1. Introduction
In 2019, the European Commission declared an ambitious goal by 2050 to become the first climate-neutral continent in the world [
1]. Such a transition will require comprehensive changes and improvements at every element of the energy system including its generation, storage, and utilization. Naturally, the current 20% share of the renewable energy sources in EU energy generation [
2] will have to be substantially increased. In addition, a wide diversity of carbon-neutral technologies will have to be applied to balance the mismatch of energy generation and demand (both in time and site) [
3]. Power-to-gas systems can be used to transform renewable electrical energy into chemical energy in the form of substitute natural gas (SNG) and is among the top candidate technologies to balance the mismatch [
4,
5,
6]. In comparison to electricity, SNG has two important advantages. First, it can store energy for the long periods of time. Second, existing infrastructure makes it immediately available to be used as fuel for transport and a convenient measure to provide energy for remote energy consumers.
One of the most environmentally friendly power-to-gas solutions is to generate hydrogen (e.g., by the electrolysis of water or via a reversed PEM fuel cell) and then to react it with CO
2 through the Sabatier reaction [
3]. Though there are still some disputes over the mechanisms of the individual steps [
7] an overall Sabatier process Equation (1) can be described as a two stage (Equations (2) and (3)) reaction [
8]:
Gao et al. [
9] reported that based on free Gibbs energy calculations, the optimal temperature (in respect to CH
4 selectivity and CO
x conversion) for the Sabatier reaction (1) is 300–400 °C. However, due to the endothermic reaction (2), relatively high activation barrier [
3], and limited kinetics, at this temperature range significant methanation reaction is observed only with the presence of suitable catalyst [
10]. The need for the catalyst is lessened when temperature exceeds 500 °C and reverse water gas shift reaction (2) becomes exothermic. But at high temperatures the competing Bosch reaction takes place and the efficiency of methanation process (1) is starting to decrease [
9]. The presence of conventional heterogeneous catalysts (for example Ni, Fe, Co or Ru [
11]) do not help either because in addition to promoting CO
2 methanation they also serve as catalytic drivers for the competing reaction (4) which produces elemental carbon and poisons the catalysts [
12]:
Looking for the solutions to avoid carbon build up at elevated temperatures Gao et al. [
9] and Jurgensen et al. [
12] investigated the role of CO
2:H
2 and CO:H
2 gas ratios. The researchers determined that higher gas ratios have positive effects on lowering down carbon production and that preferable CO
2:H
2 gas ratios should be at least 1:6 [
9]. Additionally, it was demonstrated that by increasing gas mixture pressure from 1 to 11 bars, the starting temperature for carbon generation rises from 365 to 515 °C [
12]. The other way to minimize carbon formation and the poisoning of the catalyst is to find new materials that have a higher selectivity for CH
4 production and do not support the Bosch reaction.
In 1990, Selvam et al. [
13] investigated the interaction between CO
2, hydrogen storage alloys, and compounds including LaNi
5, CaNi
5, Mg
2Ni, Mg
2Cu, and FeTi. They reported that during exposure to air, all these compounds form surface oxides and hydroxides that, in turn, actively adsorb atmospheric CO
2 and favor the formation of carbonate species on the top few layers of the surface. In a subsequent study of air-exposed Mg
2NiH
4 hydride [
14], Selvam et al. reached conclusion that later hydride also undergoes similar processes and forms surface carbonate species, especially in the presence of moisture.
Two decades later, Kato et al. [
6] argued that the high absorptivity of CO
2 on an Mg
2NiH
4 surface could be beneficial for the Sabatier type methanation reaction. Their comprehensive study on the catalytic interactions of the hydride surface of Mg
2NiH
4 powder with CO
2 provided valuable insights into material disproportionation and oxidation during cyclic hydrogen absorption and hydrogen desorption under CO
2 (at temperatures up to 500 °C). Based on their findings, during the first few dehydriding cycles in a CO
2-containing atmosphere, the simultaneous disproportionation of Mg
2NiH
4 and the selective oxidation of Mg take place. As a result, a layered structure consisting of Mg
2NiH
4/Mg
2Ni/Ni/MgO was formed. According to the authors, when it was active, Ni helped to dissociate CO
2 and CO molecules and promoted methanation [
6]. Eventually, after approximately 20 cycles, Mg
2NiH
4 and Mg
2Ni were no longer formed and only MgO and Ni phases were observed.
In two more recent studies, Grasso et al. reported experimental data on CO
2 methanation processes using as-sintered monoclinic [
15] and as-milled cubic Mg
2NiH
4 [
16] powders. In these studies, Mg
2NiH
4 powders served as the sole hydrogen sources and “providers” of catalytic sites for the promotion of the conversion of CO
2 to CH
4. To proceed with the experiments, a certain amount of hydride powder was placed into a stainless steel reactor connected to Sieverts volumetric equipment. The reactor was provided a specific CO
2 pressure and heated to 400 °C at a rate of 10 °C/min. The mass of the used Mg
2NiH
4 powder and the applied CO
2 pressure (approximately 1.2 bars [
16]) were proportioned in such a way that the calculated total molar quantity of H
2 released by the complete decomposition of Mg
2NiH
4 would ensure an H
2:CO
2 molar ratio of 4:1. Interestingly, despite the nearly identical reaction conditions, the authors found evidence for two slightly different reaction mechanisms. After 10 h of the as-sintered monoclinic Mg
2NiH
4‘s reaction with CO
2 at 400 °C, the complete decomposition of Mg
2NiH
4 was not reached, although CO
2 was totally consumed and no carbon deposition was observed [
15]. Considering the intermediate reaction products that were obtained after 1 and 5 h (namely, Mg
2NiH
4, MgH
2, Mg
2Ni, MgO, and CO), Grasso et al. concluded that the methanation of CO
2 using Mg
2NiH
4 involves two simultaneous processes: (i) the catalytic conversion of CO
2 through reactions (2) and (3) and (ii) the direct reduction of CO
2 by the reducing effect of MgH
2 (Equation (5)).
The chemical activity of the as-milled cubic polymorph form of Mg
2NiH
4 was found to be considerably higher, and its reaction with CO
2 at 400 °C was completed in just 5 h [
16]. The observed intermediate and final products of the methanation process were slightly different from the formerly described case. Therefore, the authors concluded that under a CO
2 atmosphere, as–milled Mg
2NiH
4 rapidly decomposes directly to the Ni-Mg
2Ni-MgNi
2-/MgO (all of this phase remains after the reaction is complete) catalytic system Equation (6) which promotes the methanation reaction Equation (7).
Reports by Kato et al. [
6] and Grasso et al. [
15,
16] provided clear evidence that during its interaction with CO
2 gas at 400–500 °C, Mg
2NiH
4 powder disproportionates into catalytically active compounds that efficiently promote the generation of methane. However, in all three studies, different reaction products were observed and divergent mechanisms were proposed. Grasso et al. assumed that at least some of the observed variations could have related to the specific forms and preparation methods of the Mg
2NiH
4 hydride [
16].
Potentially, an Mg
2NiH
4 hydride-based CO
2 methanation reaction catalyst can be used in powder, as well as in the supported film (coating) form, but up to now, all of the reported studies were only conducted with Mg
2NiH
4 powders. Previous studies of Mg
2NiH
4 hydride films [
17,
18] indicated that characteristic strains caused by the film–substrate interaction can introduce noticeable changes of properties in comparison to corresponding powders. Therefore, there is a possibility that one form of Mg
2NiH
4 hydride might be better suited for CO
2 methanation catalysis than the other.
Accordingly, in the current study, we investigated the structural transformations of Mg2NiH4 hydride films under a CO2 atmosphere and examined whether they correlated with the ones reported for Mg2NiH4 powders. In addition, our interest to investigate Mg2NiH4 films had two more motives. Firstly, film condensation from physical vapor allows one to synthesize very uniform samples with strictly controlled component ratios that prevent the formation of undesirable phases. This makes them particularly suitable as the subject for in-situ structural analysis, and this is useful for resolving discrepancies between the proposed reaction pathways. Secondly, films are eligible to be approximated as 1D objects, which is a clear advantage for the observation and evaluation of surface changes, including the analysis of surface region depth profiles.
In addition, we synthesized Mg2NiH4 in powder and investigated its efficiency for the methanation of CO2 in order to compare processes in different forms of Mg2NiH4 hydride and to see how this material develops during the CO2 methanation reaction.
3. Results and Discussions
As is typical for metallic films, the as-deposited Mg
2Ni coatings had mirror like appearances. After hydriding, they became transparent and had a bright orange hue. Both of these features are characteristic for the low temperature (LT) phase of Mg
2NiH
4 [
22,
23,
24]. The prevalence of the monoclinic LT phase of Mg
2NiH
4 was subsequently confirmed by XRD (
Figure 2). In addition to the LT phase, small fractions of the so-called pseudo-cubic high temperature (HT) phase of Mg
2NiH
4 [
17,
25] were observed at room temperature.
During the in-situ heating of the Mg
2NiH
4 film under the CO
2 atmosphere (
Figure 2), several temperature ranges related to substantial composition changes could be identified. Starting from the room temperature up till 240 °C, the LT and HT phases of Mg
2NiH
4 co-existed, and their ratio in the film did not change noticeably. Above 240 °C, the fraction of LT phase gradually decreased, and at 300 °C, only the HT phase of Mg
2NiH
4 persisted. The initiation of Mg
2NiH
4 decomposition was observed at 350–360 °C when weak peaks attributed to Mg
2Ni arose. At 410 °C, the transition from Mg
2NiH
4 to Mg
2Ni was complete, and at 420 °C, only peaks of Mg
2Ni were observed. A further increase of the temperature was followed by the gradual disproportionation of Mg
2Ni and the crystallization of MgO oxide. The crystalline MgO phase was at first observed at 450 °C and remained as the main phase at the end of the measurement at 500 °C.
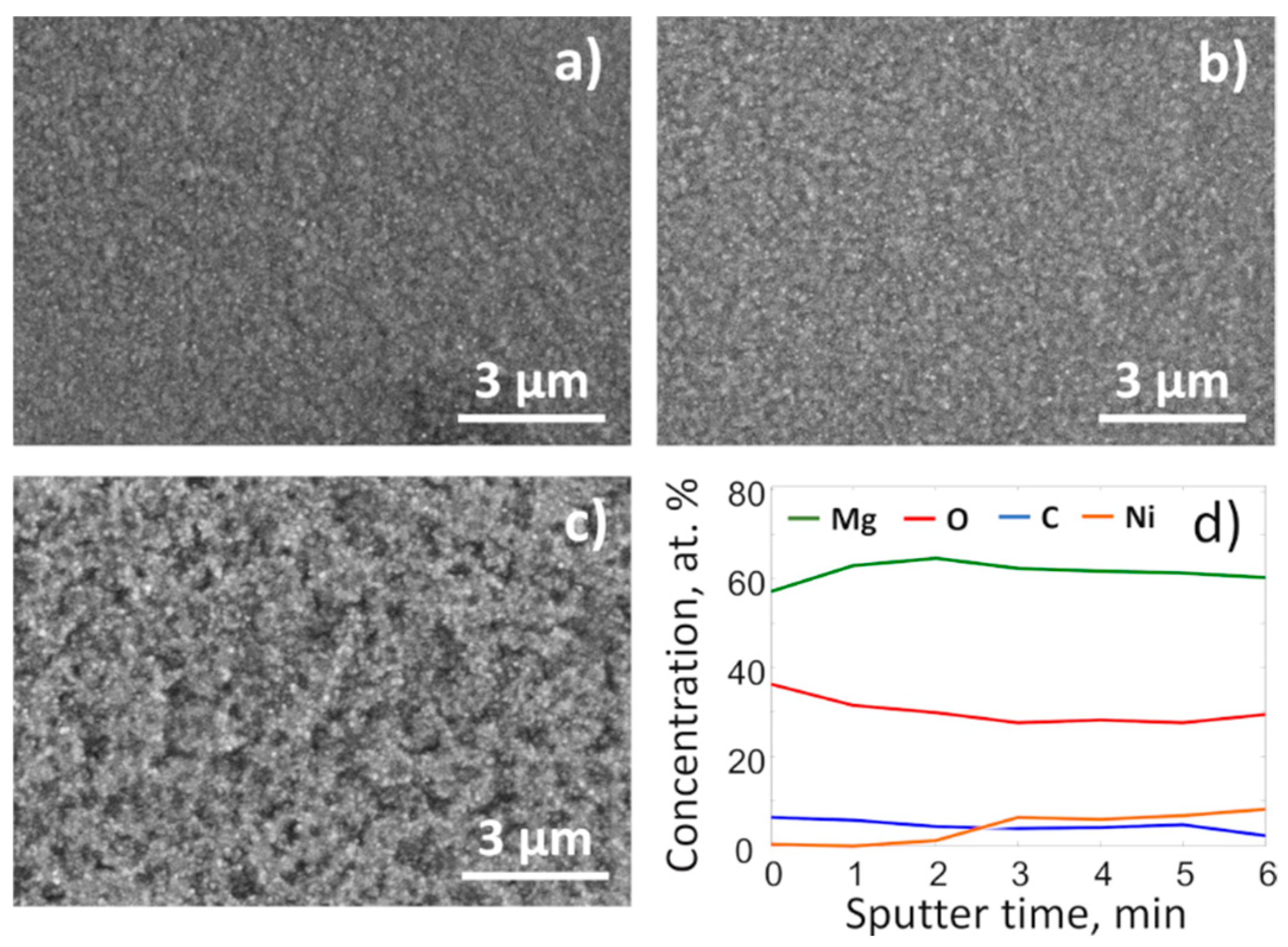
Surface images made by SEM and the near surface region depth profile recorded by XPS complemented the findings of in-situ XRD and highlighted structural changes of the film that occurred due to the heating in the CO
2 atmosphere. More specifically, the SEM images (
Figure 3) show that after heating in the CO
2 atmosphere, the surface of the films became considerably rougher, which indicated significant mass transfer processes within the film. At the same time, an XPS depth profile (
Figure 3d) specified that the Ni concentration at the surface was less than 1/6 of Mg. This was clear evidence that during the disproportionation of Mg
2Ni, magnesium separated from Ni, oxidized, and had a tendency to segregate at the near surface region.
By comparing earlier reports by Kato et al. [
6] and Grasso et al. [
15,
16], who worked with powders, with current in-situ XRD data that were observed for film, an outstanding feature was found: in the later case, there were no signs of the crystalline MgNi
2 and Ni phases. On one hand, this could be considered a sign of a slightly different reaction pathway for the film samples in comparison to the powder equivalent. On the other hand, it seems that a full-scale comparison does not provide substantial proof for that. For example, the observed quantitative concentration values, as well as their qualitative changes at the surface of the film (
Figure 3d) in great proximity, resembled the data presented by Kato et al. [
6], who measured the XPS depth profile of Mg
2NiH
4 powders that were cyclically exposed to a CO
2 atmosphere at a temperature of 500 °C. Furthermore, several tests with Mg
2NiH
4 powder that had an MgNi
2 phase (more details are provided in the following paragraphs) showed that there was a very close agreement between on-set process temperatures for powder and film samples. Altogether, we presume that the absence of MgNi
2 and Ni phases in in-situ XRD data was the result of insufficient crystallization rather than different reaction pathways.
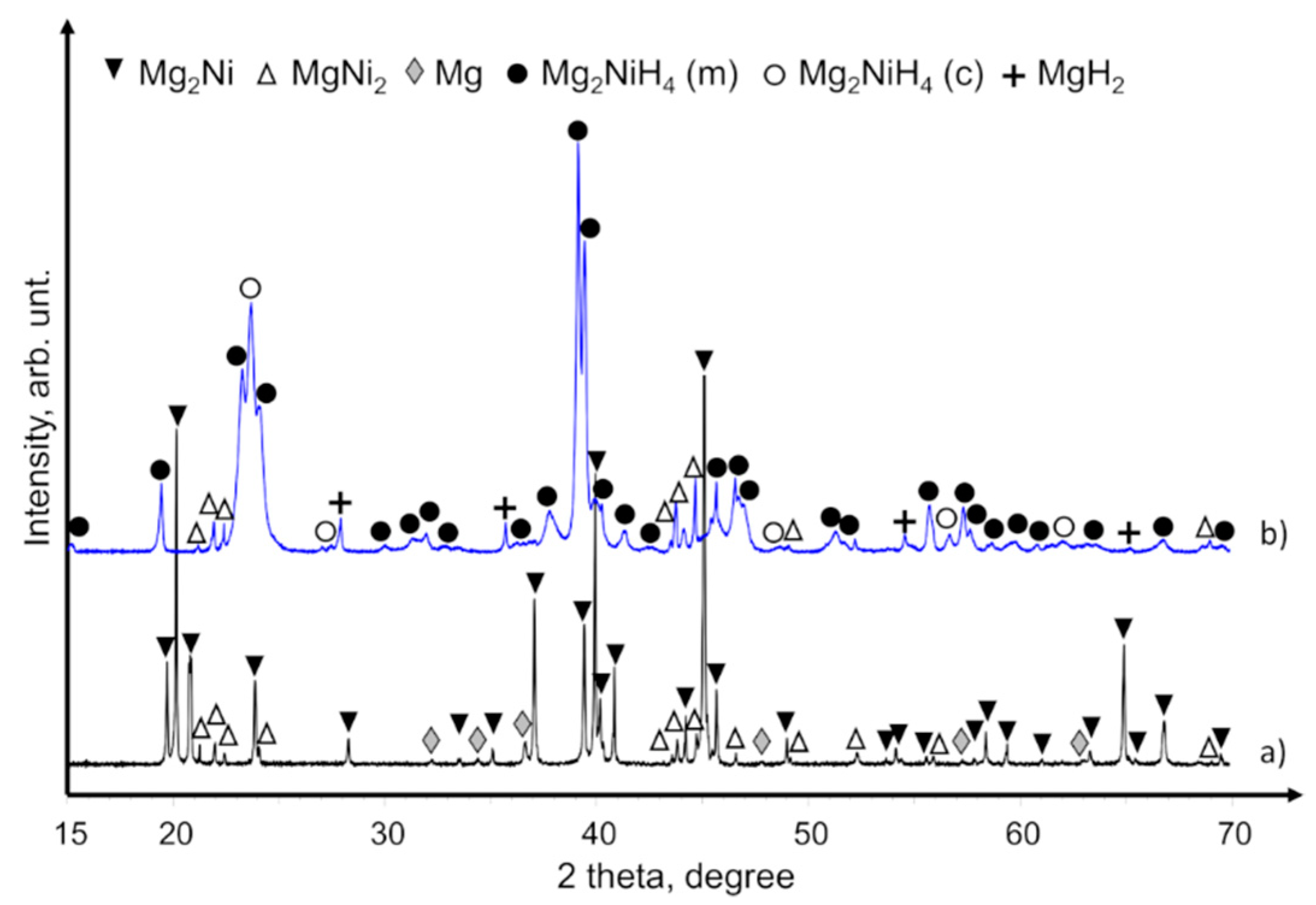
XRD patterns of commercial Mg
2Ni powders that contained Mg and MgNi
2 components are provided in
Figure 4a. After hydriding these powders, the crystal phases of Mg
2NiH
4, MgH
2, and MgNi
2 were obtained (
Figure 4b). The XRD patterns of the hydrided powders had no peaks of unreacted Mg and Mg
2Ni phases, so we assume that the loss of hydrogen concentration (see DSC/TGA analyses below) was caused by the aggregative effect of hydrogen non-adsorbing phases like MgNi
2. MgO was not observed by XRD, but we nevertheless assume that it also contributed to the lower hydrogen concentration because commercial Mg–Ni grains were delivered in an air-containing canister and had successive handling in air.
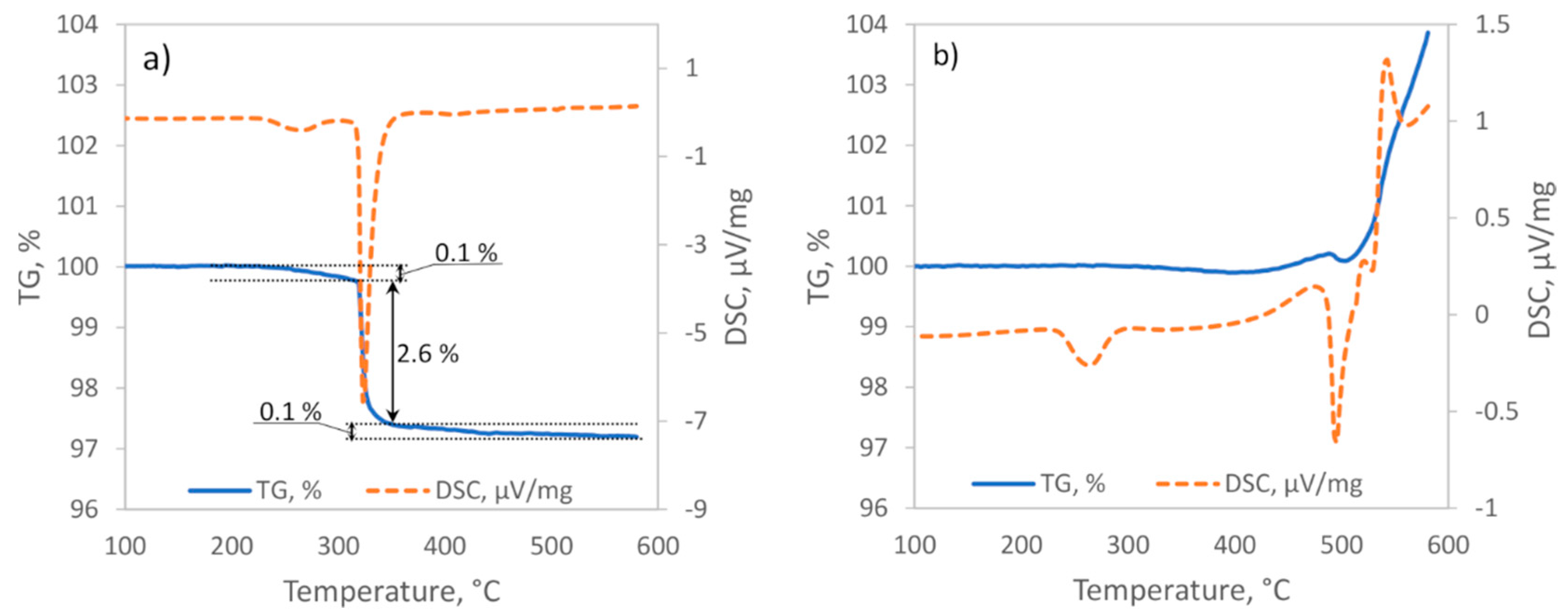
The DSC analysis of hydrided powders under Ar flow indicated the presence of two distinct thermal events (
Figure 5a). The first endothermal event started at approximately 235 °C and was attributed to the LT–HT phase transition of Mg
2NiH
4 [
26,
27]. Close to the beginning of the phase transition, the sample started to lose some mass (up to 0.2 wt.%), and this indicated the partial decomposition of Mg
2NiH
4. Proceeding further, most of the sample mass (≈2.6 wt.%) was lost during the second endothermal event starting at approximately 315–320 °C. This event represented the rapid hydrogen release during the supposedly full dehydriding of Mg
2NiH
4. Lastly, there was a lengthy mass loss of approximately 0.1 wt.% that led to a total mass loss of nearly 2.8 wt.%. Accordingly, we attributed the last phase of mass loss to the dehydriding of MgH
2.
The first thermal event during hydrided powder heating under the Ar and CO
2 gas flow started with the LT–HT phase transition at 235 °C (
Figure 5b). A nearly identical onset temperature and width of the Mg
2NiH
4 phase transition range (235–300 °C) were also observed for the Mg
2NiH
4 film (
Figure 2) and powder samples (
Figure 5a) that were tested under CO
2 and Ar gas atmospheres, respectively. This indicated that below the phase transition temperature, the activation barrier for Mg
2NiH
4 hydride decomposition and/or oxidation was relatively high, and at near-atmosphere pressures, Mg
2NiH
4 hydride demonstrated enduring stability regardless of its form and surrounding gas phase composition.
Disparities between Mg
2NiH
4 hydride behavior under the inert and reactive gases arose soon after start of the LT–HT phase transition. Namely, under the CO
2 and Ar gas flow, there were no mass change events up to approximately 330 °C. An analysis of the results of the in-situ XRD of Mg
2NiH
4 film heating under CO
2 gas (
Figure 2) suggested that at later temperatures, the dehydriding of Mg
2NiH
4 phase should have taken place. The decomposition reaction of Mg
2NiH
4 was expected to result in approximately 2.7 wt.% mass loss and a strong endothermic minimum in the TGA and DSC curves, respectively (
Figure 5a). Instead, under the CO
2 and Ar gas flow, a prolonged small (≈0.1 wt.%) decline of mass and a low intensity exothermic upswing was observed. Therefore, it is clear that the endothermal reaction (8) [
28] was simultaneously accompanied by some additional reactions.
Kato et al. [
6] reported that under a CO
2 and H
2 gas flow over Mg
2NiD
4 covered with surface oxide layers, the conversion of CO
2 to CO (Equation (2)) and methane (Equation (3)) commenced at 330 °C. The sum of these reactions was exothermic, and, considering that there was some hydrogen released from the Mg
2NiH
4 methanation of CO/CO
2, this could explain the lack of an endothermic minimum in the DSC curve. However, the CO
2 methanation through Reactions (2) and (3) does not explain the lack of the mass loss. The Bosch reaction (reaction (4)), carbon monoxide reduction (reaction (9)), and Boudouard Reaction (10) could potentially lead to solid carbon build up [
29,
30]. However, most of these reactions become depressed as temperature is increased [
9]. Therefore, without the complete denial of carbonization, we suggest that the disproportionation of Mg
2Ni powder surface and its oxidation were the most dominant processes in our experiments. A similar conclusion was drawn up by other researchers [
6,
15,
16] and was supported by the eventual observation of an MgO phase by in-situ XRD. In this reaction pathway, finite oxygen adsorption compensated the mass of the desorbed hydrogen; meanwhile, the strongly exothermic formation of MgO overcame the endothermic decomposition of Mg
2NiH
4. To reaffirm this view, we note that the TGA curve reached its minimum slightly above 400 °C, which was consistent with the disappearance of Mg
2NiH
4 XRD peaks at 410–420 °C (
Figure 2). The exhaustion of hydrogen sources should have stopped all of the above-mentioned carbon production reactions, stabilized sample mass, and translated into severe changes of the DSC curve. Instead, both curves maintained small increases, thus indicating the superior role of other processes.
A strong endothermal event at 475 °C was accompanied by a 0.1% mass loss that was equal to the hydrogen desorption in the last section of the sample heating under Ar (
Figure 5a). Accordingly, following the same reasoning as above, we attribute this event to the decomposition of MgH
2. The higher than usual decomposition temperature can be explained by two factors: (i) MgH
2 decomposition is usually kinetically limited [
31] and the heating rate during DSC/TGA analysis was relatively high (10 °C/min); (ii) the dehydriding temperature is known to be postponed by surface oxides [
15,
32]. After MgH
2 decomposition, the powder reaction with CO
2 was finalized by some more thermal events, thus representing rapid oxidation of Mg that was produced by the dehydriding of MgH
2 and continued the disproportionation of Mg
2Ni and, eventually, MgNi
2.
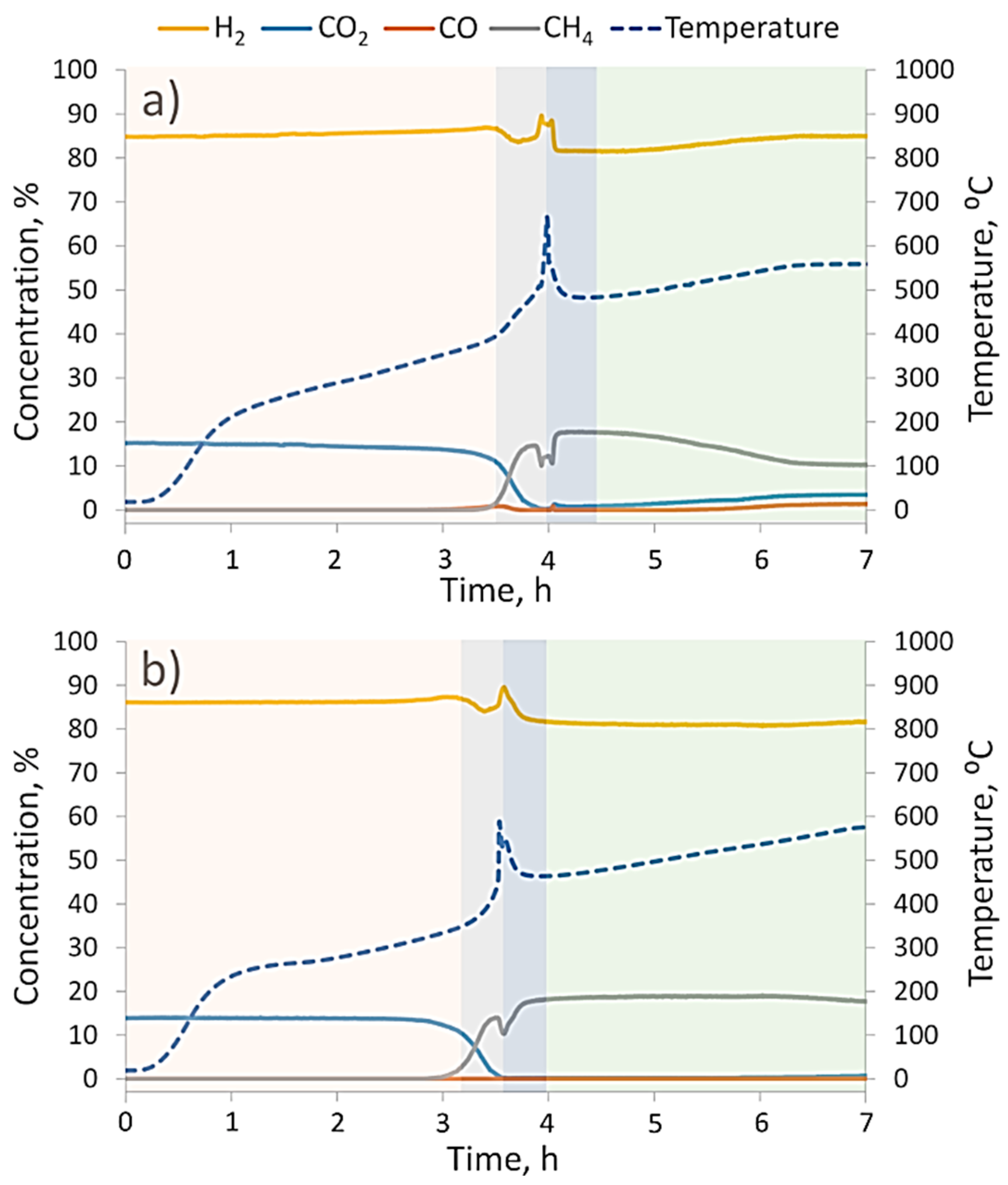
The CO
2 methanation reaction dynamics at 1 and 10 bars of gas pressure are provided in
Figure 6a,b, respectively. Both charts have temperature peaks that correspond to the fast decomposition of hydrides (Mg
2NiH
4 and MgH
2) and the oxidation of Mg
2Ni disproportionation products. After the heat burst, we found a new catalyst with substantially different properties.
By comparing methanation dynamics at different pressures, it could be noticed that at 1 bar in a low temperature region (prior to the temperature burst), there was a slight increase in H
2 (and a decrease in CO
2) concentration, whereas at 10 bars, the H
2:CO
2 ratio remained stable. At 1 bar of pressure, such behavior was not surprising because a similar hydrogen release was also observed in the TGA pattern (
Figure 5a) and was attributed to the plateau pressure levels of Mg
2NiH
4 [
33,
34]. On the other hand, Mg
2NiH
4 stability at 10 bars of pressure demonstrated that high partial hydrogen pressure was a stronger factor for the stabilization of Mg
2NiH
4 than the oxidative potential of CO
2, even though at low pressure Mg
2NiH
4 decomposition and partial oxidation happened at the same time (see
Figure 5b and comments above).
The second thing to notice from
Figure 6 is that at 10 bars of pressure, the methanation reaction started at approximately 50 °C lower (330 °C versus 380 °C), and as soon as temperature reached 420–430 °C, there were almost no unreacted CO
2 or intermediate products from reaction (2), namely CO. A nearly 100% CO
2 conversion was maintained up to approximately 530 °C, with an estimated CH
4 yield of approximately 75%. On the other hand, methanation at 1 bar of pressure was able to reach a 100% CO
2 conversion within a narrow time frame when the hydrogen desorption from powders significantly increased the hydrogen concentration in the gas flow. When the hydrogen source was depleted, the methanation reaction lost its temporarily boosted efficiency (approximately 90% at 500 °C) and maintained a downtrend as temperature rose to 550 °C. At both investigated pressures, CO
2 methanation by the reaction product of Mg
2NiH
4 powder had lower CH
4 yields than were achieved with standard commercial Ni-based catalysts at 10 bar (H
2:CO
2 ratio of 4:1) and 30 bar (H
2:CO
2 ratio of 6:1) [
9]. When operating at 10 bars of pressure, we did not detect CO or other carbon-containing species in gaseous reaction products; therefore, we assumed that relative difference between initial CO
2 and CH
4 product fluxes qualitatively reflected the actual solid C yields. This was quite surprising because theoretical and experimental study by Gao et al. [
9] estimated that at the specifically used gas pressure and H
2:CO
2 ratios, carbonization should have been prohibited. The cause of such a high carbon yield has not yet been identified and will be subject of our future studies.
A study by Kato et al. [
6] demonstrated that the properties of powder catalysts can be significantly improved after they are cycled through H
2 (25 bar and 350 °C), CO
2 (1 bar and 500 °C), and vacuum (500 °C). They reported that after 18 cycles, the methanation process was initiated at 250–260 °C, whereas an uncycled high purity Mg
2NiH
4 catalyst initiated a methanation reaction at approximately 330–340 °C (in comparison to the current study with a relatively low grade Mg
2NiH
4 catalyst that initiated methanation at 330 °C for 1 bar of pressure and 380 °C for 10 bars of pressure).
After considering the potential benefits of catalyst cycling after the first methanation run-up at 10 bars of pressure was finished, we gradually decreased furnace temperature down to 250 °C. During the cool-down period and 30 min after the target temperature was reached, the container was constantly flushed with hydrogen. Subsequently, the container was filled up with hydrogen to 10 bars of pressure and confined for 16 h at 250 °C. The next day, we repeated the methanation process (
Figure 7) with same parameters as were used before (10 bars of pressure and H
2:CO
2 ratio of 6:1). This time, the methanation reaction started immediately at 250 °C (close to the lowest reported values [
35,
36]), reached a 100% CO
2 conversion at 320–330 °C, and continued without the significant deterioration of CO
2 conversion up to the maximum tested temperature of 600 °C. The CH
4 yield at 500 °C was slightly higher and reached just over 80%. Looking at the methanation dynamics (
Figure 7a), one can notice that there was no temperature burst or hydrogen concentration increase that could be attributed to the decomposition of Mg
2NiH
4 or MgH
2 hydride. This suggested that during the first methanation experiment, Mg
2NiH
4 and MgH
2 hydrides completely disproportionated, and only the MgO and Ni phases were formed. Accordingly, such a powder catalyst did not adsorb any additional hydrogen and was able to provide an enhanced performance after one just methanation cycle.
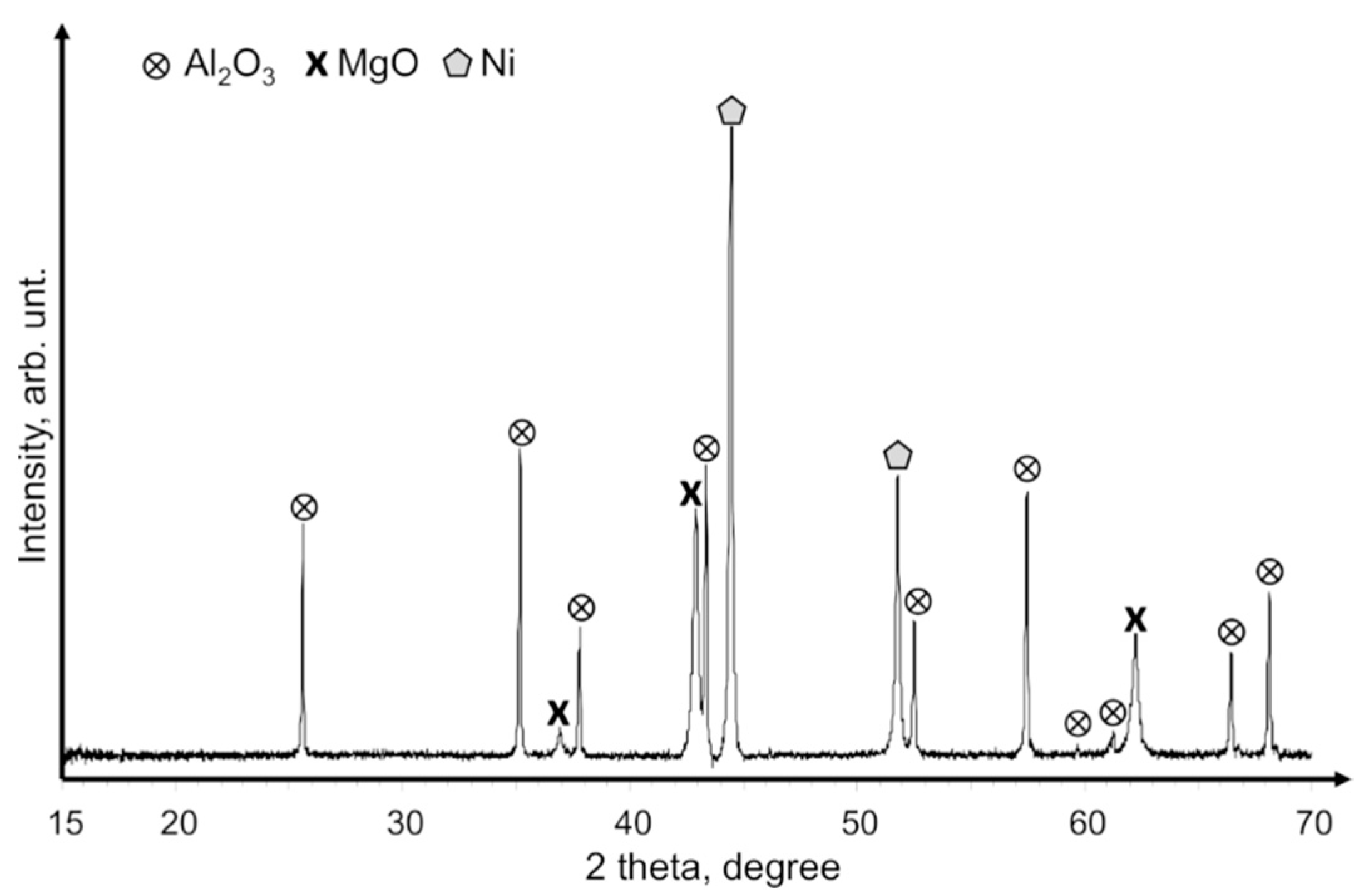
A comparison of SEM images of the as-ground Mg
2Ni alloy grains (
Figure 8b) and the final catalyst powder (
Figure 8c) provided some more evidence regarding how specific features of hydrides could become beneficial for the formation of efficient catalysts for the methanation reaction. The initially ground Mg
2Ni alloy grains were relatively course, and their size ranged from 10 to 50 µm. During hydride formation (hydriding/dehydriding cycling), the catalyst material underwent huge volume changes (up to 30% [
37]) that introduced large amount of structural defects, widened up grain boundaries, and ultimately fractured initial grains into sub-micrometer-size fine powders (
Figure 8c). The first temperature run-up under CO
2 and H
2 gas flow promoted CO
2 methanation and initiated hydride decomposition and oxidation. These highly energetic chemical reactions empowered Mg
2Ni disproportionation and MgO–Ni phase separation (
Figure 9). As the result, MgO particles decorated by nanocrystalline Ni were formed. Such a catalyst is able to promote CH
4 formation at a relatively low temperature (250 °C) and an do this with a significantly higher CH
4 output than that not oxidized hydride powder. The superior catalytic properties of disproportionated MgO–Ni systems can be attributed to the role of MgO because MgO does not just work as an inert support for catalytic Ni nanoparticles but also actively changes the methanation pathway for Ni [
38]. For example, studies have shown that MgO enhances the adsorption and activation of CO
2 [
39]. On the other hand, MgO was also found to promote H
2O formation and its desorption from a catalyst [
15].
In-situ XRD, TGA/DSC, and methanation reaction products analyses showed that in order for all these reactions to complete, it is recommended to heat a hydride powder above 500 °C (in a CO
2-containing atmosphere) or, for a short period of time, as high as 600 °C. When such high temperature conditioning is induced (or in the current study case, a spontaneous temperature burst is not suppressed), the full development of the catalyst can be obtained in one cycle within 1 h. Meanwhile, the catalyst development from a Mg
2NiH
4 powder kept below 500 °C might take more than 20 hydriding/dehydriding cycles [
6] or up to 10-fold more time for the reaction to complete [
15].

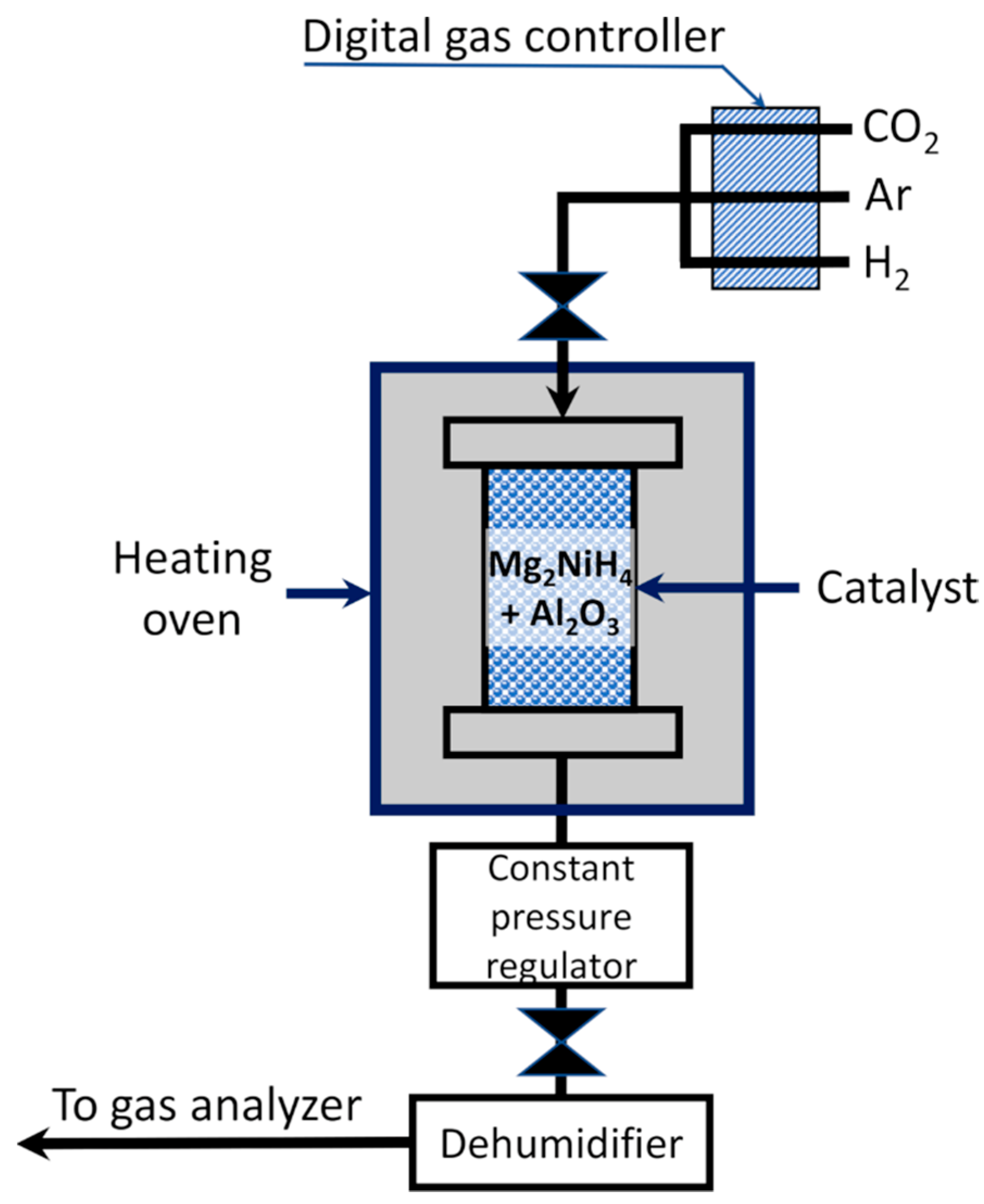{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








