Synergies with and Resistance to Membrane-Active Peptides



Abstract
:1. Introduction
2. Defining Antimicrobial Synergy and Resistance
2.1. Minimum Inhibitory Concentration Assays
2.2. Antimicrobial Activity Assays
3. Synergy and Resistance to MAPs
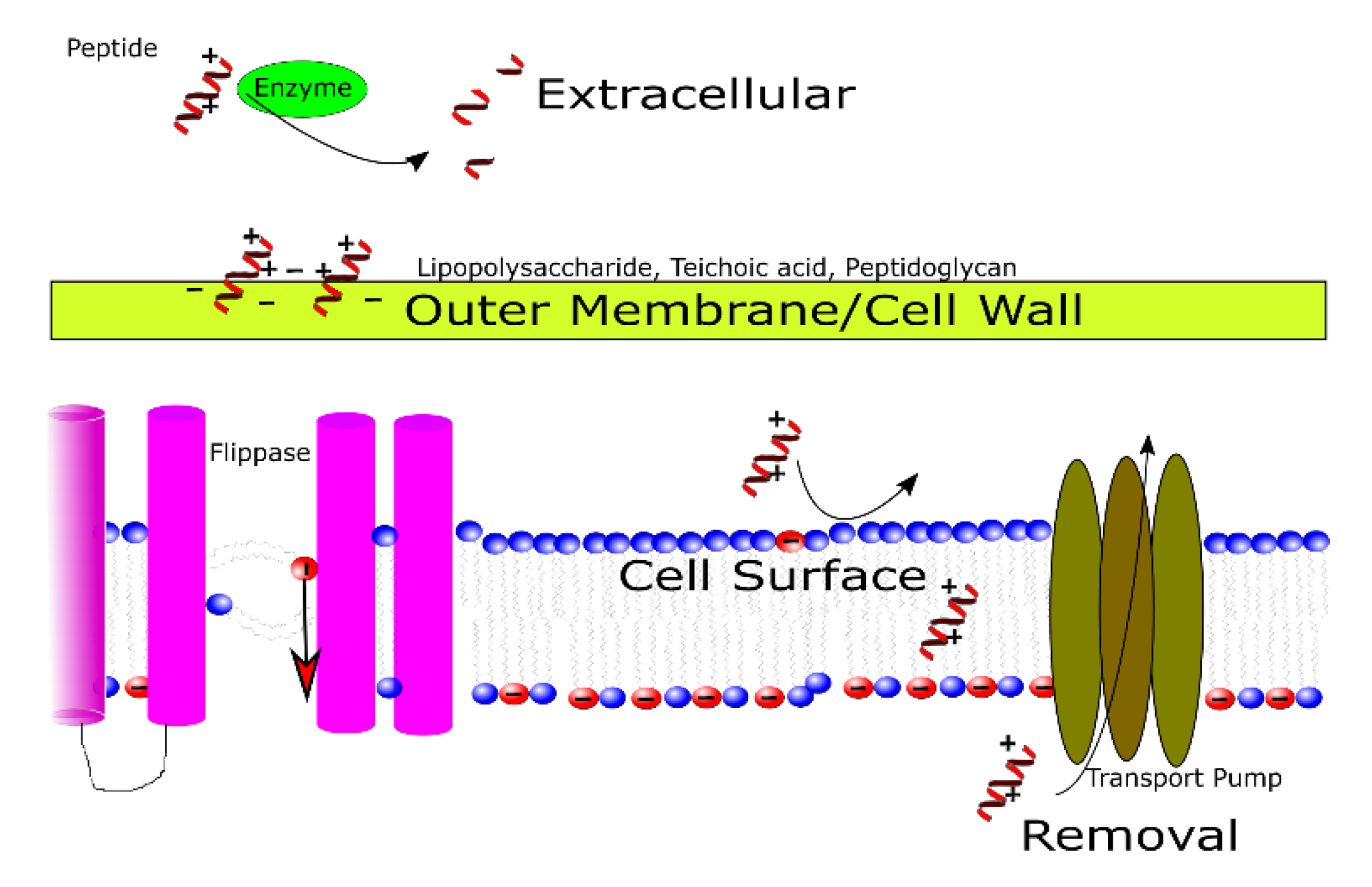
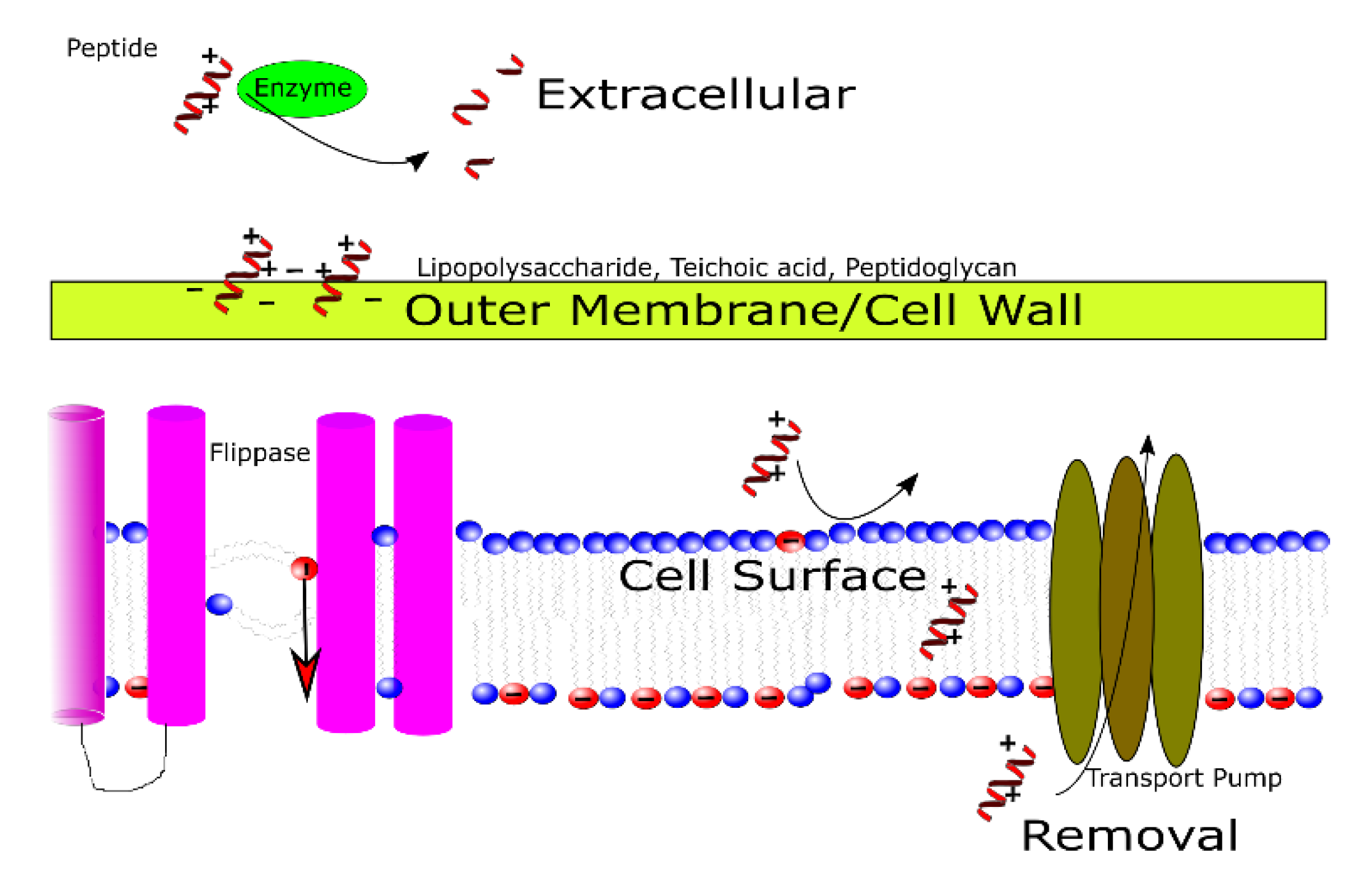
3.1. Outside of the Cell
3.2. In the Cell Wall and Outer Membrane
3.3. At the Cell Membrane
3.4. At the Point of Removal and/or Efflux
4. Examples of Natural Synergies
4.1. Peptides and Mixtures
4.2. Peptide Prodrugs Susceptible to Proteolytic Enzymes
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avci, F.G.; Akbulut, B.S.; Ozkirimli, E. Membrane active peptides and their biophysical characterization. Biomolecules 2018, 8, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehbach, J.; Craik, D.J. The Vast Structural Diversity of Antimicrobial Peptides. Trends Pharmacol. Sci. 2019, 40, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, E216–E230. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E.W. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef]
- Friedrich, C.L.; Moyles, D.; Beveridge, T.J.; Hancock, R.E.W. Antibacterial action of structurally diverse cationic peptides on gram-positive bacteria. Antimicrob. Agents Chemother. 2000, 44, 2086–2092. [Google Scholar] [CrossRef] [Green Version]
- Oren, Z.; Shai, Y. Mode of action of linear amphipathic α-helical antimicrobial peptides. Pept. Sci. 1998, 47, 451–463. [Google Scholar] [CrossRef]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolym. Pept. Sci. Sect. 2002, 66, 236–248. [Google Scholar] [CrossRef]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [Green Version]
- Wiedman, G.; Fuselier, T.; He, J.; Searson, P.C.; Hristova, K.; Wimley, W.C. Highly efficient macromolecule-sized poration of lipid bilayers by a synthetically evolved peptide. J. Am. Chem. Soc. 2014, 136, 4724–4731. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Koh, J.J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane active antimicrobial peptides: Translating mechanistic insights to design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Sun, L.C.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q.Y. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Rončević, T.; Puizina, J.; Tossi, A. Antimicrobial peptides as anti-infective agents in pre-post-antibiotic era? Int. J. Mol. Sci. 2019, 20, 5713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.H.; Myrold, D.D.; Mueller, R.S. Distributions of extracellular peptidases across prokaryotic genomes reflect phylogeny and habitat. Front. Microbiol. 2019, 10, 413. [Google Scholar] [CrossRef] [Green Version]
- Tannert, A.; Pohl, A.; Pomorski, T.; Herrmann, A. Protein-mediated transbilayer movement of lipids in eukaryotes and prokaryotes: The relevance of ABC transporters. Int. J. Antimicrob. Agents 2003, 22, 177–187. [Google Scholar] [CrossRef]
- Dawson, R.J.P.; Locher, K.P. Structure of a bacterial multidrug ABC transporter. Nature 2006, 443, 180–185. [Google Scholar] [CrossRef]
- Contreras, F.X.; Sánchez-Magraner, L.; Alonso, A.; Goñi, F.M. Transbilayer (flip-flop) lipid motion and lipid scrambling in membranes. FEBS Lett. 2010, 584, 1779–1786. [Google Scholar] [CrossRef] [Green Version]
- Van der Mark, V.A.; Elferink, R.P.J.O.; Paulusma, C.C. P4 ATPases: Flippases in health and disease. Int. J. Mol. Sci. 2013, 14, 7897–7922. [Google Scholar] [CrossRef] [Green Version]
- Chiang, W.C.; Pamp, S.J.; Nilsson, M.; Givskov, M.; Tolker-Nielsen, T. The metabolically active subpopulation in Pseudomonas aeruginosa biofilms survives exposure to membrane-targeting antimicrobials via distinct molecular mechanisms. FEMS Immunol. Med. Microbiol. 2012, 65, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.A.E.G.E.-S.; Zhong, L.L.; Shen, C.; Yang, Y.; Doi, Y.; Tian, G.B. Colistin and its role in the Era of antibiotic resistance: An extended review (2000–2019). Emerg. Microbes Infect. 2020, 9, 868–885. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.H.; Yu, C.M.; Yu, V.L.; Chow, J.W. Synergy assessed by checkerboard a critical analysis. Diagn. Microbiol. Infect. Dis. 1993, 16, 343–349. [Google Scholar] [CrossRef]
- Tilton, R.C.; Lieberman, L.; Gerlach, E.H. Microdilution antibiotic susceptibility test: Examination of certain variables. J. Appl. Microbiol. 1973, 26, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Ebbensgaard, A.; Mordhorst, H.; Overgaard, M.T.; Nielsen, C.G.; Aarestrup, F.M.; Hansen, E.B. Comparative evaluation of the antimicrobial activity of different antimicrobial peptides against a range of pathogenic bacteria. PLoS ONE 2015, 10, e0144611. [Google Scholar] [CrossRef] [Green Version]
- Kraszewska, J.; Beckett, M.C.; James, T.C.; Bond, U. Comparative analysis of the antimicrobial activities of plant defensin-like and ultrashort peptides against food-spoiling bacteria. Appl. Environ. Microbiol. 2016, 82, 4288–4298. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.R.; Bajaksouzian, S.; Palavecino-Fasola, E.L.; Holoszyc, H.M.; Appelbaum, P.C. Determination of penicillin MICs for Streptococcus pneumoniae by using a two- or three-disk diffusion procedure. J. Clin. Microbiol. 1998, 36, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Clinical Laboratory Standards Institute Why Use CLSI Breakpoints? Available online: https://clsi.org/meetings/rationale-for-using-clsi-breakpoints/ (accessed on 1 August 2020).
- European Committe on Antimicrobial Susceptability Testing Clinical Breakpoints and Dosing of Antibiotics. Available online: https://eucast.org/clinical_breakpoints/ (accessed on 1 August 2020).
- Mercer, D.K.; Torres, M.D.T.; Duay, S.S.; Lovie, E.; Simpson, L.; von Kockritz-Blickwede, M.; de la Fuente-Nunez, C.; O’Neil, D.A.; Angeles-Boza, A.M. Antimicrobial Susceptibility Testing of Antimicrobial Peptides to Better Predict Efficacy. Front. Cell. Infect. Microbiol. 2020, 10, 326. [Google Scholar] [CrossRef]
- Brook, I. Inoculum Effect. Rev. Infect. Dis. 1989, 11, 361–368. [Google Scholar] [CrossRef]
- Ge, Y.; MacDonald, D.L.; Holroyd, K.J.; Thornsberry, C.; Wexler, H.; Zasloff, M. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob. Agents Chemother. 1999, 43, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Jepson, A.K.; Schwarz-Linek, J.; Ryan, L.; Ryadnov, M.G.; Poon, W.C.K. What is the ‘Minimum Inhibitory Concentration’ (MIC) of pexiganan acting on Escherichia coli?—A cautionary case study. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2016; Volume 915, pp. 33–48. [Google Scholar]
- Van Belkum, A.; Burnham, C.A.D.; Rossen, J.W.A.; Mallard, F.; Rochas, O.; Dunne, W.M. Innovative and rapid antimicrobial susceptibility testing systems. Nat. Rev. Microbiol. 2020, 18, 299–311. [Google Scholar] [CrossRef]
- Al Nahas, K.; Cama, J.; Schaich, M.; Hammond, K.; Deshpande, S.; Dekker, C.; Ryadnov, M.G.; Keyser, U.F. A microfluidic platform for the characterisation of membrane active antimicrobials. Lab Chip 2019, 19, 837–844. [Google Scholar] [CrossRef] [Green Version]
- Sonesson, A.; Przybyszewska, K.; Eriksson, S.; Mörgelin, M.; Kjellström, S.; Davies, J.; Potempa, J.; Schmidtchen, A. Identification of bacterial biofilm and the Staphylococcus aureus derived protease, staphopain, on the skin surface of patients with atopic dermatitis. Sci. Rep. 2017, 7, 8689. [Google Scholar] [CrossRef]
- Rompikuntal, P.K.; Vdovikova, S.; Duperthuy, M.; Johnson, T.L.; Åhlund, M.; Lundmark, R.; Oscarsson, J.; Sandkvist, M.; Uhlin, B.E.; Wai, S.N. Outer membrane vesicle-mediated export of processed PrtV protease from Vibrio cholerae. PLoS ONE 2015, 10, e0134098. [Google Scholar] [CrossRef] [Green Version]
- Loughran, A.J.; Atwood, D.N.; Anthony, A.C.; Harik, N.S.; Spencer, H.J.; Beenken, K.E.; Smeltzer, M.S. Impact of individual extracellular proteases on Staphylococcus aureus biofilm formation in diverse clinical isolates and their isogenic sarA mutants. Microbiologyopen 2014, 3, 897–909. [Google Scholar] [CrossRef] [Green Version]
- Wessel, E.M.; Tomich, J.M.; Todd, R.B. Biodegradable Drug-Delivery Peptide Nanocapsules. ACS Omega 2019, 4, 20059–20063. [Google Scholar] [CrossRef] [Green Version]
- Gimza, B.D.; Larias, M.I.; Budny, B.G.; Shaw, L.N. Mapping the Global Network of Extracellular Protease Regulation in Staphylococcus aureus. mSphere 2019, 4, e00676-19. [Google Scholar] [CrossRef] [Green Version]
- Joo, H.S.; Otto, M. Mechanisms of resistance to antimicrobial peptides in staphylococci. Biochim. Biophys. Acta Biomembr. 2015, 1848, 3055–3061. [Google Scholar] [CrossRef] [Green Version]
- Schneider, B.A.; Balskus, E.P. Discovery of small molecule protease inhibitors by investigating a widespread human gut bacterial biosynthetic pathway. Tetrahedron 2018, 74, 3215–3230. [Google Scholar] [CrossRef]
- LaRock, C.N.; Nizet, V. Cationic antimicrobial peptide resistance mechanisms of streptococcal pathogens. Biochim. Biophys. Acta Biomembr. 2015, 1848, 3047–3054. [Google Scholar] [CrossRef] [Green Version]
- Samykannu, G.; Vijayababu, P.; Natarajan, J. Substrate specificities in Salmonella typhi outer membrane protease (PgtE) from omptin family—An in silico proteomic approach. Informatics Med. Unlocked 2019, 16, 100237. [Google Scholar] [CrossRef]
- Brannon, J.R.; Burk, D.L.; Leclerc, J.M.; Thomassin, J.L.; Portt, A.; Berghuis, A.M.; Gruenheid, S.; Le Moual, H. Inhibition of outer membrane proteases of the omptin family by aprotinin. Infect. Immun. 2015, 83, 2300–2311. [Google Scholar] [CrossRef] [Green Version]
- Osmolovskiy, A.A.; Kurakov, A.V.; Kreyer, V.G.; Baranova, N.A.; Egorov, N.S. Ability of extracellular proteinases of micromycetes Aspergillus flavipes, Aspergillus fumigatus, and Aspergillus sydowii to affect proteins of the human haemostatic system. Moscow Univ. Biol. Sci. Bull. 2017, 72, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Zawrotniak, M.; Bochenska, O.; Karkowska-Kuleta, J.; Seweryn-Ozog, K.; Aoki, W.; Ueda, M.; Kozik, A.; Rapala-Kozik, M. Aspartic proteases and major cell wall components in candida albicans trigger the release of neutrophil extracellular traps. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Shemesh, E.; Hanf, B.; Hagag, S.; Attias, S.; Shadkchan, Y.; Fichtman, B.; Harel, A.; Krüger, T.; Brakhage, A.A.; Kniemeyer, O.; et al. Phenotypic and proteomic analysis of the Aspergillus fumigatus ΔPrtT, ΔXprG and ΔXprG/ΔPrtT protease-deficient mutants. Front. Microbiol. 2017, 8, 2490. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Liu, Y.; Wu, S.; Han, G.; Tu, J.; Dong, G.; Liu, N.; Sheng, C. Targeting fungal virulence factor by small molecules: Structure-based discovery of novel secreted aspartic protease 2 (SAP2) inhibitors. Eur. J. Med. Chem. 2020, 201, 112515. [Google Scholar] [CrossRef]
- Thomsen, P.; Bjursten, L.M.; Ahlstedt, S.; Bagge, U.; Björkstén, B. Inhibitory effect of honey bee venom on immune complex mediated leukocyte migration into rabbit knee-joints. Agents Actions 1984, 14, 662–666. [Google Scholar] [CrossRef]
- Lubawy, J.; Urbański, A.; Mrówczyńska, L.; Matuszewska, E.; Światły-Błaszkiewicz, A.; Matysiak, J.; Rosiński, G. The influence of bee venom melittin on the functioning of the immune system and the contractile activity of the insect heart—A preliminary study. Toxins 2019, 11, 494. [Google Scholar] [CrossRef] [Green Version]
- Saeed, W.S.E.; Khalil, E.A.G. Immune Response Modifying Effects of Bee Venom Protein [Melittin]/Autoclaved L. donovani complex in CD1 Mice: The Search for New Vaccine Adjuvants. J. Vaccines Vaccin. 2017, 8. [Google Scholar] [CrossRef]
- Philibert, C.; Pinzani, V.; Foglia, K.; Coccini, C.; Thompson-Bos, M.A.; Hillaire-Buys, D. Nonsteroidal anti-inflammatory drugs (NSAIDs) and necrotizing fasciitis: Even when used by local route. Fundam. Clin. Pharmacol. 2017, 31, 67–68. [Google Scholar]
- Boöttger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, e0178943. [Google Scholar] [CrossRef]
- Seymour, C.W.; Gesten, F.; Prescott, H.C.; Friedrich, M.E.; Iwashyn, T.J.; Phillips, G.S.; Lemeshow, S.; Osborn, T.; Terry, K.M.; Levy, M.M. Time to treatment and mortality during mandated emergency care for sepsis. N. Engl. J. Med. 2017, 376, 2235–2244. [Google Scholar] [CrossRef]
- Ferrer, R.; Martin-Loeches, I.; Phillips, G.; Osborn, T.M.; Townsend, S.; Dellinger, R.P.; Artigas, A.; Schorr, C.; Levy, M.M. Empiric antibiotic treatment reduces mortality in severe sepsis and septic shock from the first hour: Results from a guideline-based performance improvement program. Crit. Care Med. 2014, 42, 1749–1755. [Google Scholar] [CrossRef]
- Werle, M.; Bernkop-Schnürch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Li, Y.; Liu, T.; Liu, Y.; Tan, Z.; Ju, Y.; Yang, Y.; Dong, W. Antimicrobial activity, membrane interaction and stability of the D-amino acid substituted analogs of antimicrobial peptide W3R6. J. Photochem. Photobiol. B Biol. 2019, 200. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.P.; Lin, Q.; Chen, C.; Montelaro, R.C.; Doi, Y.; Deslouches, B. Enhanced therapeutic index of an antimicrobial peptide in mice by increasing safety and activity against multidrug-resistant bacteria. Sci. Adv. 2020, 6, 18. [Google Scholar] [CrossRef]
- Lam, S.J.; O’Brien-Simpson, N.M.; Pantarat, N.; Sulistio, A.; Wong, E.H.H.; Chen, Y.Y.; Lenzo, J.C.; Holden, J.A.; Blencowe, A.; Reynolds, E.C.; et al. Combating multidrug-resistant Gram-negative bacteria with structurally nanoengineered antimicrobial peptide polymers. Nat. Microbiol. 2016, 1, 16162. [Google Scholar] [CrossRef]
- Huang, C.; Jin, H.; Qian, Y.; Qi, S.; Luo, H.; Luo, Q.; Zhang, Z. Hybrid melittin cytolytic peptide-driven ultrasmall lipid nanoparticles block melanoma growth in vivo. ACS Nano 2013, 7, 5791–5800. [Google Scholar] [CrossRef]
- Zong, J.; Cobb, S.L.; Cameron, N.R. Peptide-functionalized gold nanoparticles: Versatile biomaterials for diagnostic and therapeutic applications. Biomater. Sci. 2017, 5, 872–886. [Google Scholar] [CrossRef] [Green Version]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [Green Version]
- Papo, N.; Shai, Y. A molecular mechanism for lipopolysaccharide protection of gram-negative bacteria from antimicrobial peptides. J. Biol. Chem. 2005, 280, 10378–10387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanram, H.; Bhattacharjya, S. Resurrecting inactive antimicrobial peptides from the lipopolysaccharide trap. Antimicrob. Agents Chemother. 2014, 58, 1987–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleifer, K.H.; Kandler, O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 1972, 36, 407–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, A.J.; Keck, W. Peptidoglycan as a barrier to transenvelope transport. J. Bacteriol. 1996, 178, 5555–5562. [Google Scholar] [CrossRef] [Green Version]
- Savini, F.; Loffredo, M.R.; Troiano, C.; Bobone, S.; Malanovic, N.; Eichmann, T.O.; Caprio, L.; Canale, V.C.; Park, Y.; Mangoni, M.L.; et al. Binding of an antimicrobial peptide to bacterial cells: Interaction with different species, strains and cellular components. Biochim. Biophys. Acta Biomembr. 2020, 1862. [Google Scholar] [CrossRef]
- Portlock, S.H.; Clague, M.J.; Cherry, R.J. Leakage of internal markers from erythrocytes and lipid vesicles induced by melittin, gramicidin S and alamethicin: A comparative study. BBA Biomembr. 1990, 1030, 1–10. [Google Scholar] [CrossRef]
- Lohner, K.; Blondelle, S. Molecular Mechanisms of Membrane Perturbation by Antimicrobial Peptides and the Use of Biophysical Studies in the Design of Novel Peptide Antibiotics. Comb. Chem. High Throughput Screen. 2005, 8, 241–256. [Google Scholar] [CrossRef]
- Hovakeemian, S.G.; Liu, R.; Gellman, S.H.; Heerklotz, H. Correlating antimicrobial activity and model membrane leakage induced by nylon-3 polymers and detergents. Soft Matter 2015, 11, 6840–6851. [Google Scholar] [CrossRef] [Green Version]
- Suntharalingam, P.; Senadheera, M.D.; Mair, R.W.; Levesque, C.M.; Cvitkovitch, D.G. The LiaFSR system regulates the cell envelope stress response in streptococcus mutans. J. Bacteriol. 2009, 191, 2973–2984. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Davlieva, M.; Panesso, D.; Rincon, S.; Miller, W.R.; Diaz, L.; Reyes, J.; Cruz, M.R.; Pemberton, O.; Nguyen, A.H.; et al. Antimicrobial sensing coupled with cell membrane remodeling mediates antibiotic resistance and virulence in Enterococcus faecalis. Proc. Natl. Acad. Sci. USA 2019, 116, 26925–26932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Vila-Farres, X.; Inoyama, D.; Ternei, M.; Cohen, L.J.; Gordon, E.A.; Reddy, B.V.B.; Charlop-Powers, Z.; Zebroski, H.A.; Gallardo-Macias, R.; et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol. 2016, 12, 1004–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Vila-Farres, X.; Inoyama, D.; Gallardo-Macias, R.; Jaskowski, M.; Satish, S.; Freundlich, J.S.; Brady, S.F. Human Microbiome Inspired Antibiotics with Improved β-Lactam Synergy against MDR Staphylococcus aureus. ACS Infect. Dis. 2018, 4, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sham, L.T.; Butler, E.K.; Lebar, M.D.; Kahne, D.; Bernhardt, T.G.; Ruiz, N. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 2014, 345, 220–222. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, T.T.; Baldridge, R.D.; Xu, P.; Graham, T.R. Phospholipid flippases: Building asymmetric membranes and transport vesicles. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 1068–1077. [Google Scholar] [CrossRef] [Green Version]
- Puts, C.F.; Panatala, R.; Hennrich, H.; Tsareva, A.; Williamson, P.; Holthuis, J.C.M. Mapping functional interactions in a heterodimeric phospholipid pump. J. Biol. Chem. 2012, 287, 30529–30540. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Liao, G.; Baker, G.M.; Wang, Y.; Lau, R.; Paderu, P.; Perlin, D.S.; Xue, C. Lipid flippase subunit Cdc50 mediates drug resistance and virulence in Cryptococcus neoformans. MBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Marques, R.L.; Theorin, L.; Palmgren, M.G.; Pomorski, T.G. P4-ATPases: Lipid flippases in cell membranes. Pflugers Arch. Eur. J. Physiol. 2014, 466, 1227–1240. [Google Scholar] [CrossRef] [Green Version]
- Lenoir, G.; Williamson, P.; Puts, C.F.; Holthuis, J.C.M. Cdc50p plays a vital role in the ATPase reaction cycle of the putative aminophospholipid transporter Drs2p. J. Biol. Chem. 2009, 284, 17956–17967. [Google Scholar] [CrossRef] [Green Version]
- Shor, E.; Wang, Y.; Perlin, D.S.; Xue, C. Cryptococcus flips its lid—Membrane phospholipid asymmetry modulates antifungal drug resistance and virulence. Microb. Cell 2016, 3, 358–360. [Google Scholar] [CrossRef] [Green Version]
- Blanco, P.; Hernando-Amado, S.; Reales-Calderon, J.; Corona, F.; Lira, F.; Alcalde-Rico, M.; Bernardini, A.; Sanchez, M.; Martinez, J. Bacterial Multidrug Efflux Pumps: Much More Than Antibiotic Resistance Determinants. Microorganisms 2016, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Hassanzadeh, S.; Ganjloo, S.; Pourmand, M.R.; Mashhadi, R.; Ghazvini, K. Epidemiology of efflux pumps genes mediating resistance among Staphylococcus aureus; A systematic review. Microb. Pathog. 2020, 139. [Google Scholar] [CrossRef]
- Ghosh, A.; Roymahapatra, G.; Paul, D.; Mandal, S.M. Theoretical analysis of bacterial efflux pumps inhibitors: Strategies in-search of competent molecules and develop next. Comput. Biol. Chem. 2020, 87, 107275. [Google Scholar] [CrossRef]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef]
- Zheng, S.; Sonomoto, K. Diversified transporters and pathways for bacteriocin secretion in gram-positive bacteria. Appl. Microbiol. Biotechnol. 2018, 102, 4243–4253. [Google Scholar] [CrossRef]
- Annunziato, G. Strategies to overcome antimicrobial resistance (AMR) making use of non-essential target inhibitors: A review. Int. J. Mol. Sci. 2019, 20, 5844. [Google Scholar] [CrossRef] [Green Version]
- Cote, C.K.; Blanco, I.I.; Hunter, M.; Shoe, J.L.; Klimko, C.P.; Panchal, R.G.; Welkos, S.L. Combinations of early generation antibiotics and antimicrobial peptides are effective against a broad spectrum of bacterial biothreat agents. Microb. Pathog. 2020, 142, 104050. [Google Scholar] [CrossRef]
- Opperman, T.J.; Nguyen, S.T. Recent advances toward a molecular mechanism of efflux pump inhibition. Front. Microbiol. 2015, 6, 421. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Espada, R.; Shahrour, H.; Pitts, B.; Stewart, P.S.; Sánchez-Gómez, S.; Martínez-de-Tejada, G. A permeability-increasing drug synergizes with bacterial efflux pump inhibitors and restores susceptibility to antibiotics in multi-drug resistant Pseudomonas aeruginosa strains. Sci. Rep. 2019, 9, 3452. [Google Scholar] [CrossRef] [Green Version]
- Goli, H.R.; Nahaei, M.R.; Rezaee, M.A.; Hasani, A.; Kafil, H.S.; Aghazadeh, M.; Nikbakht, M.; Khalili, Y. Role of MexAB-OprM and MexXY-OprM efflux pumps and class 1 integrons in resistance to antibiotics in burn and Intensive Care Unit isolates of Pseudomonas aeruginosa. J. Infect. Public Health 2018, 11, 364–372. [Google Scholar] [CrossRef]
- Blodkamp, S.; Kadlec, K.; Gutsmann, T.; Quiblier, C.; Naim, H.Y.; Schwarz, S.; von Köckritz-Blickwede, M. Effects of SecDF on the antimicrobial functions of cathelicidins against Staphylococcus aureus. Vet. Microbiol. 2017, 200, 52–58. [Google Scholar] [CrossRef]
- Ohene-Agyei, T.; Mowla, R.; Rahman, T.; Venter, H. Phytochemicals increase the antibacterial activity of antibiotics by acting on a drug efflux pump. Microbiologyopen 2014. [Google Scholar] [CrossRef]
- Ferrer-Espada, R.; Sánchez-Gómez, S.; Pitts, B.; Stewart, P.S.; Martínez-de-Tejada, G. Permeability enhancers sensitize β-lactamase-expressing Enterobacteriaceae and Pseudomonas aeruginosa to β-lactamase inhibitors, thereby restoring their β-lactam susceptibility. Int. J. Antimicrob. Agents 2020, 105986. [Google Scholar] [CrossRef]
- Grafskaia, E.N.; Nadezhdin, K.D.; Talyzina, I.A.; Polina, N.F.; Podgorny, O.V.; Pavlova, E.R.; Bashkirov, P.V.; Kharlampieva, D.D.; Bobrovsky, P.A.; Latsis, I.A.; et al. Medicinal leech antimicrobial peptides lacking toxicity represent a promising alternative strategy to combat antibiotic-resistant pathogens. Eur. J. Med. Chem. 2019, 180, 143–153. [Google Scholar] [CrossRef]
- Habermann, E. Bee and wasp venoms. Science 1972, 177, 314–322. [Google Scholar] [CrossRef]
- Lee, M.T.; Sun, T.L.; Hung, W.C.; Huang, H.W. Process of inducing pores in membranes by melittin. Proc. Natl. Acad. Sci. USA 2013, 110, 14243–14248. [Google Scholar] [CrossRef] [Green Version]
- Munjal, D.; Elliott, W.B. Studies of antigenic fractions in honey-bee (Apis mellifera) venom. Toxicon 1971, 9, 229–236. [Google Scholar] [CrossRef]
- Galdiero, E.; Siciliano, A.; Gesuele, R.; Di Onofrio, V.; Falanga, A.; Maione, A.; Liguori, R.; Libralato, G.; Guida, M. Melittin inhibition and eradication activity for resistant polymicrobial biofilm isolated from a dairy industry after disinfection. Int. J. Microbiol. 2019, 2019, 125654442. [Google Scholar] [CrossRef]
- Kokot, Z.J.; Matysiak, J.; Kłos, J.; Kȩdzia, B.; Hołderna-Kȩdzia, E. Application of principal component analysis for evaluation of chemical and antimicrobial properties of honey bee (Apis mellifera) venom. J. Apic. Res. 2009, 48, 168–175. [Google Scholar] [CrossRef]
- Uvnäs, B.; Wold, J.K. Isolation of a Mast Cell Degranulating Polypeptide from Ascaris suis. Acta Physiol. Scand. 1967, 70, 269–276. [Google Scholar] [CrossRef]
- Thompson, A.R. Isolation and characterisation of a mast cell degranulating substance from Ascaris suum. Biochim. Biophys. Acta BBA Gen. Subj. 1972, 261, 245–257. [Google Scholar] [CrossRef]
- Stansfeld, C.E.; Marsh, S.J.; Parcej, D.N.; Dolly, J.O.; Brown, D.A. Mast cell degranulating peptide and dendrotoxin selectively inhibit a fast-activating potassium current and bind to common neuronal proteins. Neuroscience 1987, 23, 893–902. [Google Scholar] [CrossRef]
- Ohizumi, Y. Application of physiologically active substances isolated from natural resources to pharmacological studies. Jpn. J. Pharmacol. 1997, 73, 263–289. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Guan, S.; Liu, J.; Ng, C.C.W.; Chan, G.H.H.; Sze, S.C.W.; Zhang, K.Y.; Naude, R.; Rolka, K.; Wong, J.H.; et al. Activities of Venom Proteins and Peptides with Possible Therapeutic Applications from Bees and WASPS. Protein Pept. Lett. 2016, 23, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.P.d.S.; Arcisio-Miranda, M.; da Costa, L.C.; de Souza, B.M.; Costa, S.T.B.; Palma, M.S.; Neto, J.R.; Procopio, J. Interactions of mast cell degranulating peptides with model membranes: A comparative biophysical study. Arch. Biochem. Biophys. 2009, 486, 1–11. [Google Scholar] [CrossRef]
- Kudo, I.; Murakami, M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002, 68, 3–58. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 biochemistry. Cardiovasc. Drugs Ther. 2009, 23, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Dennis, E.A. Diversity of group types, regulation, and function of phospholipase A2. J. Biol. Chem. 1994, 269, 13057–13060. [Google Scholar]
- Harwig, S.S.L.; Tan, L.; Qu, X.D.; Cho, Y.; Eisenhauer, P.B.; Lehrer, R.I. Bactericidal properties of murine intestinal phospholipase A2. J. Clin. Investig. 1995, 95, 603–610. [Google Scholar] [CrossRef]
- Dubouix, A.; Campanac, C.; Fauvel, J.; Simon, M.F.; Salles, J.P.; Roques, C.; Chap, H.; Marty, N. Bactericidal properties of group IIa secreted phospholipase A2 against Pseudomonas aeruginosa clinical isolates. J. Med. Microbiol. 2003, 52, 1039–1045. [Google Scholar] [CrossRef] [Green Version]
- Steinbrecher, U.P.; Parthasarathy, S.; Leake, D.S.; Witztum, J.L.; Steinberg, D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc. Natl. Acad. Sci. USA 1984, 81, 3883–3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esterbauer, H.; Muskiet, F.; Horrobin, D.F. Cytotoxicity and genotoxicity of lipid-oxidation products. Am. J. Clin. Nutr. 1993, 57, 779S–786S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, J.; Lomonte, B. Phospholipase A2 myotoxins from Bothrops snake venoms. Toxicon 1995, 33, 1405–1424. [Google Scholar] [CrossRef]
- Argiolas, A.; Pisano, J.J. Facilitation of phospholipase A2 activity by mastoparans, a new class of mast cell degranulating peptides from wasp venom. J. Biol. Chem. 1983, 258, 13697–13702. [Google Scholar]
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.A.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu. Rev. Genom. Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef] [Green Version]
- Köhler, G.A.; Brenot, A.; Haas-Stapleton, E.; Agabian, N.; Deva, R.; Nigam, S. Phospholipase A2 and Phospholipase B activities in fungi. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2006, 1761, 1391–1399. [Google Scholar] [CrossRef] [Green Version]
- Forde, É.; Shafiy, G.; Fitzgerald-Hughes, D.; Strömstedt, A.A.; Devocelle, M. Action of antimicrobial peptides and their prodrugs on model and biological membranes. J. Pept. Sci. 2018, 24, e3086. [Google Scholar] [CrossRef]
- Forde, E.; Devocelle, M. Pro-moieties of antimicrobial peptide prodrugs. Molecules 2015, 20, 1210–1227. [Google Scholar] [CrossRef]
- Shi, W.J.; Zhang, S.F.; Zhang, C.X.; Cheng, J.A. Cloning and comparative analysis of the venom prepromelittin genes from four wasp species. Acta Genet. Sin. 2003, 30, 555–559. [Google Scholar]
- KREIL, G.; HAIML, L.; SUCHANEK, G. Stepwise Cleavage of the Pro Part of Promelittin by Dipeptidylpeptidase IV: Evidence for a New Type of Precursor—Product Conversion. Eur. J. Biochem. 1980, 111, 49–58. [Google Scholar] [CrossRef]
- Pathak, N.; Salas-Auvert, R.; Ruche, G.; Janna, M.-h.; McCarthy, D.; Harrison, R.G. Comparison of the effects of hydrophobicity, amphiphilicity, and α-helicity on the activities of antimicrobial peptides. Proteins Struct. Funct. Bioinform. 1995, 22, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Forde, É.; Schütte, A.; Reeves, E.; Greene, C.; Humphreys, H.; Mall, M.; Fitzgerald-Hughes, D.; Devocelle, M. Differential in vitro and in vivo toxicities of antimicrobial peptide prodrugs for potential use in cystic fibrosis. Antimicrob. Agents Chemother. 2016, 60, 2813–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devocelle, M. Targeted antimicrobial peptides. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, L.L. A Gestalt approach to Gram-negative entry. Bioorganic Med. Chem. 2016, 24, 6379–6389. [Google Scholar] [CrossRef] [PubMed]
- Cama, J.; Voliotis, M.; Metz, J.; Smith, A.; Iannucci, J.; Keyser, U.F.; Tsaneva-Atanasova, K.; Pagliara, S. Single-cell microfluidics facilitates the rapid quantification of antibiotic accumulation in Gram-negative bacteria. Lab Chip 2020, 20, 2765–2775. [Google Scholar] [CrossRef]

{kind=link}
| Peptide | Sequence |
|---|---|
| Melittin | GIGAVLKVLTTGLPALISWIKRKRQQ |
| Pixiganan | GIGKFLKKAKKFGKAFVKILKK |
| CAP18 | GLRKRLRKFRNKIKEKLKKIGQKIQGLLPKLAPRTDY |
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES |
| K5L7 | KLLLKLKLKLLK |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kmeck, A.; Tancer, R.J.; Ventura, C.R.; Wiedman, G.R. Synergies with and Resistance to Membrane-Active Peptides. Antibiotics 2020, 9, 620. https://doi.org/10.3390/antibiotics9090620
Kmeck A, Tancer RJ, Ventura CR, Wiedman GR. Synergies with and Resistance to Membrane-Active Peptides. Antibiotics. 2020; 9(9):620. https://doi.org/10.3390/antibiotics9090620
Chicago/Turabian StyleKmeck, Adam, Robert J. Tancer, Cristina R. Ventura, and Gregory R. Wiedman. 2020. "Synergies with and Resistance to Membrane-Active Peptides" Antibiotics 9, no. 9: 620. https://doi.org/10.3390/antibiotics9090620
APA StyleKmeck, A., Tancer, R. J., Ventura, C. R., & Wiedman, G. R. (2020). Synergies with and Resistance to Membrane-Active Peptides. Antibiotics, 9(9), 620. https://doi.org/10.3390/antibiotics9090620