Amidochelocardin Overcomes Resistance Mechanisms Exerted on Tetracyclines and Natural Chelocardin

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. CHD and CDCHD Were Effective against Clinically Relevant Uropathogens and Displayed Resistance-Breaking Properties In Vitro
2.2. CHD Resistance Development in Target Pathogens Revealed Mutation of ramR in K. pneumoniae
2.3. Absence of Cross-Resistance to CDCHD
2.4. Resistance to CHD in K. pneumoniae Was Based on Efflux
2.5. ChdR Was Found to be the Primary Self-Resistance Factor in Natural CHD Producer A. sulphurea
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Antimicrobial Screening
4.3. In Vitro Selection of CHD-Resistant Klebsiella Mutants
4.4. Construction of a K. pneumoniae ramR Deletion Mutant
4.5. Construction of A. sulphurea Double Mutant and Complementation Vectors
4.6. In Vitro Selection of CHD-Resistant Amycolatopsis Mutants
4.7. Comparative Whole-Genome Sequencing
4.8. Quantification of Gene Expression by qPCR
4.9. Gene Expression Analysis by RNA-Seq
4.10. Preparation of an A. sulphurea Genomic Cosmid Library and Its Expression in A. mediterranei
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. The Review on Antimicrobial Resistance; UK Government and Wellcome Trust: London, UK, 2016.
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Procopio, R.E.d.L.; Silva, I.R.D.; Martins, M.K.; Azevedo, J.L.D.; Araujo, J.M.D. Antibiotics produced by Streptomyces. Braz. J. Infect. Dis. 2012, 16, 466–471. [Google Scholar] [CrossRef] [Green Version]
- Berdy, J. Bioactive microbial metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Watve, M.G.; Tickoo, R.; Jog, M.M.; Bhole, B.D. How many antibiotics are produced by the genus Streptomyces? Arch. Microbiol. 2001, 176, 386–390. [Google Scholar] [CrossRef]
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Pogue, J.M.; Marchaim, D.; Kaye, D.; Kaye, K.S. Revisiting “older” antimicrobials in the era of multidrug resistance. Pharmacotherapy 2011, 31, 912–921. [Google Scholar] [CrossRef]
- Poulakou, G.; Bassetti, M.; Righi, E.; Dimopoulos, G. Current and future treatment options for infections caused by multidrug-resistant Gram-negative pathogens. Future Microbiol. 2014, 9, 1053–1069. [Google Scholar] [CrossRef]
- Cassir, N.; Rolain, J.-M.; Brouqui, P. A new strategy to fight antimicrobial resistance: The revival of old antibiotics. Front. Microbiol. 2014, 5, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirst, H.A. Developing new antibacterials through natural product research. Expert Opin. Drug Discov. 2013, 8, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Lešnik, U.; Lukežič, T.; Podgoršek, A.; Horvat, J.; Polak, T.; Šala, M.; Jenko, B.; Harmrolfs, K.; Ocampo-Sosa, A.; Martínez-Martínez, L.; et al. Construction of a new class of tetracycline lead structures with potent antibacterial activity through biosynthetic engineering. Angew. Chem. Int. Ed. Engl. 2015, 54, 3937–3940. [Google Scholar] [CrossRef] [PubMed]
- Oliver, T.J.; Sinclair, A.C. Antibiotic M-319. US Patent US3155582, 3 November 1964. [Google Scholar]
- Oliva, B.; Gordon, G.; McNicholas, P.; Ellestad, G.A.; Chopra, I. Evidence that Tetracycline Analogs Whose Primary Target Is Not the Bacterial Ribosome Cause Lysis of Escherichia Coli. Antimicrob. Agents Chemother. 1992, 36, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, B.; Noller, H.F.; Daubresse, G.; Oliva, B.; Misulovin, Z.; Rothstein, D.M.M.; Ellestad, G.A.; Gluzman, Y.; Tally, F.P.; Chopra, I. Molecular Basis of Tetracycline Action Identification of Analogs Whose Primary Target Is Not the Bacterial Ribosome. Antimicrob. Agents Chemother. 1991, 35, 2306–2311. [Google Scholar] [CrossRef] [Green Version]
- Proctor, R.; Craig, W.; Kunin, C. Cetocycline, Tetracycline Analog: In Vitro Studies of Antimicrobial Activity, Serum Binding, Lipid Solubility, and Uptake by Bacteria. Antimicrob. Agents Chemother. 1978, 13, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Stepanek, J.J.; Lukezic, T.; Teichert, I.; Petkovic, H.; Bandow, J.E. Dual mechanism of action of the atypical tetracycline chelocardin. Biochim. Biophys. Acta 2016, 1864, 645–654. [Google Scholar] [CrossRef]
- Chopra, I. Tetracycline analogs whose primary target is not the bacterial ribosome. Antimicrob. Agents Chemother. 1994, 38, 637–640. [Google Scholar] [CrossRef] [Green Version]
- Oliva, B.; Chopra, I. Tet determinants provide poor protection against some tetracyclines: Further evidence for division of tetracyclines into two classes. Antimicrob. Agents Chemother. 1992, 36, 876–878. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, F.; Starosta, A.L.; Arenz, S.; Sohmen, D.; Donhofer, A.; Wilson, D.N. Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 2014, 395, 559–575. [Google Scholar] [CrossRef]
- Markley, J.L.; Wencewicz, T.A. Tetracycline-Inactivating Enzymes. Front. Microbiol. 2018, 9, 1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molnar, V.; Matkociv, Z.; Tambic, T.; Kozma, C. Klinicko-farmakolosko ispitivanje kelokardina u bolesnika s infekcijom mokraćnih putova [Clinico-pharmacological investigation of chelocardine in patients suffering from urinary tract infection (author’s transl)]. Lijec Vjesn. 1977, 99, 560–562. [Google Scholar] [PubMed]
- Lukežič, T.; Fayad, A.A.; Bader, C.; Harmrolfs, K.; Bartuli, J.; Groß, S.; Lešnik, U.; Hennessen, F.; Herrmann, J.; Pikl, Š.; et al. Engineering Atypical Tetracycline Formation in Amycolatopsis sulphurea for the Production of Modified Chelocardin Antibiotics. ACS Chem. Biol. 2019, 14, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Grandclaudon, C.; Birudukota, N.V.S.; Elgaher, W.A.M.; Jumde, R.P.; Yahiaoui, S.; Arisetti, N.; Hennessen, F.; Hüttel, S.; Stadler, M.; Herrmann, J.; et al. Semisynthesis and biological evaluation of amidochelocardin derivatives as broad-spectrum antibiotics. Eur. J. Med. Chem. 2020, 188, 112005. [Google Scholar] [CrossRef] [PubMed]
- Mitscher, L.A.; Juvarkar, J.V.; Rosenbrook, W., Jr.; Andres, W.W.; Schenk, J.; Egan, R.S. Structure of chelocardin, a novel tetracycline antibiotic. J. Am. Chem. Soc. 1970, 92, 6070–6071. [Google Scholar] [CrossRef] [PubMed]
- Millner, R.; Becknell, B. Urinary Tract Infections. Pediatr. Clin. N. Am. 2019, 66, 1–13. [Google Scholar] [CrossRef]
- Flores-Mireles, A.L.; Walker, J.N.; Caparon, M.; Hultgren, S.J. Urinary tract infections: Epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 2015, 13, 269–284. [Google Scholar] [CrossRef]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3322–3327. [Google Scholar] [CrossRef] [Green Version]
- Köhler, T.; Michéa-Hamzehpour, M.; Henze, U.; Gotoh, N.; Curty, L.K.; Pechère, J.C. Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 1997, 23, 345–354. [Google Scholar] [CrossRef]
- Li, X.Z.; Livermore, D.M.; Nikaido, H. Role of efflux pump(s) in intrinsic resistance of Pseudomonas aeruginosa: Resistance to tetracycline, chloramphenicol, and norfloxacin. Antimicrob. Agents Chemother. 1994, 38, 1732–1741. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.-W.; Lam, I.; Chang, H.-Y.; Tsai, S.-F.; Palsson, B.O.; Charusanti, P. Capsule deletion via a lambda-Red knockout system perturbs biofilm formation and fimbriae expression in Klebsiella pneumoniae MGH 78578. BMC Res. Notes 2014, 7, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abouzeed, Y.M.; Baucheron, S.; Cloeckaert, A. ramR Mutations Involved in Efflux-Mediated Multidrug Resistance in Salmonella enterica Serovar Typhimurium. Antimicrob. Agents Chemother. 2008, 52, 2428–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, N.; Sharma, P.; Ricci, V.; Piddock, L. Regulation of the AcrAB-TolC efflux pump in Enterobacteriaceae. Res. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Chen, Q.; Shi, K.; Li, X.; Shi, Q.; He, F.; Zhou, J.; Yu, Y.; Hua, X. Step-Wise Increase in Tigecycline Resistance in Klebsiella pneumoniae Associated with Mutations in ramR, lon and rpsJ. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Hentschke, M.; Wolters, M.; Sobottka, I.; Rohde, H.; Aepfelbacher, M. ramR mutations in clinical isolates of Klebsiella pneumoniae with reduced susceptibility to tigecycline. Antimicrob. Agents Chemother. 2010, 54, 2720–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, E.; Leautaud, V.; Demple, B. The redox-regulated SoxR protein acts from a single DNA site as a repressor and an allosteric activator. EMBO J. 1998, 17, 2629–2636. [Google Scholar] [CrossRef]
- Pérez, A.; Poza, M.; Aranda, J.; Latasa, C.; Medrano, F.J.; Tomás, M.; Romero, A.; Lasa, I.; Bou, G. Effect of transcriptional activators SoxS, RobA, and RamA on expression of multidrug efflux pump AcrAB-TolC in Enterobacter cloacae. Antimicrob. Agents Chemother. 2012, 56, 6256–6266. [Google Scholar] [CrossRef] [Green Version]
- Grimsey, E.M.; Weston, N.; Ricci, V.; Stone, J.W.; Piddock, L.J.V. Overexpression of RamA, Which Regulates Production of the Multidrug Resistance Efflux Pump AcrAB-TolC, Increases Mutation Rate and Influences Drug Resistance Phenotype. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Martin, R.G.; Jair, K.W.; Wolf, R.E.; Rosner, J.L. Autoactivation of the marRAB multiple antibiotic resistance operon by the MarA transcriptional activator in Escherichia coli. J. Bacteriol. 1996, 178, 2216–2223. [Google Scholar] [CrossRef] [Green Version]
- Chollet, R.; Chevalier, J.; Bollet, C.; Pages, J.-M.; Davin-Regli, A. RamA is an alternate activator of the multidrug resistance cascade in Enterobacter aerogenes. Antimicrob. Agents Chemother. 2004, 48, 2518–2523. [Google Scholar] [CrossRef] [Green Version]
- Ricci, V.; Blair, J.M.A.; Piddock, L.J.V. RamA, which controls expression of the MDR efflux pump AcrAB-TolC, is regulated by the Lon protease. J. Antimicrob. Chemother. 2014, 69, 643–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, K.L.; Shah, I.M.; Wolf, R.E. Proteolytic degradation of Escherichia coli transcription activators SoxS and MarA as the mechanism for reversing the induction of the superoxide (SoxRS) and multiple antibiotic resistance (Mar) regulons. Mol. Microbiol. 2004, 51, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Lukežič, T.; Lešnik, U.; Podgoršek, A.; Horvat, J.; Polak, T.; Šala, M.; Jenko, B.; Raspor, P.; Herron, P.R.; Hunter, I.S.; et al. Identification of the chelocardin biosynthetic gene cluster from Amycolatopsis sulphurea: A platform for producing novel tetracycline antibiotics. Microbiology 2013, 159, 2524–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prija, F.; Srinivasan, P.; Das, S.; Kattusamy, K.; Prasad, R. DnrI of Streptomyces peucetius binds to the resistance genes, drrAB and drrC but is activated by daunorubicin. J. Basic Microbiol. 2017, 57, 862–872. [Google Scholar] [CrossRef]
- Srinivasan, P.; Palani, S.N.; Prasad, R. Daunorubicin efflux in Streptomyces peucetius modulates biosynthesis by feedback regulation. FEMS Microbiol. Lett. 2010, 305, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Microbial Threats to Health: Emergence, Detection, and Response; Smolinski, M.S.; Hamburg, M.A.; Lederberg, J. (Eds.) National Academies Press (US): Washington, DC, USA, 2003. [Google Scholar]
- Blaskovich, M.A. Antibiotics Special Issue: Challenges and Opportunities in Antibiotic Discovery and Development. ACS Infect. Dis. 2020, 6, 1286–1288. [Google Scholar] [CrossRef]
- Zavala-Cerna, M.G.; Segura-Cobos, M.; Gonzalez, R.; Zavala-Trujillo, I.G.; Navarro-Perez, S.F.; Rueda-Cruz, J.A.; Satoscoy-Tovar, F.A. The Clinical Significance of High Antimicrobial Resistance in Community-Acquired Urinary Tract Infections. Can. J. Infect. Dis. Med. Microbiol. 2020, 2020, 2967260. [Google Scholar] [CrossRef]
- Mazzariol, A.; Bazaj, A.; Cornaglia, G. Multi-drug-resistant Gram-negative bacteria causing urinary tract infections: A review. J. Chemother. 2017, 29, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Nickel, J.C. Urinary Tract Infections and Resistant Bacteria: Highlights of a Symposium at the Combined Meeting of the 25th International Congress of Chemotherapy (ICC) and the 17th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), March 31–April 3, 2007, Munich, Germany. Rev. Urol. 2007, 9, 78–80. [Google Scholar]
- Stamm, W.E.; Norrby, S.R. Urinary Tract Infections: Disease Panorama and Challenges. J. Infect. Dis. 2001, 183, S1–S4. [Google Scholar] [CrossRef]
- Claeys, K.C.; Hopkins, T.L.; Vega, A.D.; Heil, E.L. Fluoroquinolone Restriction as an Effective Antimicrobial Stewardship Intervention. Curr. Infect. Dis. Rep. 2018, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Disabling and Potentially Permanent Side Effects Lead to Suspension or Restrictions of Quinolone and Fluoroquinolone Antibiotics, EMA/175398/2019, 2019. Available online: https://www.ema.europa.eu/en/documents/referral/quinolone-fluoroquinolone-article-31-referral-disabling-potentially-permanent-side-effects-lead_en.pdf (accessed on 3 August 2020).
- Du, D.; Wang, Z.; James, N.R.; Voss, J.E.; Klimont, E.; Ohene-Agyei, T.; Venter, H.; Chiu, W.; Luisi, B.F. Structure of the AcrAB-TolC multidrug efflux pump. Nature 2014, 509, 512–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pos, K.M. Drug transport mechanism of the AcrB efflux pump. Biochim. Biophys. Acta 2009, 1794, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Greer, N.D. Tigecycline (Tygacil): The first in the glycylcycline class of antibiotics. Bayl. Univ. Med. Cent. Proc. 2006, 19, 155–161. [Google Scholar] [CrossRef]
- Garrison, M.W.; Neumiller, J.J.; Setter, S.M. Tigecycline: An investigational glycylcycline antimicrobial with activity against resistant gram-positive organisms. Clin. Ther. 2005, 27, 12–22. [Google Scholar] [CrossRef]
- Fluit, A.C.; Florijn, A.; Verhoef, J.; Milatovic, D. Presence of Tetracycline Resistance Determinants and Susceptibility to Tigecycline and Minocycline. Antimicrob. Agents Chemother. 2005, 49, 1636–1638. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Lin, Z.; Bai, B.; Xu, G.; Li, P.; Yu, Z.; Deng, Q.; Shang, Y.; Zheng, J. Eravacycline susceptibility was impacted by genetic mutation of 30S ribosome subunits, and branched-chain amino acid transport system II carrier protein, Na/Pi cotransporter family protein in Staphylococcus aureus. BMC Microbiol. 2020, 20, 189. [Google Scholar] [CrossRef]
- Gasparrini, A.J.; Markley, J.L.; Kumar, H.; Wang, B.; Fang, L.; Irum, S.; Symister, C.T.; Wallace, M.; Burnham, C.-A.D.; Andleeb, S.; et al. Tetracycline-inactivating enzymes from environmental, human commensal, and pathogenic bacteria cause broad-spectrum tetracycline resistance. Commun. Biol. 2020, 3, 241. [Google Scholar] [CrossRef]
- Ruzin, A.; Keeney, D.; Bradford, P.A. AcrAB Efflux Pump Plays a Role in Decreased Susceptibility to Tigecycline in Morganella morganii. Antimicrob. Agents Chemother. 2005, 49, 791–793. [Google Scholar] [CrossRef] [Green Version]
- Pournaras, S.; Koumaki, V.; Spanakis, N.; Gennimata, V.; Tsakris, A. Current perspectives on tigecycline resistance in Enterobacteriaceae: Susceptibility testing issues and mechanisms of resistance. Int. J. Antimicrob. Agents 2016, 48, 11–18. [Google Scholar] [CrossRef]
- Burgos, R.M.; Rodvold, K.A. Omadacycline: A novel aminomethylcycline. Infect. Drug Resist. 2019, 12, 1895–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, B.; Lin, Z.; Pu, Z.; Xu, G.; Zhang, F.; Chen, Z.; Sun, X.; Zheng, J.; Li, P.; Deng, Q.; et al. In vitro Activity and Heteroresistance of Omadacycline against Clinical Staphylococcus aureus Isolates From China Reveal the Impact of Omadacycline Susceptibility by Branched-Chain Amino Acid Transport System II Carrier Protein, Na/Pi Cotransporter Family Protein, and Fibronectin-Binding Protein. Front. Microbiol. 2019, 10, 2546. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Hua, X.; Xu, Q.; Yang, Y.; Zhang, L.; He, J.; Mu, X.; Hu, L.; Leptihn, S.; Yu, Y. Mechanism of eravacycline resistance in Acinetobacter baumannii mediated by a deletion mutation in the sensor kinase adeS, leading to elevated expression of the efflux pump AdeABC. Infect. Genet. Evol. 2020, 80, 104185. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Shang, Y.; Xu, G.; Pu, Z.; Lin, Z.; Bai, B.; Chen, Z.; Zheng, J.; Deng, Q.; Yu, Z. Mechanism of Eravacycline Resistance in Clinical Enterococcus faecalis Isolates From China. Front. Microbiol. 2020, 11, 916. [Google Scholar] [CrossRef] [PubMed]
- Bialek-Davenet, S.; Marcon, E.; Leflon-Guibout, V.; Lavigne, J.-P.; Bert, F.; Moreau, R.; Nicolas-Chanoine, M.-H. In vitro selection of ramR and soxR mutants overexpressing efflux systems by fluoroquinolones as well as cefoxitin in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2011, 55, 2795–2802. [Google Scholar] [CrossRef] [Green Version]
- Kehrenberg, C.; Cloeckaert, A.; Klein, G.; Schwarz, S. Decreased fluoroquinolone susceptibility in mutants of Salmonella serovars other than Typhimurium: Detection of novel mutations involved in modulated expression of ramA and soxS. J. Antimicrob. Chemother. 2009, 64, 1175–1180. [Google Scholar] [CrossRef] [Green Version]
- Oethinger, M.; Podglajen, I.; Kern, W.V.; Levy, S.B. Overexpression of the marA or soxS regulatory gene in clinical topoisomerase mutants of Escherichia coli. Antimicrob. Agents Chemother. 1998, 42, 2089–2094. [Google Scholar] [CrossRef] [Green Version]
- O’Regan, E.; Quinn, T.; Pagès, J.-M.; McCusker, M.; Piddock, L.; Fanning, S. Multiple regulatory pathways associated with high-level ciprofloxacin and multidrug resistance in Salmonella enterica serovar enteritidis: Involvement of RamA and other global regulators. Antimicrob. Agents Chemother. 2009, 53, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Farhadieh, B. Stable Chelocardin Composition. US Patent US4025654A, 24 May 1977. [Google Scholar]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef] [Green Version]
- EUCAST. Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth microdilution. European Committee on Antimicrobial Susceptibility Testing. EUCAST Discussion Document E.Def 5.1. Clin. Microbiol. Infect. 2003, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Stickler, D.J.; Morris, N.S.; Winters, C. Simple physical model to study formation and physiology of biofilms on urethral catheters. Methods Enzymol. 1999, 310, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Hoshino, Y.; Ishino, K.; Kogure, T.; Mikami, Y.; Uehara, Y.; Ishikawa, J. Construction of a pair of practical Nocardia-Escherichia coli shuttle vectors. Jpn. J. Infect. Dis. 2007, 60, 45–47. [Google Scholar] [PubMed]
- Wilkinson, C.J.; Hughes-Thomas, Z.A.; Martin, C.J.; Böhm, I.; Mironenko, T.; Deacon, M.; Wheatcroft, M.; Wirtz, G.; Staunton, J.; Leadlay, P.F. Increasing the efficiency of heterologous promoters in actinomycetes. J. Mol. Microbiol. Biotechnol. 2002, 4, 417–426. [Google Scholar] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Jackman, S.D.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Yeo, S.; Hammond, S.A.; Jahesh, G.; Khan, H.; Coombe, L.; Warren, R.L.; et al. ABySS 2.0: Resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 2017, 27, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bierman, M.; Logan, R.; O’Brien, K.; Seno, E.T.; Nagaraja Rao, R.; Schoner, B.E. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 1992, 116, 43–49. [Google Scholar] [CrossRef]
- Muth, G. The pSG5-based thermosensitive vector family for genome editing and gene expression in actinomycetes. Appl. Microbiol. Biotechnol. 2018, 102, 9067–9080. [Google Scholar] [CrossRef]
- Madoń, J.; Hütter, R. Transformation system for Amycolatopsis (Nocardia) mediterranei: Direct transformation of mycelium with plasmid DNA. J. Bacteriol. 1991, 173, 6325–6331. [Google Scholar] [CrossRef] [Green Version]
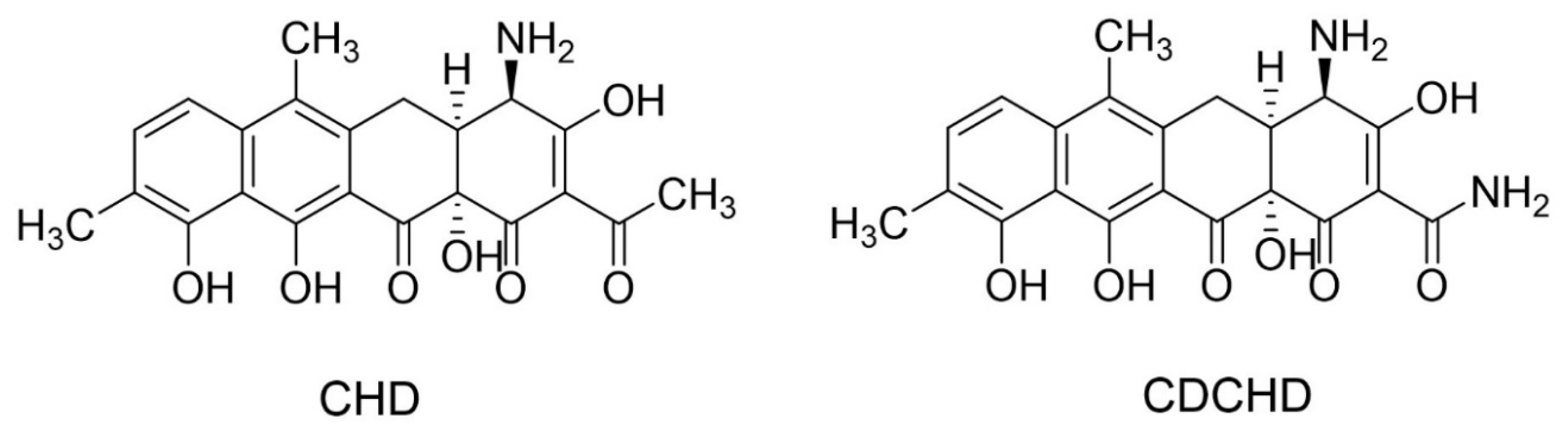

{kind=link}
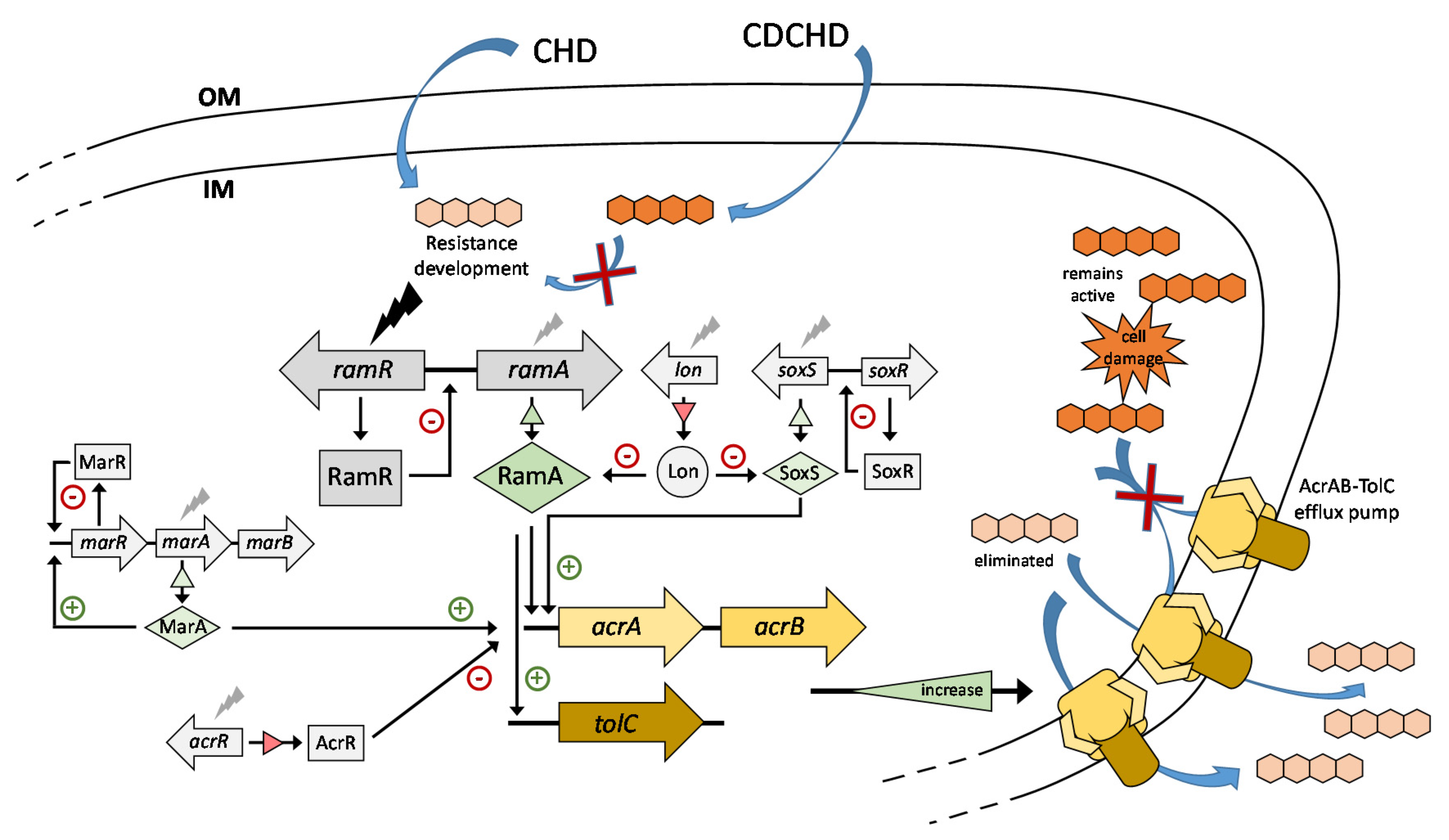
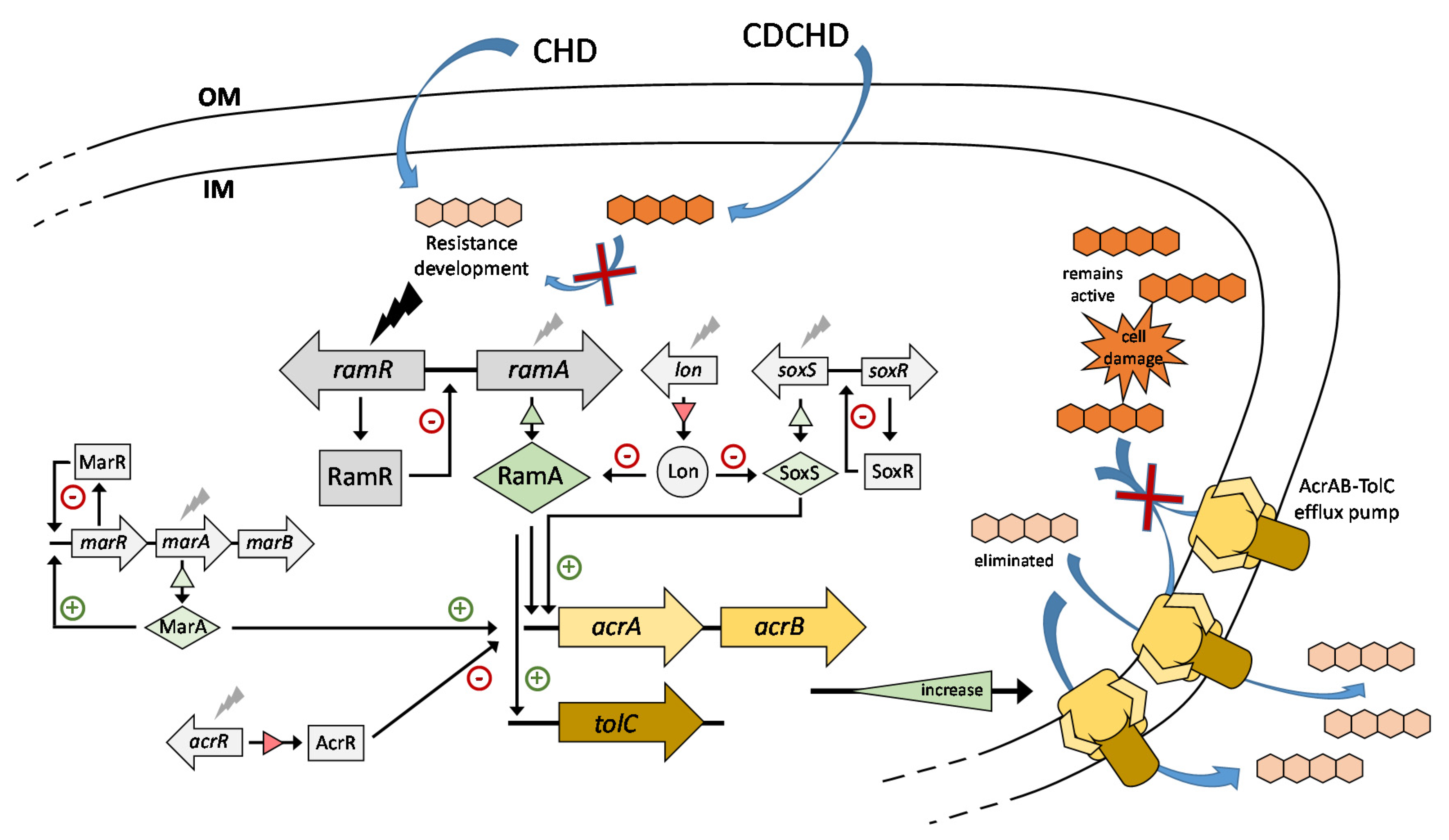{kind=link}
| Isolate (n = Number of Strains) | MIC (µg/mL) | MBC (µg/mL) | ||
|---|---|---|---|---|
| CHD | CDCHD | CHD | CDCHD | |
| Escherichia coli * | ||||
| Sensitive (15) | 2 | 4 | 16 | 4 |
| TEM β-lactamase (8) | 2–4 | 8 | 16 | 8 |
| ESBL (7) | 4 | 4 | 32 | 8 |
| Colistin-resistant (5) | 8 | 8 | 32 | 16 |
| Enterobacter spp. * (8) | 4 | 4 | 16 | 8–16 |
| Klebsiella spp. * | ||||
| Sensitive (7) | 4 | 4 | 16 | 8 |
| ESBL (2) | 4 | 4–8 | 32 | 8–16 |
| Carbapenem-resistant (2) | 4 | 4–8 | 32–64 | 8 |
| Enterococcus faecalis (19) | 8 | 16 | >64 | 16 |
| Enterococcus faecium (6) | 4 | 8 | 32 | 16 |
| Proteus spp. | ||||
| Sensitive (7) | 4 | 8 | 8–16 | 64 |
| ESBL (2) | 4 | 8–32 | 8–16 | 16–32 |
| Pseudomonas aeruginosa (10) | >64 | 32–64 | >64 | >64 |
| Test Condition | Isolate (n=) | MIC (µg/mL) | MBC (µg/mL) | ||
|---|---|---|---|---|---|
| CHD | CDCHD | CHD | CDCHD | ||
| Artificial urine | Escherichia coli (7) | 1 | 1 | 8 | 2 |
| Klebsiella pneumoniae (3) | 1 | 1 | 8 | 4 | |
| CAMHB | Escherichia coli (7) | 4 | 4 | 32 | 8 |
| Klebsiella pneumoniae (3) | 4 | 4 | 32 | 8 | |
| TET-Resistant Strain | Resistance Gene | Resistance Mechanism | MIC (µg/mL) | |
|---|---|---|---|---|
| CDCHD | TET | |||
| E. coli | tetB | Efflux | 4 | >64 |
| E. coli | tetM | Ribosomal protection | 0.5 | 64 |
| E. coli | tetW | Ribosomal protection | 1 | >64 |
| E. coli 49 | tetB | Efflux | 4 | >64 |
| E. coli 74 | tetB | Efflux | 8 | >64 |
| Serratia liquefaciens | tetB; tet34 | Efflux; enzymatic inactivation | 2 | >64 |
| Pseudomonas pseudoalcaligenes | tetB; tet34 | Efflux; enzymatic inactivation | 2 | >64 |
| K. pneumoniae 3 | tetA | Efflux | 8 | >64 |
| K. pneumoniae 8 | tetA | Efflux | 32 | >64 |
| K. pneumoniae 24 | tetA | Efflux | 8 | >64 |
| Gene | Mt8.1 | Mt8.2 | Mt8.3 | Mt8.4 | Mt8.5 | Mt8.6 | Mt8.7 | Mt8.8 |
|---|---|---|---|---|---|---|---|---|
| Efflux | ||||||||
| acrA | 2.65 | 1.72 | 1.97 | 2.16 | 2.03 | 2.33 | 1.98 | 2.88 |
| acrB | 3.27 | 2.82 | 2.01 | 2.70 | 2.88 | 2.48 | 3.03 | 2.31 |
| tolC | 2.97 | 2.00 | 2.25 | 2.15 | 2.10 | 3.39 | 2.12 | 3.96 |
| Regulators | ||||||||
| ramR | 5.80 | 4.44 | 4.35 | 6.78 | 4.89 | 5.59 | 4.98 | 3.01 |
| ramA | 16.57 | 11.79 | 10.47 | 13.40 | 9.89 | 13.73 | 10.91 | 6.11 |
| soxR | 1.02 | 1.03 | 3.22 | 0.70 | 0.93 | 3.83 | 0.81 | 0.73 |
| soxS | 1.13 | 1.09 | 18.09 | 0.81 | 1.12 | 16.80 | 0.86 | 0.91 |
| marR | 5.22 | 1.49 | 17.79 | 4.16 | 1.13 | 20.16 | 1.11 | 2.93 |
| marA | 2.58 | 1.14 | 11.69 | 2.65 | 0.83 | 11.66 | 0.75 | 0.78 |
| marB | 2.84 | 1.71 | 17.47 | 3.86 | 1.23 | 13.69 | 1.30 | 1.30 |
| acrR | 0.63 | 1.09 | 1.01 | 0.67 | 0.95 | 0.76 | 1.10 | 0.41 |
| Protease | ||||||||
| lon | 0.78 | 0.86 | 0.28 | 0.76 | 0.87 | 0.35 | 0.84 | 0.92 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hennessen, F.; Miethke, M.; Zaburannyi, N.; Loose, M.; Lukežič, T.; Bernecker, S.; Hüttel, S.; Jansen, R.; Schmiedel, J.; Fritzenwanker, M.; et al. Amidochelocardin Overcomes Resistance Mechanisms Exerted on Tetracyclines and Natural Chelocardin. Antibiotics 2020, 9, 619. https://doi.org/10.3390/antibiotics9090619
Hennessen F, Miethke M, Zaburannyi N, Loose M, Lukežič T, Bernecker S, Hüttel S, Jansen R, Schmiedel J, Fritzenwanker M, et al. Amidochelocardin Overcomes Resistance Mechanisms Exerted on Tetracyclines and Natural Chelocardin. Antibiotics. 2020; 9(9):619. https://doi.org/10.3390/antibiotics9090619
Chicago/Turabian StyleHennessen, Fabienne, Marcus Miethke, Nestor Zaburannyi, Maria Loose, Tadeja Lukežič, Steffen Bernecker, Stephan Hüttel, Rolf Jansen, Judith Schmiedel, Moritz Fritzenwanker, and et al. 2020. "Amidochelocardin Overcomes Resistance Mechanisms Exerted on Tetracyclines and Natural Chelocardin" Antibiotics 9, no. 9: 619. https://doi.org/10.3390/antibiotics9090619
APA StyleHennessen, F., Miethke, M., Zaburannyi, N., Loose, M., Lukežič, T., Bernecker, S., Hüttel, S., Jansen, R., Schmiedel, J., Fritzenwanker, M., Imirzalioglu, C., Vogel, J., Westermann, A. J., Hesterkamp, T., Stadler, M., Wagenlehner, F., Petković, H., Herrmann, J., & Müller, R. (2020). Amidochelocardin Overcomes Resistance Mechanisms Exerted on Tetracyclines and Natural Chelocardin. Antibiotics, 9(9), 619. https://doi.org/10.3390/antibiotics9090619