Design Strategy for Self-Healing Epoxy Coatings

 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Preparation of Self-Healing Coatings
2.3. Characterization
2.3.1. Microscopy Characterization
2.3.2. Thermal Characterization
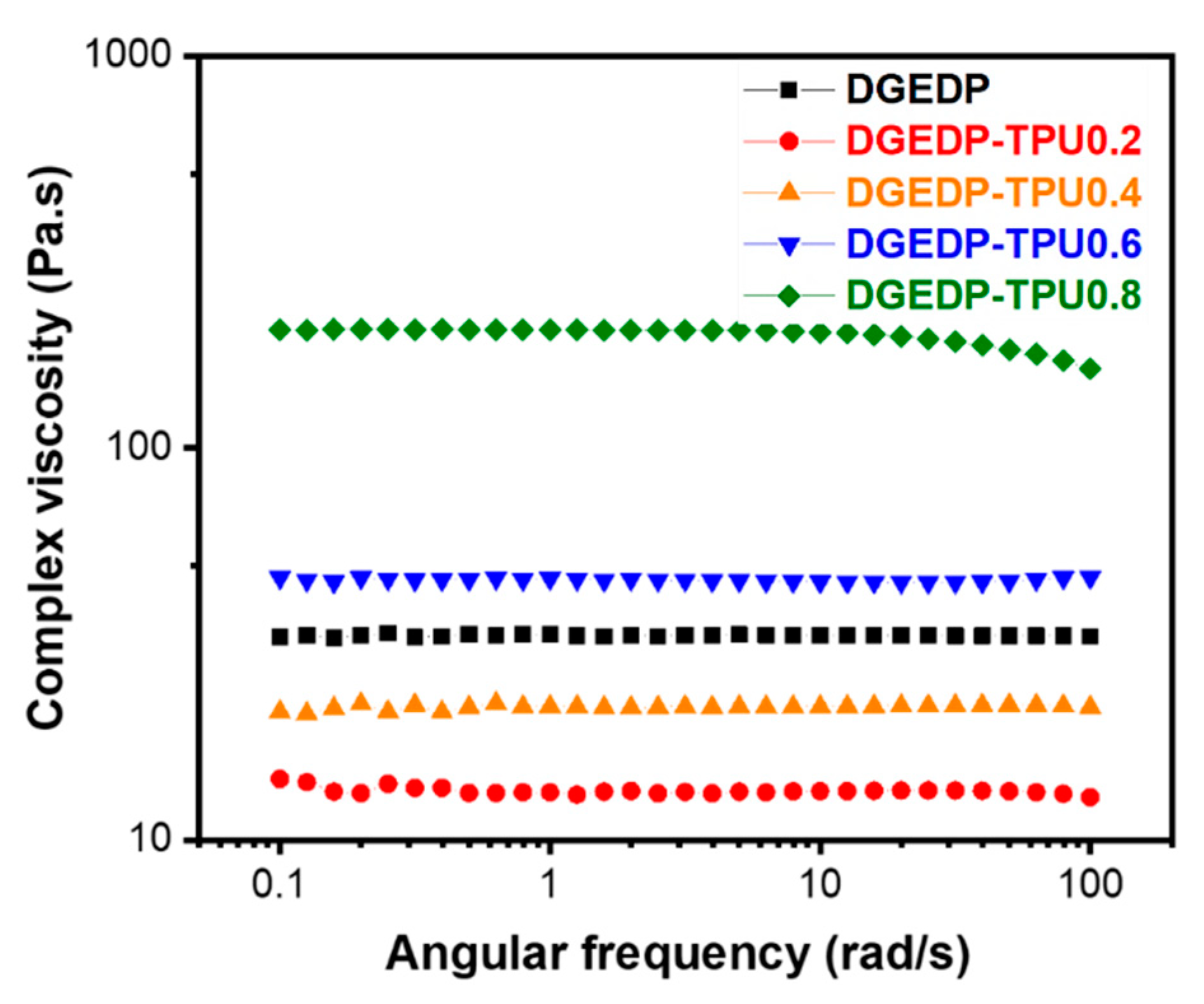
2.3.3. Rheological Characterization
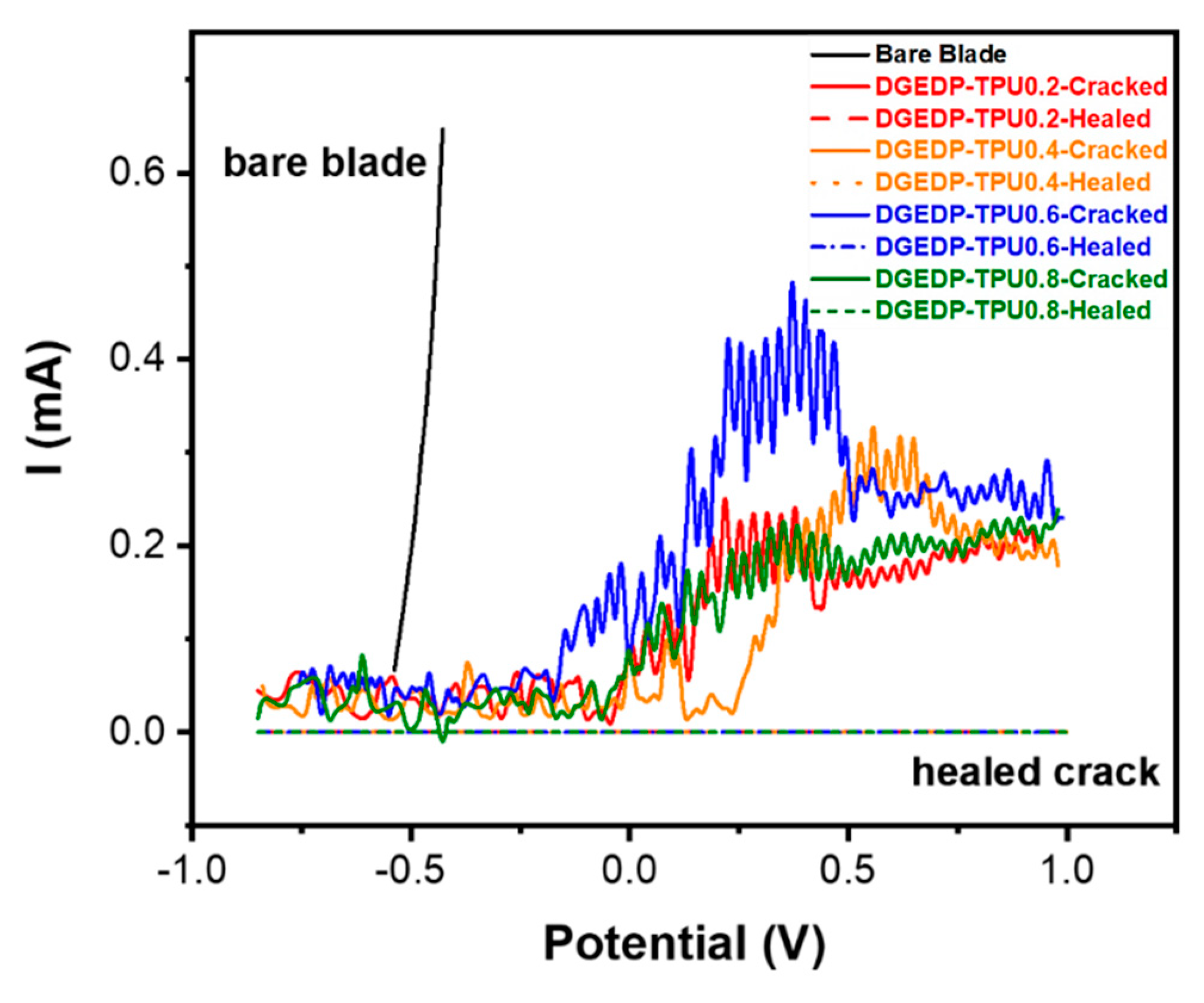
2.3.4. Anti-Corrosion Behavior
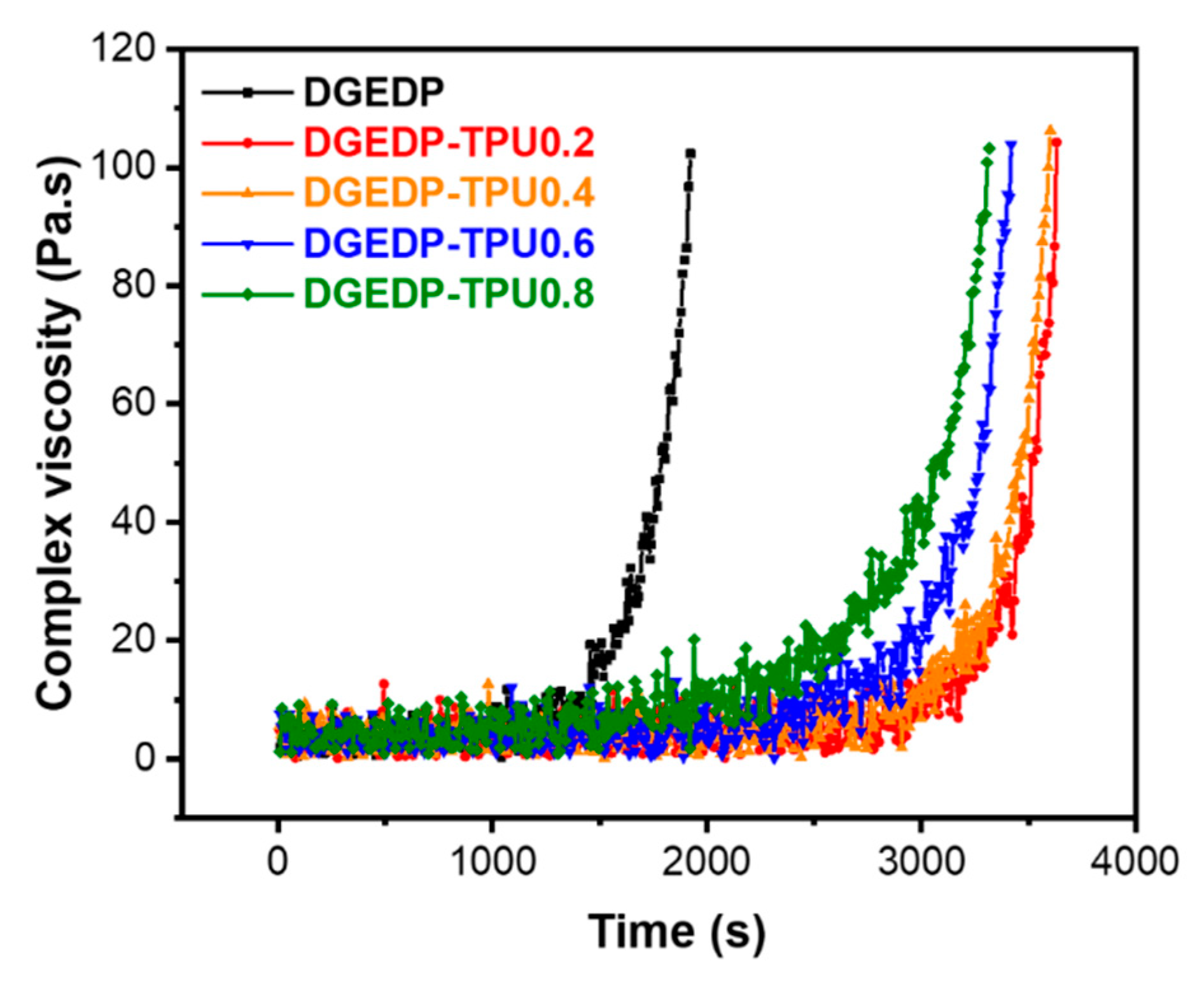
3. Results and Discussion
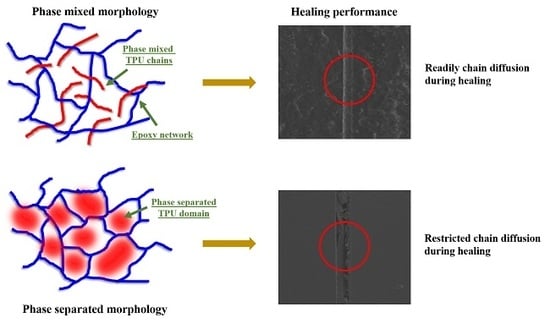
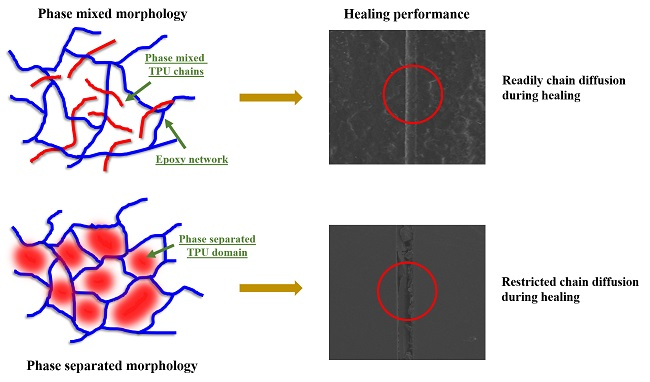
3.1. Self-Healing Performance of the Coatings
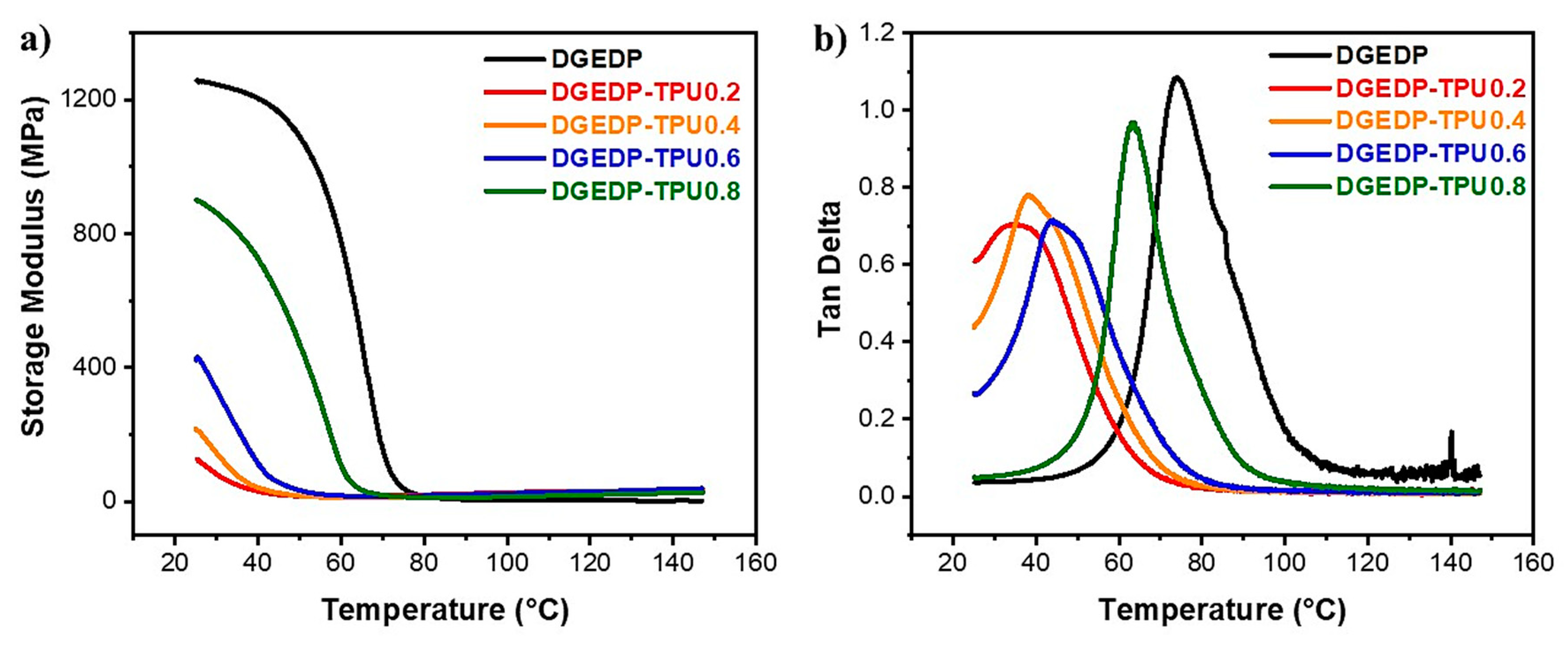
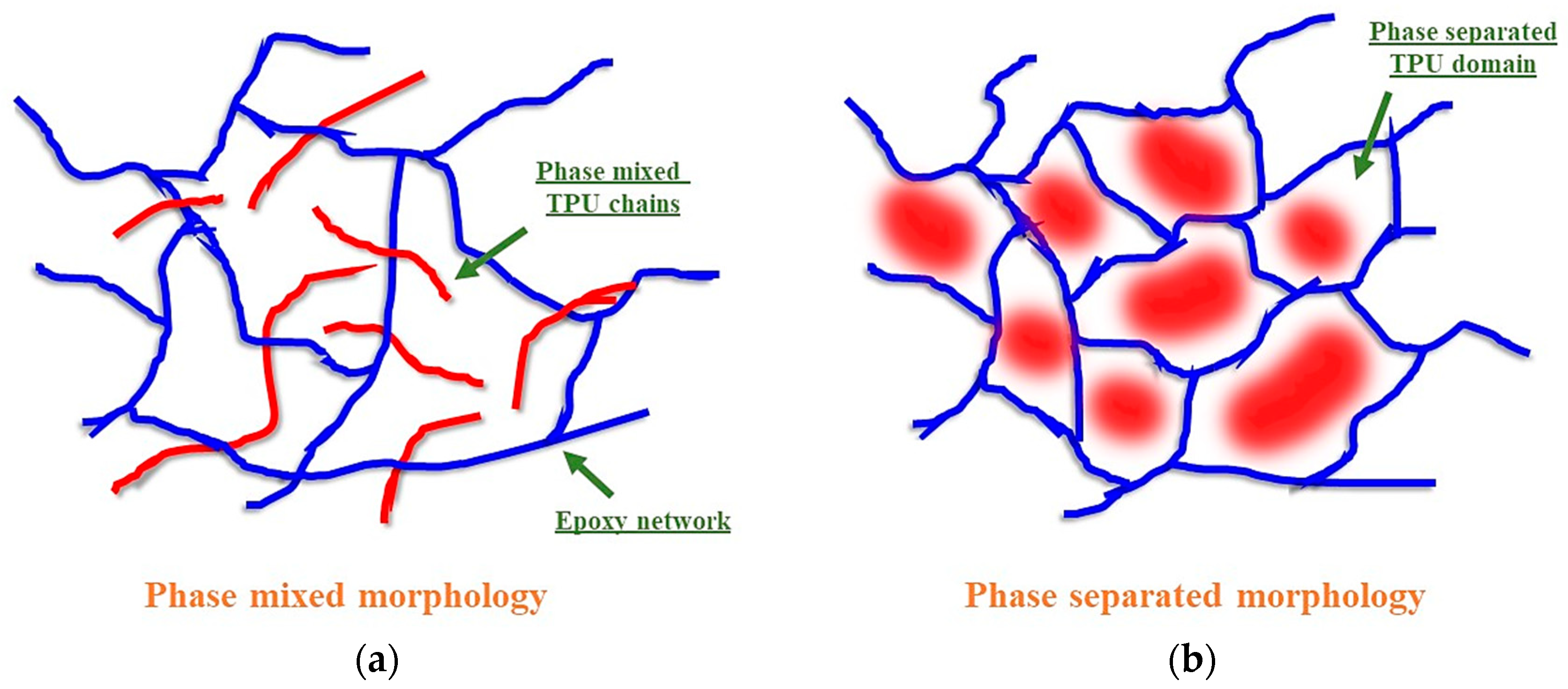
3.2. Structure–Property Relationship for the Coatings
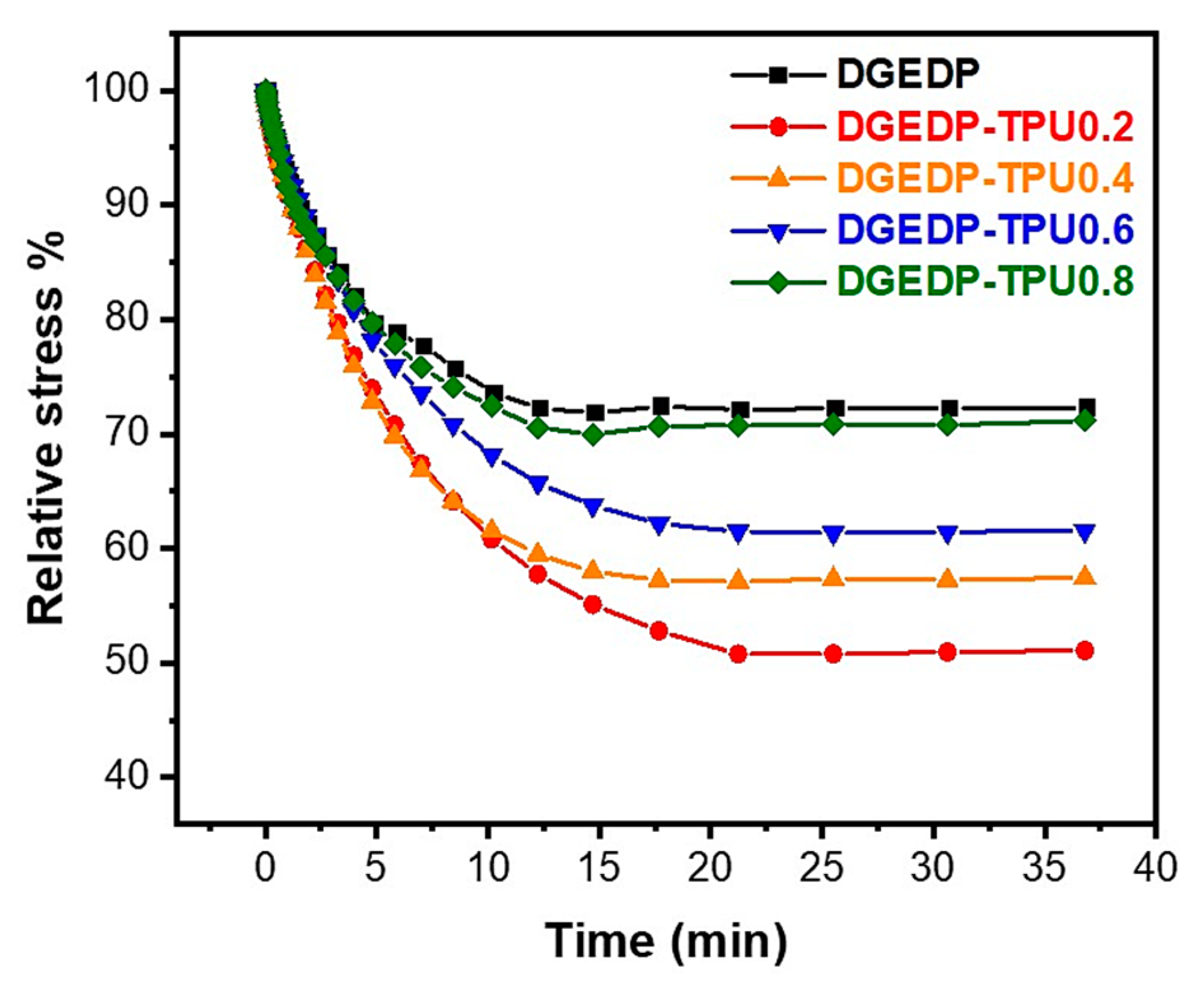
3.3. Stress Relaxation and Self-Diffusion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hager, M.D.; Greil, P.; Leyens, C.; Van Der Zwaag, S.; Schubert, U.S. Self-healing materials. Adv. Mater. 2010, 22, 5424–5430. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. Redox-responsive self-healing materials formed from host–guest polymers. Nat. Commun. 2011, 2, 511. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.Y.; Meure, S.; Solomon, D. Self-healing polymeric materials: A review of recent developments. Prog. Polym. Sci. 2008, 33, 479–522. [Google Scholar] [CrossRef]
- Wool, R.P. Self-healing materials: A review. Soft Matter 2008, 4, 400–418. [Google Scholar] [CrossRef]
- Van der Kooij, H.M.; Susa, A.; García, S.J.; van der Zwaag, S.; Sprakel, J. Imaging the Molecular Motions of Autonomous Repair in a Self-Healing Polymer. Adv. Mater. 2017, 29, 1701017. [Google Scholar] [CrossRef] [PubMed]
- Geitner, R.; Legesse, F.-B.; Kuhl, N.; Bocklitz, T.W.; Zechel, S.; Vitz, J.; Hager, M.; Schubert, U.S.; Dietzek, B.; Schmitt, M.; et al. Do You Get What You See? Understanding Molecular Self-Healing. Chem. A Eur. J. 2018, 24, 2493–2502. [Google Scholar] [CrossRef]
- Canadell, J.; Goossens, H.; Klumperman, B. Self-Healing Materials Based on Disulfide Links. Macromolecules 2011, 44, 2536–2541. [Google Scholar] [CrossRef]
- Ying, H.; Zhang, Y.; Cheng, J. Dynamic urea bond for the design of reversible and self-healing polymers. Nat. Commun. 2014, 5, 3218. [Google Scholar] [CrossRef]
- Imato, K.; Nishihara, M.; Kanehara, T.; Amamoto, Y.; Takahara, A.; Otsuka, H. Self-Healing of Chemical Gels Cross-Linked by Diarylbibenzofuranone-Based Trigger-Free Dynamic Covalent Bonds at Room Temperature. Angew. Chem. Int. Ed. 2012, 51, 1138–1142. [Google Scholar] [CrossRef]
- Cromwell, O.R.; Chung, J.; Guan, Z. Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds. J. Am. Chem. Soc. 2015, 137, 6492–6495. [Google Scholar] [CrossRef]
- Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guan, Z. Multivalent hydrogen bonding block copolymers self-assemble into strong and tough self-healing materials. Chem. Commun. 2014, 50, 10868–10870. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Delpierre, S.; Ke, K.; Raquez, J.-M.; Dubois, P.; Manas-Zloczower, I. Biomimetic Water-Responsive Self-Healing Epoxy with Tunable Properties. ACS Appl. Mater. Interfaces 2019, 11, 17853–17862. [Google Scholar] [CrossRef] [PubMed]
- Delpierre, S.; Willocq, B.; Manini, G.; Lemaur, V.; Goole, J.; Gerbaux, P.; Cornil, J.; Dubois, P.; Raquez, J.-M. A Simple Approach for Self-Healable and Stiff Polymer Network from Iminoboronate-Based Boroxine Chemistry. Chem. Mater. 2019. [Google Scholar] [CrossRef]
- Guimard, N.K.; Oehlenschlaeger, K.K.; Zhou, J.; Hilf, S.; Schmidt, F.G.; Barner-Kowollik, C. Current trends in the field of self-healing materials. Macromol. Chem. Phys. 2012, 213, 131–143. [Google Scholar] [CrossRef]
- Garcia, S.J. Effect of polymer architecture on the intrinsic self-healing character of polymers. Eur. Polym. J. 2014, 53, 118–125. [Google Scholar] [CrossRef]
- Van Tittelboom, K.; De Belie, N.; Van Loo, D.; Jacobs, P. Self-healing efficiency of cementitious materials containing tubular capsules filled with healing agent. Cem. Concr. Compos. 2011, 33, 497–505. [Google Scholar] [CrossRef]
- Cho, S.H.; Andersson, H.M.; White, S.R.; Sottos, N.R.; Braun, P.V. Polydimethylsiloxane-Based Self-Healing Materials. Adv. Mater. 2006, 18, 997–1000. [Google Scholar] [CrossRef]
- Jones, A.S.; Rule, J.D.; Moore, J.S.; White, S.R.; Sottos, N.R. Catalyst Morphology and Dissolution Kinetics of Self-Healing Polymers. Chem. Mater. 2006, 18, 1312–1317. [Google Scholar] [CrossRef]
- Lutterman, D.A.; Surendranath, Y.; Nocera, D.G. A Self-Healing Oxygen-Evolving Catalyst. J. Am. Chem. Soc. 2009, 131, 3838–3839. [Google Scholar] [CrossRef]
- Pingkarawat, K.; Wang, C.H.; Varley, R.J.; Mouritz, A.P. Self-healing of delamination cracks in mendable epoxy matrix laminates using poly [ethylene-co-(methacrylic acid)] thermoplastic. Compos. Part A Appl. Sci. Manuf. 2012, 43, 1301–1307. [Google Scholar] [CrossRef]
- Pingkarawat, K.; Wang, C.H.; Varley, R.J.; Mouritz, A.P. Mechanical properties of mendable composites containing self-healing thermoplastic agents. Compos. Part A Appl. Sci. Manuf. 2014, 65, 10–18. [Google Scholar] [CrossRef]
- Zhu, D.Y.; Rong, M.Z.; Zhang, M.Q. Self-healing polymeric materials based on microencapsulated healing agents: From design to preparation. Prog. Polym. Sci. 2015, 49–50, 175–220. [Google Scholar] [CrossRef]
- Pang, J.W.C.; Bond, I.P. A hollow fibre reinforced polymer composite encompassing self-healing and enhanced damage visibility. Compos. Sci. Technol. 2005, 65, 1791–1799. [Google Scholar] [CrossRef]
- Toohey, K.S.; Sottos, N.R.; Lewis, J.A.; Moore, J.S.; White, S.R. Self-healing materials with microvascular networks. Nat. Mater. 2007, 6, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; Trask, R.; Bond, I. A self-healing carbon fibre reinforced polymer for aerospace applications. Compos. Part A Appl. Sci. Manuf. 2007, 38, 1525–1532. [Google Scholar] [CrossRef]
- Mansfeld, F. Use of electrochemical impedance spectroscopy for the study of corrosion protection by polymer coatings. J. Appl. Electrochem. 1995, 25, 187–202. [Google Scholar] [CrossRef]
- Tan, C.K.; Blackwood, D.J. Corrosion protection by multilayered conducting polymer coatings. Corros. Sci. 2003, 45, 545–557. [Google Scholar] [CrossRef]
- Grundmeier, G.; Schmidt, W.; Stratmann, M. Corrosion protection by organic coatings: Electrochemical mechanism and novel methods of investigation. Electrochim. Acta 2000, 45, 2515–2533. [Google Scholar] [CrossRef]
- Cho, S.H.; White, S.R.; Braun, P.V. Self-healing polymer coatings. Adv. Mater. 2009, 21, 645–649. [Google Scholar] [CrossRef]
- Samadzadeh, M.; Boura, S.H.; Peikari, M.; Kasiriha, S.M.; Ashrafi, A. A review on self-healing coatings based on micro/nanocapsules. Prog. Org. Coat. 2010, 68, 159–164. [Google Scholar] [CrossRef]
- Huang, M.; Yang, J. Facile microencapsulation of HDI for self-healing anticorrosion coatings. J. Mater. Chem. 2011, 21, 11123–11130. [Google Scholar] [CrossRef]
- Park, J.H.; Braun, P.V. Coaxial electrospinning of self-healing coatings. Adv. Mater. 2010, 22, 496–499. [Google Scholar] [CrossRef]
- Luo, X.; Mather, P.T. Shape Memory Assisted Self-Healing Coating. ACS Macro Lett. 2013, 2, 152–156. [Google Scholar] [CrossRef]
- Andreeva, D.V.; Fix, D.; Möhwald, H.; Shchukin, D.G. Self-Healing Anticorrosion Coatings Based on pH-Sensitive Polyelectrolyte/Inhibitor Sandwichlike Nanostructures. Adv. Mater. 2008, 20, 2789–2794. [Google Scholar] [CrossRef]
- Doan, T.Q.; Leslie, L.S.; Kim, S.Y.; Bhargava, R.; White, S.R.; Sottos, N.R. Characterization of core-shell microstructure and self-healing performance of electrospun fiber coatings. Polymer 2016, 107, 263–272. [Google Scholar] [CrossRef]
- Kumar, G.S. Self-Healing Materials: Fundamentals, Design Strategies, and Applications; Wiley-vch: Weinheim, Germany, 2009; ISBN 9783527318292. [Google Scholar]
- Yang, T.; Du, Y.; Li, Z.M.; Wang, C.H. Mechanical Properties of Self-Healing Carbon Fiber-Epoxy Composite Stitched with Mendable Polymer Fiber. Polym. Polym. Compos. 2014, 22, 329–336. [Google Scholar] [CrossRef]
- Varley, R.J.; Craze, D.A.; Mouritz, A.P.; Wang, C.H. Thermoplastic Healing in Epoxy Networks: Exploring Performance and Mechanism of Alternative Healing Agents. Macromol. Mater. Eng. 2013, 298, 1232–1242. [Google Scholar] [CrossRef]
- Grigoriev, D.; Shchukina, E.; Shchukin, D.G. Nanocontainers for Self-Healing Coatings. Adv. Mater. Interfaces 2017, 4, 1600318. [Google Scholar] [CrossRef]
- Wei, H.; Wang, Y.; Guo, J.; Shen, N.Z.; Jiang, D.; Zhang, X.; Yan, X.; Zhu, J.; Wang, Q.; Shao, L.; et al. Advanced micro/nanocapsules for self-healing smart anticorrosion coatings. J. Mater. Chem. A 2015, 3, 469–480. [Google Scholar] [CrossRef]
- Yuan, D.; Bonab, V.S.; Patel, A.; Manas-Zloczower, I. Self-healing epoxy coatings with enhanced properties and facile processability. Polymer 2018, 147, 196–201. [Google Scholar] [CrossRef]
- Chrysanthos, M.; Galy, J.; Pascault, J.-P. Preparation and properties of bio-based epoxy networks derived from isosorbide diglycidyl ether. Polymer 2011, 52, 3611–3620. [Google Scholar] [CrossRef]
- Baroncini, E.A.; Kumar Yadav, S.; Palmese, G.R.; Stanzione, J.F., III. Recent advances in bio-based epoxy resins and bio-based epoxy curing agents. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Maiorana, A.; Spinella, S.; Gross, R.A. Bio-Based Alternative to the Diglycidyl Ether of Bisphenol A with Controlled Materials Properties. Biomacromolecules 2015, 16, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Mather, P.T. Shape Memory Assisted Self-Healing (SMASH) Coating. Macromolecules 2010, 6, 2146. [Google Scholar]
- Rodriguez, E.D.; Luo, X.; Mather, P.T. Linear/Network Poly(ε-caprolactone) Blends Exhibiting Shape Memory Assisted Self-Healing (SMASH). ACS Appl. Mater. Interfaces 2011, 3, 152–161. [Google Scholar] [CrossRef]
- Siddhamalli, S.K.; Kyu, T. Toughening of thermoset/thermoplastic composites via reaction-induced phase separation: Epoxy/phenoxy blends. J. Appl. Polym. Sci. 2000, 77, 1257–1268. [Google Scholar] [CrossRef]
- Fernández, B.; Corcuera, M.A.; Marieta, C.; Mondragon, I. Rheokinetic variations during curing of a tetrafunctional epoxy resin modified with two thermoplastics. Eur. Polym. J. 2001, 37, 1863–1869. [Google Scholar] [CrossRef]
- Siddhamalli, S.K. Toughening of epoxy/polycaprolactone composites via reaction induced phase separation. Polym. Compos. 2000, 21, 846–855. [Google Scholar] [CrossRef]
- Chen, J.-L.; Chang, F.-C. Phase Separation Process in Poly(ε-caprolactone)—Epoxy Blends. Macromolecules 1999, 32, 5348–5356. [Google Scholar] [CrossRef]
- Wang, M.; Yu, Y.; Wu, X.; Li, S. Polymerization induced phase separation in poly (ether imide)-modified epoxy resin cured with imidazole. Polymer 2004, 45, 1253–1259. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, M.; Gan, W.; Tao, Q.; Li, S. Polymerization-Induced Viscoelastic Phase Separation in Polyethersulfone-Modified Epoxy Systems. J. Phys. Chem. B 2004, 108, 6208–6215. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Martin, M.D.; Eceiza, A.; Oyanguren, P.; Mondragon, I. Phase separation in polysulfone-modified epoxy mixtures. Relationships between curing conditions, morphology and ultimate behavior. Polymer 2000, 41, 1027–1035. [Google Scholar] [CrossRef]
- Aizpurua, B.; Franco, M.; Corcuera, M.A.; Riccardi, C.C.; Mondragon, I. Chemorheology and ultimate behavior of epoxy-amine mixtures modified with a liquid oligomer. J. Appl. Polym. Sci. 2000, 76, 1269–1279. [Google Scholar] [CrossRef]
- Villani, M.; Deshmukh, Y.S.; Camlibel, C.; Esteves, A.C.C.; de with, G. Superior relaxation of stresses and self-healing behavior of epoxy-amine coatings. RSC Adv. 2016, 6, 245–259. [Google Scholar] [CrossRef]
- Yu, K.; Taynton, P.; Zhang, W.; Dunn, M.L.; Qi, H.J. Influence of stoichiometry on the glass transition and bond exchange reactions in epoxy thermoset polymers. RSC Adv. 2014, 4, 48682–48690. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, D.; Solouki Bonab, V.; Patel, A.; Yilmaz, T.; Gross, R.A.; Manas-Zloczower, I. Design Strategy for Self-Healing Epoxy Coatings. Coatings 2020, 10, 50. https://doi.org/10.3390/coatings10010050
Yuan D, Solouki Bonab V, Patel A, Yilmaz T, Gross RA, Manas-Zloczower I. Design Strategy for Self-Healing Epoxy Coatings. Coatings. 2020; 10(1):50. https://doi.org/10.3390/coatings10010050
Chicago/Turabian StyleYuan, Dian, Vahab Solouki Bonab, Ammar Patel, Talha Yilmaz, Richard A. Gross, and Ica Manas-Zloczower. 2020. "Design Strategy for Self-Healing Epoxy Coatings" Coatings 10, no. 1: 50. https://doi.org/10.3390/coatings10010050
APA StyleYuan, D., Solouki Bonab, V., Patel, A., Yilmaz, T., Gross, R. A., & Manas-Zloczower, I. (2020). Design Strategy for Self-Healing Epoxy Coatings. Coatings, 10(1), 50. https://doi.org/10.3390/coatings10010050