Abstract
Performance tests on caffeine’s corrosion inhibition properties and their derivatives against copper corrosion have been previously reported experimentally using gravimetric and electrochemical analyses. The test was able to measure the efficiency of their corrosion inhibition accurately. However, the caffeine and its derivatives’ structure patterns and coating mechanisms when interacting with metals during copper corrosion inhibition have not been explained in detail by experimental studies. In the present study, the theoretical density functional study (DFT), ab initio MP2, and Monte Carlo simulation approaches explain the problem. The geometrical and quantum chemical parameters of inhibitors were compared under normal and protonated conditions in the gas and aqueous environments. Theoretical studies can accurately determine the molecule’s geometrical parameters and successfully explain the quantum parameters of inhibitors. Molecular dynamics are applied to study the mechanism of interaction between inhibitors and metal surfaces in an explicit water molecule environment. The energy absorption of caffeine and its derivatives on metal surfaces was linear, with quantum parameters calculated from the density functional theory and an ab initio approach. Furthermore, these theoretical study results align with the previously reported experimental studies published by de Souza et al. The inhibition efficiency ranking of studied molecules preventing copper corrosion was caffeine > theobromine > theophylline. This theoretical approach is expected to bridge the gap in designing effective corrosion inhibitors.
1. Introduction
Corrosion is an electrochemical process in metals that damages metal structures. This process is usually triggered by corrosive environments such as hydrochloric acid and nitric acid. The corrosion process is challenging to overcome, and without prevention, corrosion will cause significant economic losses [1]. Therefore, research to find corrosion inhibitors that are environmentally friendly, economical, and of high efficiency, is still being carried out intensively. Organic compounds from natural products have a high potential to be developed as corrosion inhibitors. This compound was chosen as a corrosion inhibitor because it meets the criteria of high efficiency, economical, environmentally friendly, and non-toxic, and it does not cause harmful pollutants [2,3,4].
Many experimental studies on the efficiency of corrosion inhibition of natural product compounds have been carried out [5,6,7]. For example, alkaloids obtained from natural product extraction have high potential as good corrosion inhibitors [8,9,10]. The main factors that play a role in inhibiting corrosion are electrostatic interactions, Π electron donors, and heteroatom groups on inhibitor molecules [11]. Heterocyclic benzene and heteroatom groups such as oxygen and nitrogen in the alkaloid structure play a role in electron donors, electrostatic interactions, and electron donor acceptors in their interactions with metal surfaces. These factors will be utilized by the inhibitor to be firmly attached to the metal surface and form a thin layer that will inhibit the rate of corrosion [12].
Caffeine is an alkaloid extracted from natural products that have been tested for its corrosion inhibition efficiency [13,14,15]. Fallavena et al. examined caffeine’s ability to inhibit corrosion on copper surfaces in a solution of potassium nitrate using electrochemical techniques. The experimental results show that caffeine can inhibit corrosion by decreasing the corrosion rate in copper [16]. Combining complex caffeine-Zn2+ systems has also been studied to optimize caffeine’s corrosion inhibition efficiency against copper corrosion. The complex is capable of forming caffeine-Zn2+ film complex formation on metal surfaces [17]. Other studies also show that caffeine can coat the carbon steel surface in ethanol solvents to prevent corrosion [18]. Moreover, caffeine has also been tested to inhibit corrosion on the surface of the nickel. Ebadi et al. showed that caffeine was well adsorbed on nickel’s surface with an inhibitory efficiency of 96.8% [19]. Furthermore, caffeine derivatives have been assessed for their ability to reduce corrosion in copper. de Souza et al. showed that caffeine effectively inhibited copper corrosion than theobromine and theophylline [20].
Accurate measurement of the corrosion inhibitor performance of a compound can be done experimentally. However, this technique has a limitation: the mechanism of inhibition by inhibitors on metal surfaces may not be explained in detail. The cost and time of research is expensive. For an instant, de Souza et al. experimentally showed that caffeine more effectively inhibited copper corrosion than theobromine and theophylline [20]. However, de Souza et al. have not explained why caffeine is more efficient at inhibiting corrosion than theobromine and theophylline. The detailed pattern and mechanism of interactions between caffeine, theobromine, theophylline, and copper surfaces are not well explained. Therefore, the theoretical study supported by computer hardware and software development is a bridge to overcome this gap. The density functional theory approaches [21,22,23,24,25], ab initio [26,27,28] can explain in detail about the distribution and transfer of electrons when inhibitors interact with metals. Furthermore, the Monte Carlo simulation can explain in detail the effect of orientation and structure on the performance of each inhibitor, including the mechanism of adsorption of inhibitor molecules on metal surfaces. The density functional theory approaches [21,22,23,24,25], ab initio [26,27,28], and molecular dynamics simulation can explain in detail the effect of orientation and structure on the performance of each inhibitor, including the mechanism of adsorption of inhibitor molecules on metal surfaces [29,30]. Kaya et al. [31,32] combined DFT calculations at various theoretical and dynamic molecular levels to predict iron corrosion inhibition performance by thiazole and piperidine derivatives. It was reported that the strength of the interaction between thiazole and the surface of Fe (110) is determined by the orientation of the molecule and its donor and electron-withdrawing groups [31,32]. The theoretical studies that have been carried out show compatibility with experimental studies. The current report focuses on examining caffeine’s performance and its derivatives as a corrosion inhibitor on copper’s surface theoretically by combining the density functional theory, ab initio, and Monte Carlo simulation simultaneously. This research will also present the coating mechanism of caffeine and its derivatives on the copper surface.
2. Materials and Methods
2.1. Quantum Chemical Calculations
Quantum chemical calculations are applied to predict the structure and electron distribution of caffeine, theobromine, theophylline, and their electron transfer to the copper surface. Density functional theory is a very popular method for assessing the reactivity of molecules [33]. The ab initio method is also widely used because it has high accuracy [34]. In the quantum chemical calculation section, all calculations, including optimization of caffeine, theobromine, and theophylline, are carried out using the DFT and MP2 methods in a combination of 6-31G(d) and 6-311++G(d,p) basis sets. Geometry optimizations have been performed without any symmetry constraints. Optimized geometries are always verified as minimum energy potential surface by calculating the harmonic vibration frequencies. All quantum chemical calculations use the Gaussian 09 program [35]. Solvent effects are included using the polarized continuum model as implemented in the Gaussian code. The dielectric constant for the water solvent was taken as 78.4, and other solvents were used as in the Gaussian code. In employing polarized continuum models, the single-point calculations on gas-phase geometries are sufficient for energetics. The structure of re-optimization in the solvent’s presence was found to have a minor influence on energetics [36,37]. Therefore, the single point approach has been employed in this study, minimizing computational costs without sacrificing much accuracy in solvation energies.
The quantum chemical parameters such as the energy of the highest occupied molecular orbital (EHOMO), the energy of the lowest unoccupied molecular orbital (ELUMO), the ionization potential (I), the electron affinity (A), the absolute electronegativity (χ), hardness (η), softness (σ), the fraction of electron transferred (ΔN), and the corrosion inhibitors (IE%) efficiencies were calculated. According to Koopman’s theorem [38], ionization potential (I) and electron affinity (A), the electronegativity (χ), and global hardness (η) may be defined in terms of the EHOMO and the ELUMO. Ionization potential (I) is defined as the amount of energy required to remove an electron from a molecule [39]. It is related to the energy of the EHOMO through the Equation (1):
I = −EHOMO
Electron affinity (A) is defined as the energy released when a proton is added to a system [39]. It is related to ELUMO through the Equation (2):
A = −ELUMO
Electronegativity is the measure of the power of an atom or group of atoms to attract electrons towards itself [40]; it can be estimated by using the Equation (3):
Chemical hardness (η) measures an atom’s resistance to a charge transfer [41]; it is estimated by using the Equation (4):
According to Pearson theory [42], the fraction of transferred electrons (ΔN) from the caffeine, theobromine and theophylline to the copper atom can be calculated Equation (5):
where χCu and χinh denote the absolute electronegativity of copper and inhibitor molecule respectively, ηCu and ηinh denote the absolute hardness of copper and the inhibitor molecule, respectively. To calculate the fraction of electrons transferred, the theoretical value for bulk iron’s electronegativity was used χCu = 4.43 eV [43] and a global hardness of ηCu = 0 by assuming that for a metallic bulk I = A [44].
The Fukui indices for the nucleophilic and electrophilic sites (f+ and f−) are expressed [45] via Equations (6) and (7).
f+ = qN + 1 − qN (nucleophilic attack)
f− = qN – qN − 1 (electrophilic attack)
According to Koopman’s theorem [38], ionization potential (I) and electron affinity (A), the electronegativity (χ), and global hardness (η) may be defined in terms of the EHOMO and the ELUMO. Ionization potential (I) is defined as the amount of energy required to remove an electron from a molecule [39]. It is related to the EHOMO through the Equation (1).
2.2. Monte Carlo Simulations
The Monte Carlo simulation is very popular for investigating the interaction between the molecule inhibitor and the concerned metal surface [45,46,47]. The Monte Carlo simulation can explain a key element in the corrosion problem: the adsorption phenomenon. The most stable adsorption sites on metal surfaces with low energy. The Monte Carlo metropolis simulation methodology is used in molecular dynamics simulations performed using the adsorption locator and the Forcite code implemented in Material Studio 7.0 software [48,49]. The lowest conformational energy search is performed by adopting a Monte Carlo search of interactions between inhibitor molecules and clean copper surfaces in water. The COMPASS force field is used to optimize all the studied molecular structures. The advantages of the COMPASS force field are the ab initio force fields that allow accurate and simultaneous gas-phase predictions and condensation properties. Those properties, including structural, conformational, vibration and state equations, cohesive energy, interaction energy for a variety of organic metal molecules, metal oxides, and metal halides, use a variety of solid properties: the structure of cell units, lattice energy, and even polymers. The first step in this computational study is to optimize the geometry of the inhibitor molecule: caffeine, theobromine, and theophylline, which will adsorb next on the copper surface with minimized energy. For this purpose, the Forcite calculation was performed with the fine calculation quality using the COMPASS forcefield. Once the caffeine, theobromine, and theophylline have been optimized with the COMPASS forcefield, we simulate those corrosion inhibitors by loading with a copper surface, considering the solvent effect. The objective of this computational study is to find the low-energy adsorption sites to investigate the preferential adsorption of caffeine, theobromine, and theophylline on Cu(1 1 1) surface and to find a relationship between the effect of their molecular structure and their inhibition efficiency. The simulation protocol details follow the simulation steps that have been carried out previously [50] with the crystal surface of Cu(111) in the simulation box (20.447831 Å × 20.447831 Å × 40.43474 Å) with periodic boundary conditions to simulate the representative part of the interface without arbitrary boundary effects. The Cu plane (111) was further enlarged to supercell (8 × 8). After that, a vacuum slab with a 3.0 nm thickness was built on the Cu plane (111). The three caffeine, theobromine, and theophylline corrosion inhibitors, along with 100 water molecules, are used for simulations in each case. Water is essential in simulations because it plays a vital role in corrosion in the natural environment.
3. Results and Discussion
The theoretical study can be very close to wet laboratory experiment data, or it can also be incompatible. As a consequence of using quantum mechanical methods in theoretical calculations, it is necessary to validate the theoretical approach by comparing experimental data [51]. For this purpose, we have tested the compatibility of the 6-311++G (d,p) method and basis set options with the studied system (Figure 1). Comparisons were made between theoretical study results with the crystal structures of the three previously published inhibitor molecules: caffeine [52], theobromine [53], and theophylline [54]. It was used to test the accuracy of theoretical calculations in terms of molecular geometry parameters. The experimental results’ geometry parameters compared with the theoretical calculation using the DFT/6-311++G (d,p) method are depicted in Table 1. Table 1 shows the linear correlation between experimental X-ray and theoretical data. The difference in bond lengths and bonding angles of the X-ray structure of experiments and theoretical studies for the three compounds is 0.03 Å and 0.65°, respectively. The small difference between theoretical and experimental shows that the level of methods and the basis of the theoretical set of studies can be applied to the system being studied.
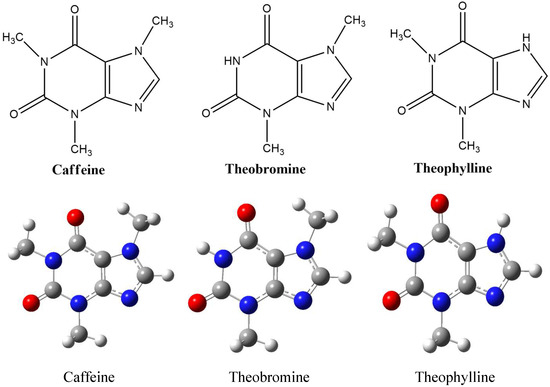
Figure 1.
Optimized geometry of caffeine, theobromine, theophylline at B3LYP/6-311++G(dp) level of theory.

Table 1.
Structural parameters of the optimized geometry of caffeine, theobromine, theophylline at B3LYP/6-311++G(d,p) level of theory.
Electron transfer between molecules can be studied by showing the conditions of molecular orbitals. The interaction between the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO) is the cause of electron transfer between molecules [39]. Electron transfer can be measured using energy values from orbitals. HOMO energy (EHOMO) indicates a molecule’s nature to donate its electrons, while LUMO energy (ELUMO) indicates the nature of a molecule to receive electrons. The greater the EHOMO or, the smaller ELUMO, the greater the electron donor so that the stronger an organic molecule is attached to the metal. As a consequence, these organic molecules will have high corrosion inhibition efficiency. Table 2, Table 3, Table 4 and Table 5 show the quantum chemical parameters for caffeine, theobromine, and theophylline, which were calculated using DFT and MP2 at theoretical levels 6-31G(d) and 6-311++G(d,p). All quantum chemical parameter data in Table 2, Table 3, Table 4 and Table 5 were calculated as given by Equations (1)–(5). Table 2, Table 3, Table 4 and Table 5 show that the increase in EHOMO value is caffeine > theobromine > theophylline. The EHOMO calculated using MP2/6-31G(d) for caffeine is −8.5411 eV, while the EHOMO for theophylline is the lowest, −8.6866 eV. These predictions from EHOMO suggest that caffeine will have the highest corrosion inhibition efficiency (IE%) compared to the other two compounds. This theoretical result is following an experimental study conducted previously by de Souza et al. They explained that the order of efficiency of corrosion inhibition of copper metal in acidic medium was caffeine > theobromine > theophylline [20]. The distribution of electrons in molecular orbitals on caffeine, theobromine, and theophylline is visualized in Figure 2. The difference in electron distribution in the three compounds is apparent where caffeine has a wider electron distribution. It strengthens the predicted EHOMO-related sequence of enhancing inhibitor performance.
Table 2.
The calculated quantum chemical parameters of neutral caffeine, theobromine, and theophylline in the gas phase at a different level of theory. (All values are in eV).
Table 3.
The calculated quantum chemical parameters of protonated caffeine, theobromine, and theophylline in the gas phase at different levels of theory. (All values are in eV).
Table 4.
The calculated quantum chemical parameters of neutral caffeine, theobromine, and theophylline in the aqueous phase at a different level of theory. (All values are in eV).
Table 5.
The calculated quantum chemical parameters of protonated caffeine, theobromine, and theophylline in the aqueous phase at different theory levels. (All values are in eV).
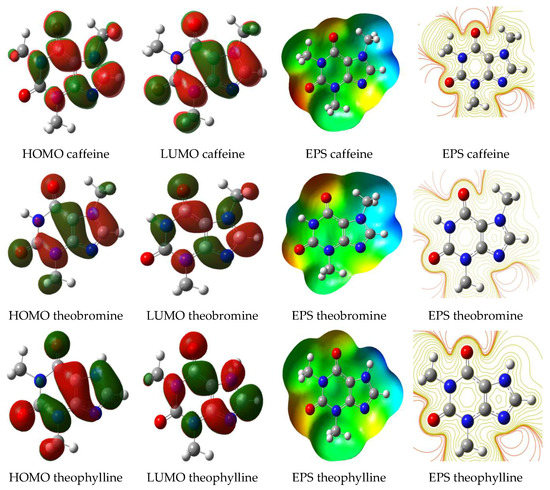
Figure 2.
Visualization of highest occupied molecular orbitals–lowest unoccupied molecular orbitals (HOMO–LUMO) energies and electrostatic potential of caffeine and its derivatives.
Ionization potential (I) can be used to measure the reactivity of a molecule. A high ionization potential indicates that the molecule has high reactivity, while a low ionization potential value indicates the molecule has a low reactivity [39]. Table 2, Table 3, Table 4 and Table 5 also show the pattern of increase in ionization potential, which follows the pattern of increase of EHOMO. The potential ionization value of caffeine calculated using MP2/6-31G (d) is 8.5411 eV, and it is lower than the potential ionization value of theobromine and theophylline. Based on this ionization potential, it can be predicted that caffeine has a higher corrosion inhibition efficiency (IE%) than theobromine and theophylline. This potential ionization value can also be used to test the accuracy of the theoretical calculation methods. The potential ionization value measured using DFT/B3LYP is far below the standard experimental value [55,56]. For example, experimentally, the value of caffeine’s ionization potential is 8.31 eV, while DFT/B3LYP/6-311++G(d,p) gives a value of 6.3310 eV. The potential ionization value of DFT/B3LYP is 2 eV lower than the experimental results. It shows the weakness of DFT, which failed to imitate the energy of the experimental results. A similar finding was found by previous researchers [57,58,59], although DFT was successful in mimicking the results of the structural parameters, as seen in Table 1. We, therefore, recommend that DFT/B3LYP not be used to measure the energy of a quantum parameter of a molecule.
Small electronegativity values cause molecules to easily reach electron equilibrium so that the molecules get more reactive. In contrast, high electronegativity values show the opposite [40]. Table 2, Table 3, Table 4 and Table 5 show how electronegativity increases are caffeine < theobromine < theophylline. The electronegativity value of caffeine that calculated using MP2/6-311++G(d,p) is the lowest (3.94 eV) compared to the electronegativity values for theobromine, and theophylline, which are 4.0261 and 4.0671 eV, respectively. Based on electronegativity data, it is predicted that caffeine has a higher corrosion inhibition efficiency (IE%) than theobromine and theophylline. Electronegativity obtained in theoretical calculations has a linear relationship with experimental studies from de Sousa et al. [20].
Table 2, Table 3, Table 4 and Table 5 show that electron transfer also affects the corrosion inhibition efficiency of caffeine and its derivatives. The fraction of electrons (∆N) quantifies the transfer of electrons from molecule to metal if ΔN > 0, and from metal to molecule if ΔN < 0 [60]. In general, the efficiency of corrosion inhibition increases as the value of the electron transfer increases, because the more electrons are transferred to the iron surface, the more electrons will coat the iron surface, so that the corrosion process can be inhibited [41]. The order of magnitude of the electron transfer for the three compounds to copper is caffeine > theobromine > theophylline. These results apply to all conditions in both the gas and solution phases, as well as for normal and protonated molecular conditions.
The local reactivity of caffeine, theobromine, and theophylline can be studied by observing the Fukui indices of each of their atoms. The Fukui indices provide more comprehensive information of the reactivity of the molecules under probe. In general, the numerical representation of the Fukui indices is the highest f− the site indication for electrophilic attack or molecule receive electron, whereas the highest f+ denotes the site for nucleophilic attack or when the molecule donates electrons [61,62].
The local reactivity of a molecule is analyzed using condensed Fukui indices as depicted in Table 6. For nucleophilic attack the most reactive sites of caffeine are O15, O16, and N24 atoms which are indicating the propensity to donate electrons to vacant molecular orbitals on the Fe (110) surface to form a coordinate bond, and the most reactive site for electrophilic attack is C17. The local softness indices also explain the comparison between reactivity of similar atoms of each part of different molecules. This result is linear with the calculation of HOMO density. For theobromine, the most nucleophilic sites are O11, O12, and N20 atoms, and the most electrophilic site is the C14 atom. For theophylline, the most nucleophilic sites are O11, O12, and N16 atoms, the most electrophilic site is the C13 atom. The electrophilic attack corresponds to the sites that most likely cause the molecule to accept electrons from the Fe (110) surface in the form of a back donation. This result corresponds to the LUMO energy values of caffeine, theobromine and theophylline.
Table 6.
Calculated Mulliken atomic charge distribution and Fukui indices for caffeine, theobromine, and theophylline in condensed phase at the B3LYP/6-311++G(dp) level.
The Monte Carlo Metropolis simulation is performed to find a possible position on the interaction between the inhibitor and the copper surface in aqueous solution. Figure 3 shows the most stable low energy adsorption configuration of the inhibitor in the Cu (111)/100H2O system using a Monte Carlo simulation. The detailed analysis results on the shortest length of the active side bond of the inhibitor with copper are less than 3.50 A, which indicates the formation of a strong adhered layer between the inhibitor and the copper surface [63,64]. This layer coats the copper surface and prevents copper from being attacked by aggressive solutions. This strong layer bond also suggests the transfer of electron density from the inhibitor’s active side to the d-orbitals of copper. This electron transfer causes strong interactions between the inhibitor and copper so that it can inhibit the rate of corrosion. Furthermore, van der Waals interactions are also involved in the adsorption of inhibitors with Cu surfaces. It is evident from observations with a bond distance above 3.50 A. Table 7 shows the adsorption energy for the most stable configurations for Cu(111)/Caffeine/100H2O, Cu(111)/Theophylline/100H2O, and Cu(111)/Theobromine/100H2O systems. Table 7 also shows the adsorption energy of water. In all cases, the adsorption inhibitors’ energy was much higher than water molecules (Table 7). It shows the possibility of gradual replacement of water molecules from the copper surface, which results in the formation of a stable layer that can protect the copper from aqueous corrosion. It can be seen from Table 6 that the energy of adsorption inhibitors on the iron surface in the presence of water decreases in the following sequence caffeine > theobromine > theophylline. This sequence is in accordance with an experimental study of caffeine, theobromine, and theophylline, which was carried out previously by de Souza et al. [20]. Caffeine has the best inhibitory ability while theophylline ranks lowest. The adsorption energy distribution of the inhibitors and water is depicted in Figure 4. It explains why caffeine is able to form a more stable layer on the copper surface than the other two inhibitors. Adsorption energy distribution in caffeine and water is separated from each other. In contrast, at some energy points of theobromine and theophylline adsorption energy distribution are almost the same as the adsorption energy distribution of water, so they form a weaker layer because, at some energy point, water is able to compete with them. Figure 5 and Figure 6 show the correlation of the adsorption energy and quantum parameters of neutral and protonated inhibitors on copper surfaces. The Pearson correlation in Figure 5 and Figure 6 indicates a linear correlation between energy adsorption and quantum parameters, especially EHOMO and ΔN. Still, it is not followed by electronegativity χ and hardness η. This shows that EHOMO and ΔN play an essential role in the inhibitor’s adsorption on the copper surface. It is generally recognized that the primary mechanism of interaction of corrosion inhibitors with copper is by adsorption. So, the adsorption energy can give us a direct tool for ranking inhibitory molecules. High negative adsorption energy indicates the system with the most stable and strong adsorption [64].
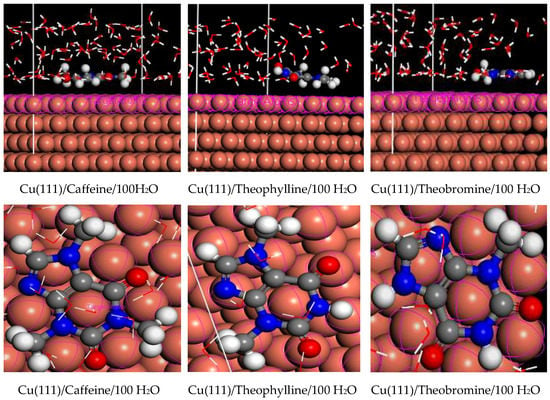
Figure 3.
Top and side views of the most stable low energy configuration for the adsorption for Cu(111)/Caffeine/100H2O, Cu(111)/Theophylline/100H2O, and Cu(111)/Theobromine/100H2O systems.
Table 7.
Adsorption Energy for the most stable configurations for Cu(111)/Caffeine/100H2O, Cu(111)/Theophylline/100H2O and Cu(111)/Theobromine/100H2O systems (all values are in kcal/mol).
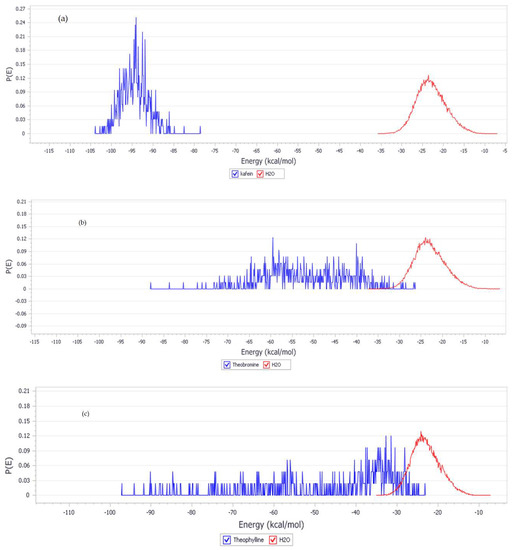
Figure 4.
Adsorption energy distributions for (a) Cu(111)/Caffeine/100H2O, (b) Cu(111)/ Theobromine /100H2O, and (c) Cu(111)/ Theophylline /100H2O systems.
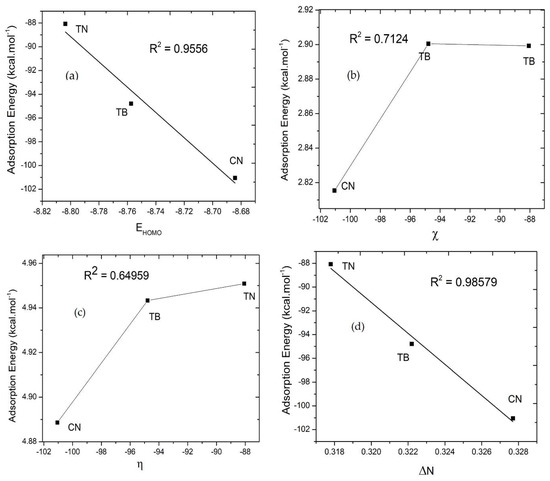
Figure 5.
Correlation of the adsorption energy and quantum parameters such as EHOMO, electronegativity, hardness, and electron transfer of neutral inhibitors (caffeine (CN), theophylline (TP), and theobromine (TB)) in copper surface. (a) The correlation between the adsorption energy and EHOMO, (b) the adsorption energy and electronegativity, (c) the adsorption energy and hardness, (d) the adsorption energy and electron transfer.
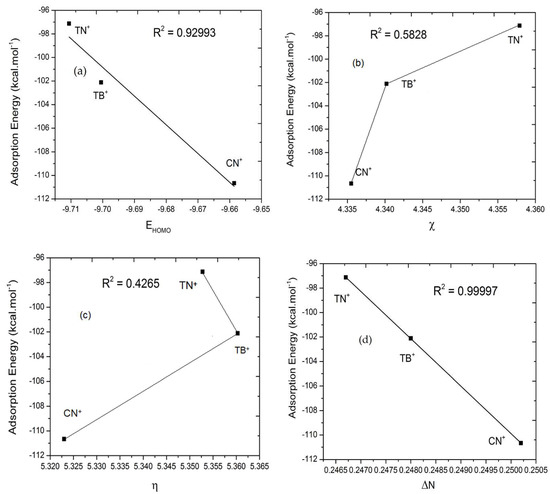
Figure 6.
Correlation of the adsorption energy and quantum parameters such as EHOMO, electronegativity, hardness, and electron transfer of protonated inhibitors (caffeine (CN+), theophylline (TP+), and theobromine (TB+)) in copper surface. (a) The correlation between the adsorption energy and EHOMO, (b) the adsorption energy and electronegativity, (c) the adsorption energy and hardness, (d) the adsorption energy and electron transfer.
4. Conclusions
This theoretical study aims to evaluate caffeine, theobromine, and theophylline’s corrosion inhibition performance against copper corrosion in gas and aqueous conditions. The density functional theory at B3LYP function and ab initio MP2 at various levels of theory and Monte Carlo simulation study were employed to evaluate the corrosion inhibition efficiency and to provide a detailed picture of the mechanism of corrosion inhibition of these three compounds on the copper surface. According to all data given in this study, caffeine, theobromine, and theophylline will be effective in preventing the corrosion of copper. The DFT, MP2 approach, and Monte Carlo simulation study showed that the inhibition efficiency ranking of studied molecules in preventing corrosion of copper as caffeine > theobromine > theophylline. The adsorption energy obtained in the study shows that the most effective inhibitor among them is caffeine. The theoretical research agrees with the results of previously published experimental studies by de Souza et al. This theoretical study will help the rational design of a more effective caffeine-based corrosion inhibitor.
Author Contributions
Conceptualization, S.H. (Saprizal Hadisaputra) and A.A.P.; methodology, N.P. and S.H. (Saprizal Hadisaputra); data curation, E.Y., S.H. (Saprini Hamdiani); writing—original draft preparation, S.H. (Saprizal Hadisaputra), N.P., and E.Y.; writing—review and editing, L.R.T.S., A.A.P., and S.H. (Saprizal Hadisaputra). All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Kemenristekdikti Republic of Indonesia, Grant No. 1730/UN18.L1/PP/2020 and The APC was funded by Kemenristekdikti Republic of Indonesia, Grant No. 1730/UN18.L1/PP/2020.
Acknowledgments
Financially supported by Kemenristekdikti Republic of Indonesia through Penelitian Dasar 2020, Grant No. 1730/UN18.L1/PP/2020, is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Umoren, S.A.; Solomon, M.M.; Obot, I.B.; Sulieman, R.K. A critical review on the recent studies on plant biomaterials as corrosion inhibitors for industrial metals. J. Ind. Eng. Chem. 2019, 76, 91–115. [Google Scholar] [CrossRef]
- Xhanari, K.; Finšgar, M.; Hrnčič, M.K.; Maver, U.; Knez, Ž.; Seiti, B. Green corrosion inhibitors for aluminium and its alloys: A review. RSC Adv. 2017, 7, 27299–27330. [Google Scholar] [CrossRef]
- Hadisaputra, S.; Purwoko, A.A.; Rahmawati; Asnawati, D.; Hamdiani, I.S.; Nuryono. Experimental and Theoretical Studies of (2R)-5-hydroxy-7-methoxy-2-phenyl-2, 3-dihydrochromen-4-one as corrosion inhibitor for Iron in Hydrochloric Acid. Int. J. Electrochem. Sci. 2019, 14, 11110–11121. [Google Scholar] [CrossRef]
- Sedik, A.; Lerari, D.; Salci, A.; Athmani, S.; Bachari, K.; Gecibesler, İ.H.; Solmaz, R. Dardagan Fruit extract as eco-friendly corrosion inhibitor for mild steel in 1 M HCl: Electrochemical and surface morphological studies. J. Taiwan Inst. Chem. Eng. 2020, 107, 189–200. [Google Scholar] [CrossRef]
- Hadisaputra, S.; Purwoko, A.A.; Ilhamsyah, I.; Hamdiani, S.; Suhendra, D.; Nuryono, N.; Bundjali, B. A combined experimental and theoretical study of (E)-ethyl 3-(4-methoxyphenyl) acrylate as corrosion inhibitor of iron in 1 M HCl solutions. Int. J. Corros. Scale Inhib. 2018, 7, 633–647. [Google Scholar] [CrossRef]
- Vu, N.S.H.; Binh, P.M.Q.; Ai, D.V.; Thu, V.T.H.; van Hien, P.; Panaitescu, C.; Nam, N.D. Combined experimental and computational studies on corrosion inhibition of Houttuynia cordata leaf extract for steel in HCl medium. J. Mol. Liq. 2020, 113787. [Google Scholar] [CrossRef]
- Verma, D.K.; Al Fantazi, A.; Verma, C.; Khan, F.; Asatkar, A.; Hussain, C.M.; Ebenso, E.E. Experimental and computational studies on hydroxamic acids as environmental friendly chelating corrosion inhibitors for mild steel in aqueous acidic medium. J. Mol. Liq. 2020, 113651. [Google Scholar] [CrossRef]
- Faiz, M.; Zahari, A.; Awang, K.; Hussin, H. Corrosion inhibition on mild steel in 1 M HCl solution by Cryptocarya nigra extracts and three of its constituents (alkaloids). RSC Adv. 2002, 10, 6547–6562. [Google Scholar] [CrossRef]
- Ngouné, B.; Pengou, M.; Nanseu-Njiki, C.P.; Ngameni, E. A comparative study using solution analysis, electrochemistry and mass change for the inhibition of carbon steel by the plant alkaloid Voacangine. Corros. Eng. Sci. Technol. 2020, 55, 138–144. [Google Scholar] [CrossRef]
- Thomas, A.; Prajila, M.; Shainy, K.M.; Joseph, A. A green approach to corrosion inhibition of mild steel in hydrochloric acid using fruit rind extract of Garcinia indica (Binda). J. Mol. Liq. 2020, 113369. [Google Scholar] [CrossRef]
- Umoren, S.A.; Solomon, M.M.; Madhankumar, A.; Obot, I.B. Exploration of natural polymers for use as green corrosion inhibitors for AZ31 magnesium alloy in saline environment. Carbohydr. Polym. 2020, 230, 115466. [Google Scholar] [CrossRef]
- Dutta, A.; Saha, S.K.; Adhikari, U.; Banerjee, P.; Sukul, D. Effect of substitution on corrosion inhibition properties of 2-(substituted phenyl) benzimidazole derivatives on mild steel in 1 M HCl solution: A combined experimental and theoretical approach. Corros. Sci. 2017, 123, 256–266. [Google Scholar] [CrossRef]
- Hamidon, T.S.; Hussin, M.H. Susceptibility of hybrid sol-gel (TEOS-APTES) doped with caffeine as potent corrosion protective coatings for mild steel in 3.5 wt.% NaCl. Prog. Org. Coat. 2020, 140, 105478. [Google Scholar] [CrossRef]
- Espinoza-Vázquez, A.; Rodríguez-Gómez, F.J. Caffeine and nicotine in 3% NaCl solution with CO2 as corrosion inhibitors for low carbon steel. RSC Adv. 2016, 6, 70226–70236. [Google Scholar] [CrossRef]
- Gudić, S.; Oguzie, E.E.; Radonić, A.; Vrsalović, L.; Smoljko, I.; Kliškić, M. Inhibition of copper corrosion in chloride solution by caffeine isolated from black tea. Maced. J. Chem. Chem. Eng. 2014, 33, 13–25. [Google Scholar] [CrossRef]
- Fallavena, T.; Antonow, M.; Gonçalves, R.S. Caffeine as non-toxic corrosion inhibitor for copper in aqueous solutions of potassium nitrate. Appl. Surf. Sci. 2006, 253, 566–571. [Google Scholar] [CrossRef]
- Rajendran, S.; Amalraj, A.J.; Joice, M.J.; Anthony, N.; Trivedi, D.C.; Sundaravadivelu, M. Corrosion inhibition by the caffeine-Zn2+ system. Corros. Rev. 2004, 22, 233–248. [Google Scholar] [CrossRef]
- da Trindade, L.G.; Goncalves, R.S. Evidence of caffeine adsorption on a low-carbon steel surface in ethanol. Corros. Sci. 2009, 51, 1578–1583. [Google Scholar] [CrossRef]
- Ebadi, M.; Basirun, W.J.; Leng, S.Y.; Mahmoudian, M.R. Investigation of corrosion inhibition properties of caffeine on nickel by electrochemical techniques. Int. J. Electrochem. Sci. 2012, 7, 8052–8063. [Google Scholar]
- de Souza, F.S.; Giacomelli, C.; Gonçalves, R.S.; Spinelli, A. Adsorption behavior of caffeine as a green corrosion inhibitor for copper. Mater. Sci. Eng. C 2012, 32, 2436–2444. [Google Scholar] [CrossRef]
- Ammouchi, N.; Allal, H.; Belhocine, Y.; Bettaz, S.; Zouaoui, E. DFT computations and molecular dynamics investigations on conformers of some pyrazinamide derivatives as corrosion inhibitors for aluminum. J. Mol. Liq. 2020, 300, 112309. [Google Scholar] [CrossRef]
- Hadisaputra, S.; Purwoko, A.A.; Wajdi, F.; Sumarlan, I.; Hamdiani, S. Theoretical study of the substituent effect on corrosion inhibition performance of benzimidazole and its derivatives. Int. J. Corros. Scale Inhib. 2019, 8, 673–688. [Google Scholar] [CrossRef]
- Obot, I.B.; Macdonald, D.D.; Gasem, Z.M. Density functional theory (DFT) as a powerful tool for designing new organic corrosion inhibitors. Part 1: An overview. Corros. Sci. 2015, 99, 1–30. [Google Scholar] [CrossRef]
- Hadisaputra, S.; Hamdiani, S.; Kurniawan, M.A.; Nuryono, N. Influence of macrocyclic ring size on the corrosion inhibition efficiency of dibenzo crown ether: A density functional study. Indones. J. Chem. 2017, 17, 431–438. [Google Scholar] [CrossRef]
- Hsissou, R.; Benhiba, F.; Abbout, S.; Dagdag, O.; Benkhaya, S.; Berisha, A.; Elharfi, A. Trifunctional epoxy polymer as corrosion inhibition material for carbon steel in 1.0 M HCl: MD simulations, DFT and complexation computations. Inorg. Chem. Commun. 2020, 107858. [Google Scholar] [CrossRef]
- Hadisaputra, S.; Purwoko, A.A.; Hakim, A.; Savalas, L.R.T.; Rahmawati, R.; Hamdiani, S.; Nuryono, N. Ab initio MP2 and DFT studies of ethyl-p-methoxycinnamate and its derivatives as corrosion inhibitors of iron in acidic medium. J. Phys. Conf. Ser. 2019, 1402, 055046. [Google Scholar] [CrossRef]
- Karakus, N.; Sayin, K. The investigation of corrosion inhibition efficiency on some benzaldehyde thiosemicarbazones and their thiole tautomers: Computational study. J. Taiwan Inst. Chem. Eng. 2015, 48, 95–102. [Google Scholar] [CrossRef]
- El Ibrahimi, B.; Jmiai, A.; Bazzi, L.; El Issami, S. Amino acids and their derivatives as corrosion inhibitors for metals and alloys. Arab J. Chem. 2020, 13, 740–771. [Google Scholar] [CrossRef]
- Sasikumar, Y.; Adekunle, A.S.; Olasunkanmi, L.O.; Bahadur, I.; Baskar, R.; Kabanda, M.M.; Ebenso, E.E. Experimental, quantum chemical and Monte Carlo simulation studies on the corrosion inhibition of some alkyl imidazolium ionic liquids containing tetrafluoroborate anion on mild steel in acidic medium. J. Mol. Liq. 2015, 211, 105–118. [Google Scholar] [CrossRef]
- Obot, I.B.; Haruna, K.; Saleh, T.A. Atomistic Simulation: A Unique and Powerful Computational Tool for Corrosion Inhibition Research. Arab J. Sci. Eng. 2019, 44, 1–32. [Google Scholar] [CrossRef]
- Kaya, S.; Kaya, C.; Guo, L.; Kandemirli, F.; Tüzün, B.; Uğurlu, İ.; Saraçoğlu, M. Quantum chemical and molecular dynamics simulation studies on inhibition performances of some thiazole and thiadiazole derivatives against corrosion of iron. J. Mol. Liq. 2016, 219, 497–504. [Google Scholar] [CrossRef]
- Kaya, S.; Guo, L.; Kaya, C.; Tüzün, B.; Obot, I.B.; Touir, R.; Islam, N. Quantum chemical and molecular dynamic simulation studies for the prediction of inhibition efficiencies of some piperidine derivatives on the corrosion of iron. J. Taiwan Inst. Chem. Eng. 2016, 65, 522–529. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Harding, L.B.; Klippenstein, S.J.; Jasper, A.W. Ab initio methods for reactive potential surfaces. Phys. Chem. Chem. Phys. 2017, 9, 4055–4070. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G. No. 3 Gaussian; Revision 02; Gaussian09, Inc.: Wallingford, UK, 2009; p. 4. [Google Scholar]
- Hadisaputra, S.; Canaval, L.R.; Pranowo, H.D.; Armunanto, R. Theoretical study of substituent effects on Cs+/Sr 2+–dibenzo-18-crown-6 complexes. Monat. Chem. 2014, 145, 737–745. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. DFT-B3LYP, NPA-, and QTAIM-based study of the physical properties of [M (II)(H2O)2(15-crown-5)](M=Mn, Fe, Co, Ni, Cu, Zn) complexes. J. Phys. Chem. A 2011, 115, 5592–5601. [Google Scholar] [CrossRef]
- Koopmans, T. Über ydie zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektronen eines atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Islam, N.; Chandra Ghosh, D. A new algorithm for the evaluation of the global hardness of polyatomic molecules. Mol. Phys. 2011, 109, 917–931. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Yang, W.; Parr, R.G. Hardness, softness, and the fukui function in the electronic theory of metals and catalysis. Proc. Natl. Acad. Sci. USA 1985, 82, 6723–6726. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases—the evolution of a chemical concept. Coord. Chem. Rev. 1990, 100, 403–425. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Sastri, V.S.; Perumareddi, J.R. Molecular orbital theoretical studies of some organic corrosion inhibitors. Corros. Sci. 1997, 53, 617–622. [Google Scholar] [CrossRef]
- Obot, I.B.; Gasem, Z.M. Theoretical evaluation of corrosion inhibition performance of some pyrazine derivatives. Corros. Sci. 2014, 83, 359–366. [Google Scholar] [CrossRef]
- Khaled, K.F. Monte Carlo simulations of corrosion inhibition of mild steel in 0.5 M sulphuric acid by some green corrosion inhibitors. J. Solid State Electrochem. 2009, 13, 1743–1756. [Google Scholar] [CrossRef]
- Khaled, K.F.; El-Maghraby, A. Experimental, Monte Carlo and molecular dynamics simulations to investigate corrosion inhibition of mild steel in hydrochloric acid solutions. Arab. J. Chem. 2014, 7, 319–326. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulations: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by simulated annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef]
- Madkour, L.H.; Kaya, S.; Obot, I.B. Computational, Monte Carlo simulation and experimental studies of some arylazotriazoles (AATR) and their copper complexes in corrosion inhibition process. J. Mol. Liq. 2018, 260, 351–374. [Google Scholar] [CrossRef]
- Tang, J.; Qiu, R.; Chen, J.; Ao, B. Diffusion behavior of hydrogen in oxygen saturated and unsaturated plutonium dioxide: An ab initio molecular dynamics study. J. Alloys Compd. 2020, 834, 155113. [Google Scholar] [CrossRef]
- Sutor, D.J. The structures of the pyrimidines and purines. VII. The crystal structure of caffeine. Acta Cryst. 1958, 11, 453–458. [Google Scholar] [CrossRef]
- Ford, K.A.; Ebisuzaki, Y.; Boyle, P.D. Methylxanthines. II. Anhydrous Theobromine. Acta Cryst. Sec. C Cryst. Struct. Commun. 1998, 54, 1980–1983. [Google Scholar] [CrossRef]
- Ebisuzaki, Y.; Boyle, P.D.; Smith, J.A. Methylxanthines. i. anhydrous theophylline. Acta Cryst. Sec. C Cryst. Struct. Commun. 1997, 53, 777–779. [Google Scholar] [CrossRef]
- Feyer, V.; Plekan, O.; Richter, R.; Coreno, M.; Prince, K.C. Photoion mass spectroscopy and valence photoionization of hypoxanthine, xanthine and caffeine. Chem. Phys. 2019, 358, 3–38. [Google Scholar] [CrossRef]
- Ajò, D.; Cingi, M.B.; Fragalá, I.; Granozzi, G. UV Phoelectron Spectra of Biological Xanthines: Theophylline, Theobromine and Caffeine. Spectrosc. Lett. 1977, 10, 757–761. [Google Scholar] [CrossRef]
- Hadisaputra, S.; Purwoko, A.A.; Wirayani, Y.; Ulfa, M.; Hamdiani, S. Density functional and perturbation calculation on the corrosion inhibition performance of benzylnicotine and its derivatives. AIP Conf. Proc. 2020, 243, 020006. [Google Scholar] [CrossRef]
- Dobeš, P.; Otyepka, M.; Strnad, M.; Hobza, P. Interaction Energies for the Purine Inhibitor Roscovitine with Cyclin-Dependent Kinase 2: Correlated Ab Initio Quantum-Chemical, DFT and Empirical Calculations. Chem. Euro. J. 2006, 12, 4297–4304. [Google Scholar] [CrossRef]
- Reimers, J.R.; Cai, Z.L.; Bilić, A.; Hush, N.S. The Appropriateness of Density-Functional Theory for the Calculation of Molecular Electronics Properties. Ann. N. Y. Acad. Sci. 2003, 1006, 235–251. [Google Scholar] [CrossRef]
- Kokalj, A.; Kovačević, N. On the consistent use of electrophilicity index and HSAB-based electron transfer and its associated change of energy parameters. Chem. Phys. Lett. 2011, 507, 181–184. [Google Scholar] [CrossRef]
- Siaka, A.A.; Eddy, N.O.; Idris, S.; Magaji, L. Experimental and computational study of corrosion potentials of penicillin G. Res. J. Appl. Sci. 2011, 6, 487–493. [Google Scholar] [CrossRef]
- Eddy, N.O.; Stoyanov, S.R.; Ebenso, E.E. Fluoroquinolones as corrosion inhibitors for mild steel in acidic medium; experimental and theoretical studies. Int. J. Electrochem. Sci. 2010, 5, 1127–1150. [Google Scholar]
- Shi, W.; Xia, M.; Lei, W.; Wang, F. Molecular dynamics study of polyether polyamino methylene phosphonates as an inhibitor of anhydrite crystal. Desalination 2013, 322, 137–143. [Google Scholar] [CrossRef]
- Umoren, S.A.; Obot, I.B.; Madhankumar, A.; Gasem, Z.M. Effect of degree of hydrolysis of polyvinyl alcohol on the corrosion inhibition of steel: Theoretical and experimental studies. J. Adhe. Sci. Tech. 2015, 29, 271–295. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).