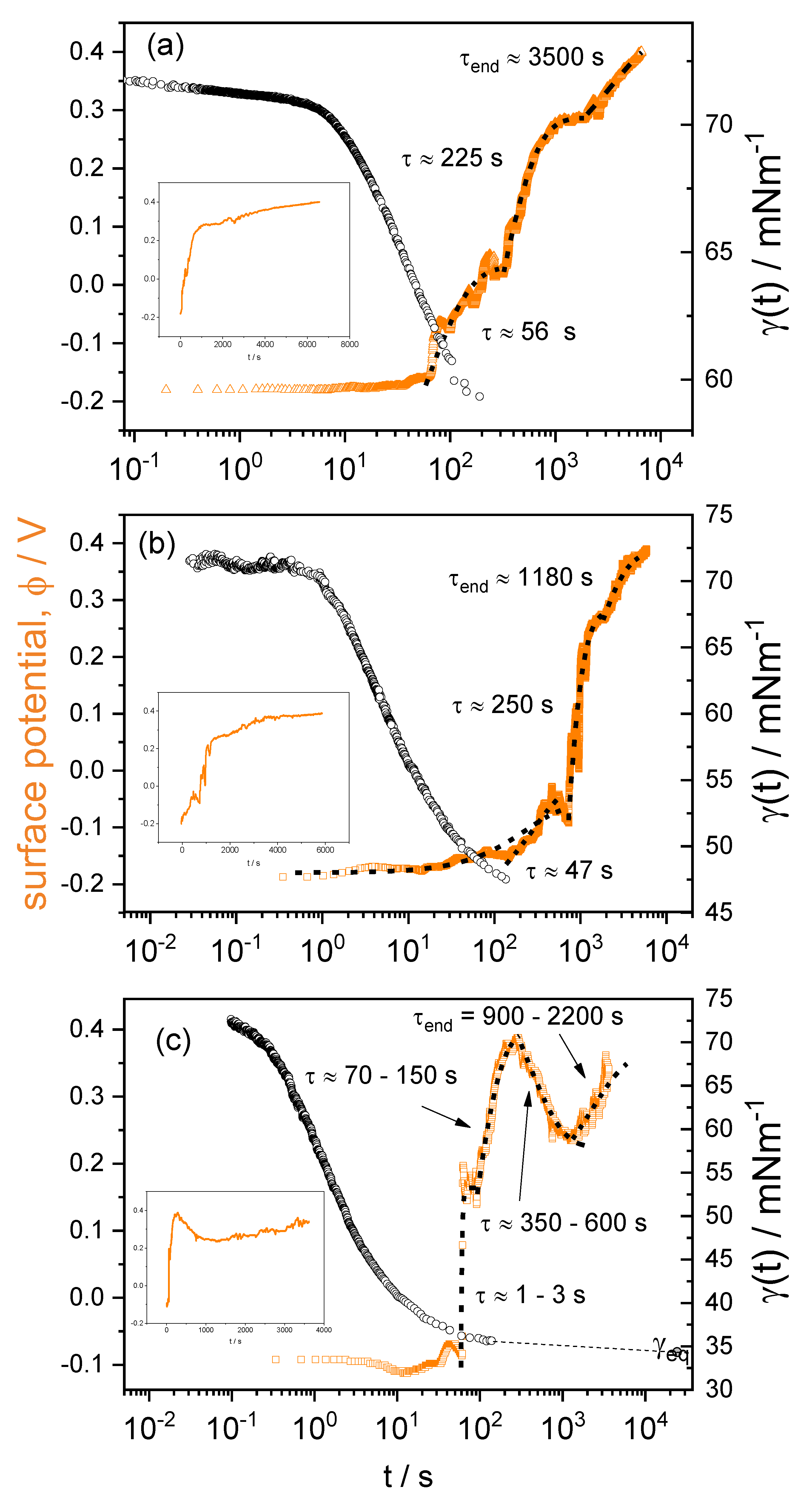
3.3. Time-Resolved Surface Potential
4. Discussion
4.1. Equilibrium Surface Tension
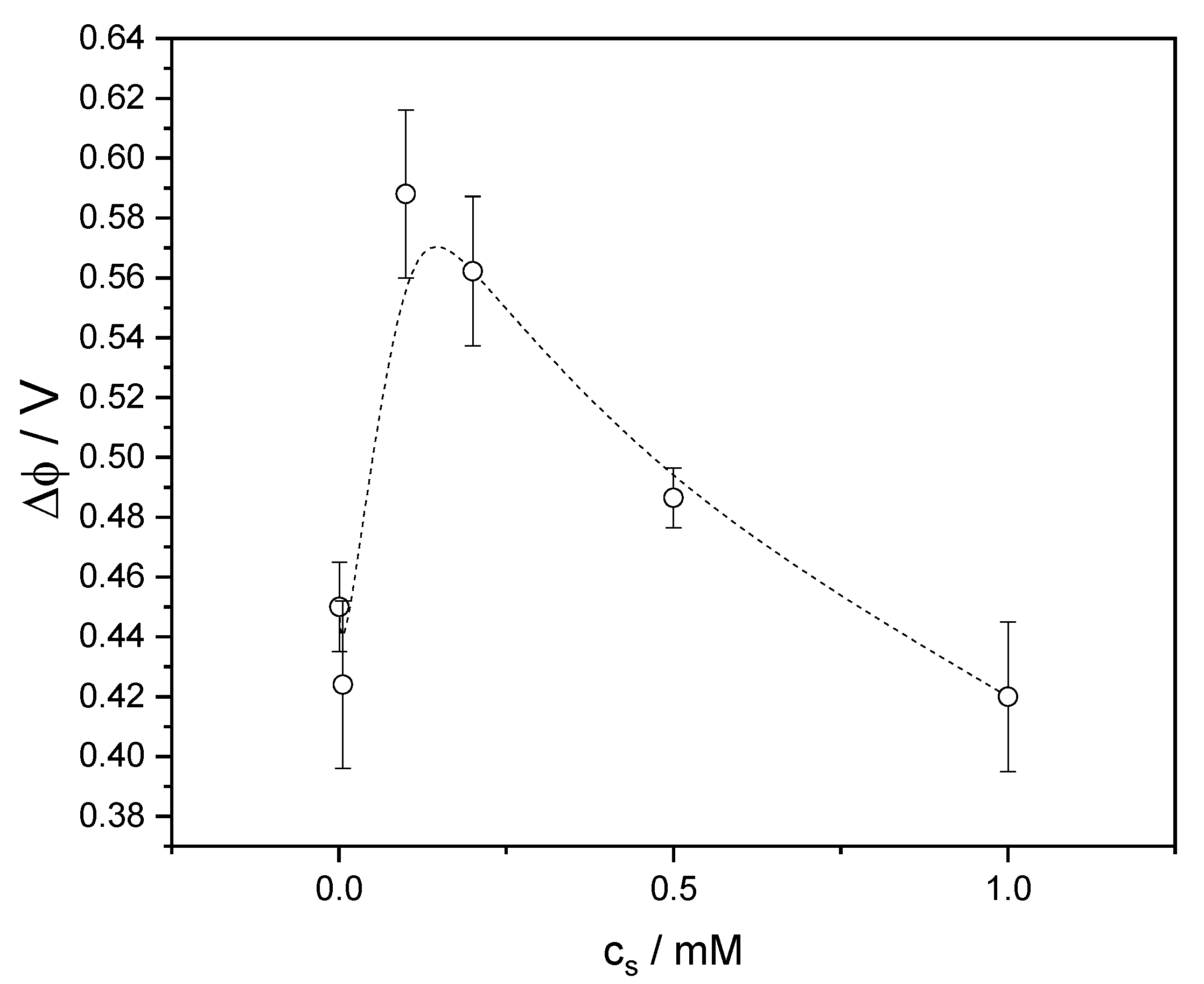
4.2. Equilibrium Surface Potential
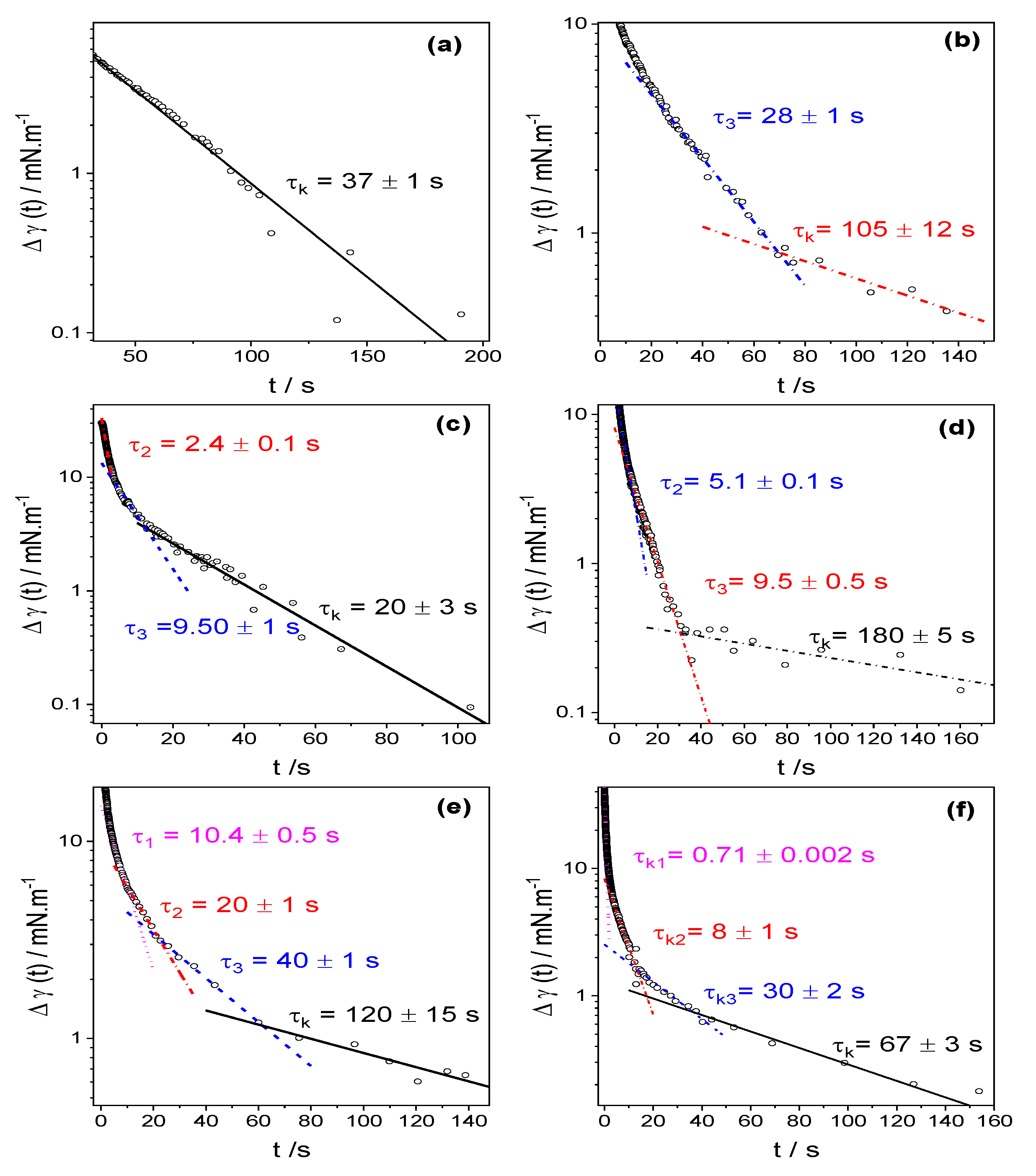
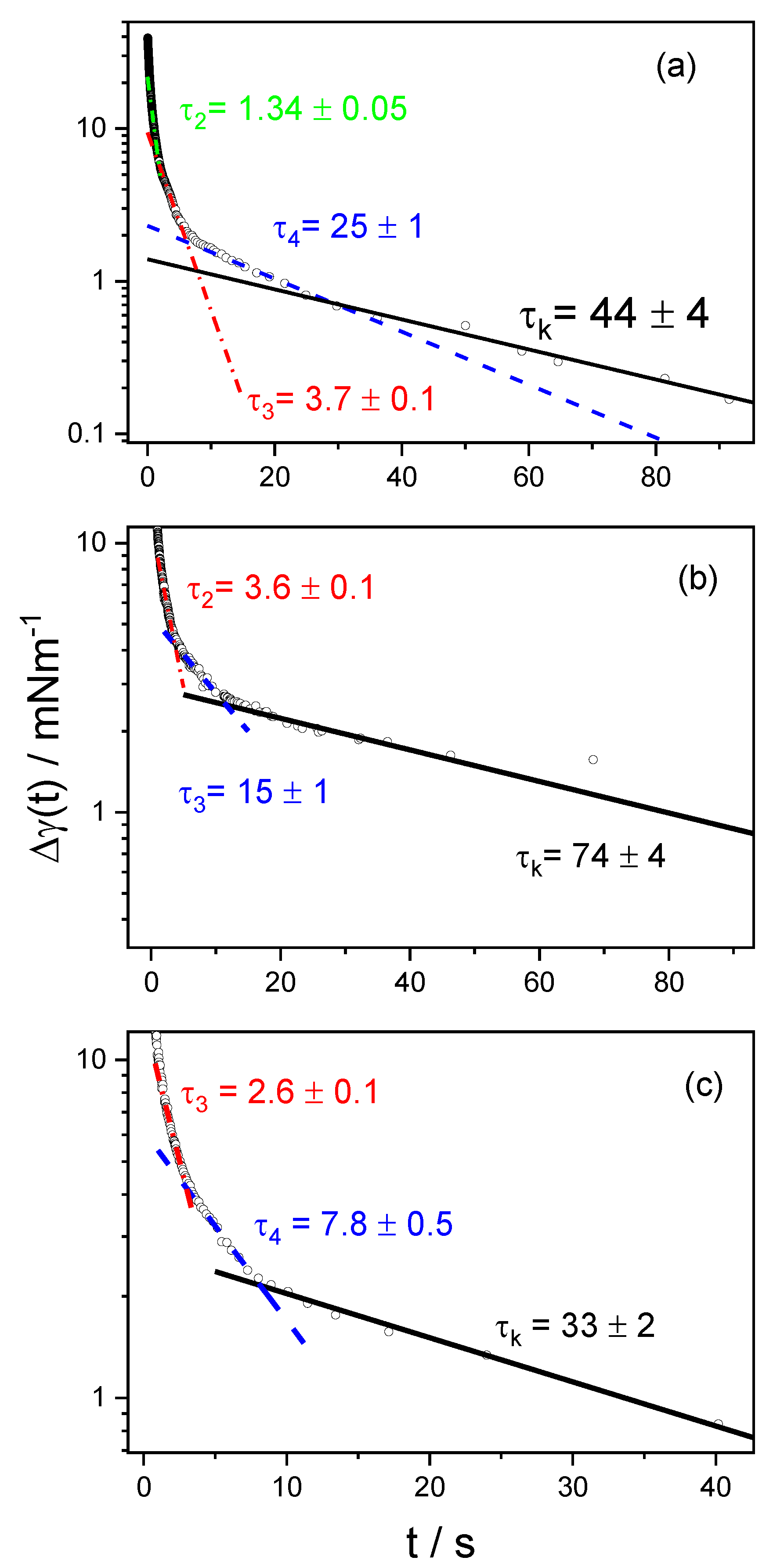
4.3. Dynamic Surface Tension
4.4. Time-Resolved Surface Potential
5. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Georgieva, D.; Schmitt, V.; Leal-Calderon, F.; Langevin, D. On the Possible Role of Surface Elasticity in Emulsion Stability. Langmuir 2009, 25, 5565–5573. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, D.; Cagna, A.; Langevin, D. Link between Surface Elasticity and Foam Stability. Soft Matter 2009, 5, 2063. [Google Scholar] [CrossRef]
- Ravera, F.; Ferrari, M.; Santini, E.; Liggieri, L. Influence of Surface Processes on the Dilational Visco-Elasticity of Surfactant Solutions. Adv. Colloid Interface Sci. 2005, 117, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Langevin, D. Rheology of Adsorbed Surfactant Monolayers at Fluid Surfaces. Annu. Rev. Fluid Mech. 2014, 46, 47–65. [Google Scholar] [CrossRef]
- He, Y.; Salonen, A.; Lecchi, A.; Veber, M.; Langevin, D. Adsorption of Gemini Surfactants at the Air-Water Surface. Colloid Polym. Sci. 2016, 294, 483–490. [Google Scholar] [CrossRef]
- Ward, A.F.H.; Tordai, L. Time-Dependence of Boundary Tensions of Solutions I. The Role of Diffusion in Time-Effects. J. Chem. Phys. 1946, 14, 453–461. [Google Scholar] [CrossRef]
- Bleys, G.; Joos, P. Adsorption Kinetics of Bolaform Surfactants at the Air/Water Interface. J. Phys. Chem. 1985, 89, 1027–1032. [Google Scholar] [CrossRef]
- Bonfillon, A.; Langevin, D. Viscoelasticity of Monolayers at Oil-Water Interfaces. Langmuir 1993, 2172–2177. [Google Scholar] [CrossRef]
- Bonfillon, A.; Langevin, D. Electrostatic Model for the Viscoelasticity of Ionic Surfactant Monolayers. Langmuir 1994, 10, 2965–2971. [Google Scholar] [CrossRef]
- Ritacco, H.; Langevin, D.; Diamant, H.; Andelman, D. Dynamic Surface Tension of Aqueous Solutions of Ionic Surfactants: Role of Electrostatics. Langmuir 2011, 27, 1009–1014. [Google Scholar] [CrossRef]
- Cuenca, V.E.; Fernández Leyes, M.; Falcone, R.D.; Correa, N.M.; Langevin, D.; Ritacco, H. Interfacial Dynamics and Its Relations with “Negative” Surface Viscosities Measured at Water–Air Interfaces Covered with a Cationic Surfactant. Langmuir 2019, 35, 8333–8343. [Google Scholar] [CrossRef] [PubMed]
- Adamson, A.; Gast, A. Physical Chemistry of Surfaces, 6th ed.; Jhon Wiley & Sons: New York, NY, USA, 1997. [Google Scholar]
- Cuenca, V.E.; Falcone, R.D.; Silber, J.J.; Correa, N.M. How the Type of Cosurfactant Impacts Strongly on the Size and Interfacial Composition in Gemini 12-2-12 RMs Explored by DLS, SLS, and FTIR Techniques. J. Phys. Chem. B 2016, 120, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Zana, R.; Benrraou, M.; Rueff, R. Alkanediyl-.Alpha.,.Omega.-Bis(Dimethylalkylammonium Bromide) Surfactants. 1. Effect of the Spacer Chain Length on the Critical Micelle Concentration and Micelle Ionization Degree. Langmuir 1991, 7, 1072–1075. [Google Scholar] [CrossRef]
- Ritacco, H.; Kurlat, D.; Langevin, D. Properties of Aqueous Solutions of Polyelectrolytes and Surfactants of Opposite Charge: Surface Tension, Surface Rheology, and Electrical Birefringence Studies. J. Phys. Chem. B 2003, 107, 9146–9158. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Miller, R. The Maximum Bubble Pressure Technique. In Drop and Bubbles in Interfacial Science; Möbius, M.E., Miller, R., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 1998; pp. 279–326. [Google Scholar]
- Espert, A.; von Klitzing, R.; Poulin, P.; Colin, A.; Zana, R.; Langevin, D. Behavior of Soap Films Stabilized by a Cationic Dimeric Surfactant. Langmuir 1998, 14, 4251–4260. [Google Scholar] [CrossRef]
- Li, Z.X.; Dong, C.C.; Thomas, R.K. Neutron Reflectivity Studies of the Surface Excess of Gemini Surfactants at the Air-Water Interface. Langmuir 1999, 15, 4392–4396. [Google Scholar] [CrossRef]
- Zana, R. Dimeric and Oligomeric Surfactants. Behavior at Interfaces and in Aqueous Solution: A Review. Adv. Colloid Interface Sci. 2002, 97, 205–253. [Google Scholar] [CrossRef]
- Diamant, H.; Ariel, G.; Andelman, D. Kinetics of Surfactant Adsorption: The Free Energy Approach. Colloids Surf. A Physicochem. Eng. Asp. 2001, 183–185, 259–276. [Google Scholar] [CrossRef]
- Diamant, H.; Andelman, D. Kinetics of Surfactant Adsorption at Fluid-Fluid Interfaces. J. Phys. Chem. 1996, 100, 13732–13742. [Google Scholar] [CrossRef]
- Almeida, J.A.S.; Pinto, S.P.R.; Wang, Y.; Marques, E.F.; Pais, A.A.C.C. Structure and Order of DODAB Bilayers Modulated by Dicationic Gemini Surfactants. Phys. Chem. Chem. Phys. 2011, 13, 13772–13782. [Google Scholar] [CrossRef]
- Manning, G.S. Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions II. Self-Diffusion of the Small Ions. J. Chem. Phys. 1969, 51, 934. [Google Scholar] [CrossRef]
- Manning, G.S. Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions I. Colligative Properties. J. Chem. Phys. 1969, 51, 924. [Google Scholar] [CrossRef]
- Nilsson, M.; Cabaleiro-Lago, C.; Valente, A.J.M.; Söderman, O. Interactions between Gemini Surfactants, 12-s-12, and β-Cyclodextrin As Investigated by NMR Diffusometry and Electric Conductometry. Langmuir 2006, 22, 8663–8669. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Monroy, F.; Giermanska Kahn, J.; Langevin, D. Dilational Viscoelasticity of Surfactant Monolayers. Colloids Surf. A Physicochem. Eng. Asp. 1998, 143, 251–260. [Google Scholar] [CrossRef]
- Bonfillon, A.; Sicoli, F.; Langevin, D. Dynamic Surface Tension of Ionic Surfactant Solutions. J. Colloid Interface Sci. 1994, 168, 497–504. [Google Scholar] [CrossRef]
- Bykov, A.G.G.; Liggieri, L.; Noskov, B.A.A.; Pandolfini, P.; Ravera, F.; Loglio, G. Surface Dilational Rheological Properties in the Nonlinear Domain. Adv. Colloid Interface Sci. 2015, 222, 110–118. [Google Scholar] [CrossRef]
- Miller, R.; Aksenenko, E.V.; Fainerman, V.B. Dynamic Interfacial Tension of Surfactant Solutions. Adv. Colloid Interface Sci. 2017, 247, 115–129. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Mys, V.D.; Makievski, A.V.; Petkov, J.T.; Miller, R. Dynamic Surface Tension of Micellar Solutions in the Millisecond and Submillisecond Time Range. J. Colloid Interface Sci. 2006, 302, 40–46. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cs/mM | τD/s | D/m2 s−1 |
|---|---|---|
| 0.1 | 0.25 ± 0.01 | 4.1 × 10−10 |
| 0.2 | 0.43 ± 0.04 | 1.1 × 10−10 |
| 0.3 | 0.048 ± 0.02 | 5.0 × 10−10 |
| 0.4 | 0.025 ± 0.01 | 5.4 × 10−10 |
| 0.5 | 0.48 ± 0.05 | 1.8 × 10−11 |
| 0.6 | 0.1 ± 0.05 | 6 × 10−11 |
| cs/mM | τ1 | τ2 | τ3 | τ4 | τk |
|---|---|---|---|---|---|
| 0.1 | -- | -- | -- | 6.5 ± 1 | 37 ± 1 |
| 0.2 | -- | -- | --- | 28.5 ± 1 | 105 ± 12 |
| 0.3 | -- | -- | 2.4 ± 0.1 | 9.5 ± 1 | 20 ± 3 |
| 0.4 | -- | -- | 5.1 ± 0.1 | 9.5 ± 0.5 | 180 ± 40 |
| 0.5 | -- | 10.4 ± 0.5 | 20 ± 1 | 40 ± 2 | 120 ± 15 |
| 0.6 | -- | 0.71 ± 0.002 | 8 ± 1 | 30 ± 2 | 67 ± 3 |
| 0.8 | 0.4 ± 0.01 | 1.34 ± 0.005 | 3.7 ± 0.1 | 25 ± 1 | 44 ± 4 |
| 1 | 0.45 ± 0.001 | -- | 3.6 ± 0.1 | 15 ± 1 | 74 ± 4 |
| 2 | 0.48 ± 0.001 | -- | 2.6 ± 0.1 | 7.8 ± 0.5 | 33 ± 2 |
| cs/mM | eψ0/kBT | eψa/kBT | |
| 0.1 | 4.96 | 11.26 | −1.34 |
| 0.2 | 6.17 | 10.93 | 1.41 |
| 0.3 | 5.75 | 10.56 | 0.94 |
| 0.4 | 7.14 | 10.27 | 4.01 |
| 0.5 | 7.15 | 10.05 | 4.25 |
| 0.6 | 7.05 | 9.87 | 4.23 |
| 0.8 | 7.12 | 9.58 | 4.66 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández Leyes, M.; Gimenez Reyes, S.; Cuenca, E.; Sánchez Morales, J.F.; Ritacco, H. Adsorption Kinetics of a Cationic Surfactant Bearing a Two-Charged Head at the Air-Water Interface. Coatings 2020, 10, 95. https://doi.org/10.3390/coatings10020095
Fernández Leyes M, Gimenez Reyes S, Cuenca E, Sánchez Morales JF, Ritacco H. Adsorption Kinetics of a Cationic Surfactant Bearing a Two-Charged Head at the Air-Water Interface. Coatings. 2020; 10(2):95. https://doi.org/10.3390/coatings10020095
Chicago/Turabian StyleFernández Leyes, Marcos, Santiago Gimenez Reyes, Ezequiel Cuenca, Jhon F. Sánchez Morales, and Hernán Ritacco. 2020. "Adsorption Kinetics of a Cationic Surfactant Bearing a Two-Charged Head at the Air-Water Interface" Coatings 10, no. 2: 95. https://doi.org/10.3390/coatings10020095
APA StyleFernández Leyes, M., Gimenez Reyes, S., Cuenca, E., Sánchez Morales, J. F., & Ritacco, H. (2020). Adsorption Kinetics of a Cationic Surfactant Bearing a Two-Charged Head at the Air-Water Interface. Coatings, 10(2), 95. https://doi.org/10.3390/coatings10020095