1. Introduction
Staphylococcus aureus is an opportunistic pathogen that can cause significant health challenges in humans and animals [
1]. Because of the ubiquity of
S.
aureus, it is difficult to prevent the involvement of the organism as secondary tissue invader following trauma or primary infections [
2]. The presence of virulence factors associated with bacterial adhesion, tissue invasion, immune evasion and toxin production may account for the frequent involvement of
S. aureus in clinical infections and food poisoning [
3]. Over the years,
S. aureus has acquired a wide variety of antimicrobial resistance properties, including methicillin resistance. The emergence and spread of methicillin-resistant
S. aureus (MRSA) strains that showed not only broad spectrum resistance to β-lactam antimicrobial agents, but often also resistance to multiple other antimicrobial agents, constitutes a great threat to the usefulness of antimicrobial agents in the treatment of staphylococcal infections, although it could lead to an increased risk of infection spread, higher medical cost and sometimes mortality [
4]. It is therefore not surprising that MRSA is one of the leading public health concerns worldwide.
MRSA first emerged as a nosocomial pathogen among hospitalized patients in healthcare facilities, but was later reported in community infections as well as in asymptomatic carriers among human populations. The subsequent detection of livestock-associated MRSA (LA-MRSA) in farm animals generated an increased interest with a OneHealth perspective [
5]. It also became pertinent to understand factors that play critical roles in the increasing incidence of the organism in different human, animal, food and environmental sources from different geographical locations. Regrettably, there are few data about MRSA, its molecular properties and the transmission routes, especially via meat production and marketing chains from low-income countries, like Nigeria. Earlier studies under separate investigations have documented the detection of MRSA in samples of human [
6,
7] and animal origin [
8,
9]. Continuous surveillance using the latest technology will provide in-depth analysis of MRSA profiles for a better understanding of the origins and dissemination patterns in human and animal populations across different geographical locations. The present study, therefore, investigated the presence and potential interrelatedness of MRSA in human, chicken and environmental samples from live bird markets in three cities located in southwestern Nigeria.
3. Discussion
This study showed the presence of MRSA in human, chicken meat and environmental samples from live bird markets/chicken slaughter centers in the study locations. No S. aureus was detected in samples from live chickens on farms, which could be due to the limited number of farms investigated. Only three farms agreed to participate in the study and they were managed by experienced veterinarians and had high-level biosecurity measures.
Live bird markets are recognized as potential sources for the spread and transmission of pathogens [
17]. The congregation of different species of poultry in these markets increases the chances for cross-contamination, a high diversity of pathogens and a possible emergence of new strains [
18,
19]. Poor hygiene and crowding facilitate the easy transmission and fast spread of pathogens via direct and indirect contact. Earlier studies have reported the presence of
S. aureus, including MRSA, in live bird and chicken carcasses at poultry markets in Zaria [
20] and Maiduguri [
9] in northern Nigeria. A high prevalence (80.0%) of
S. aureus was reported in samples from live chicken among small holder farms in Zaria [
21], while Nworie et al. [
8] reported a low MRSA prevalence (0.8%) in poultry farms in Ebonyi State, southeastern Nigeria.
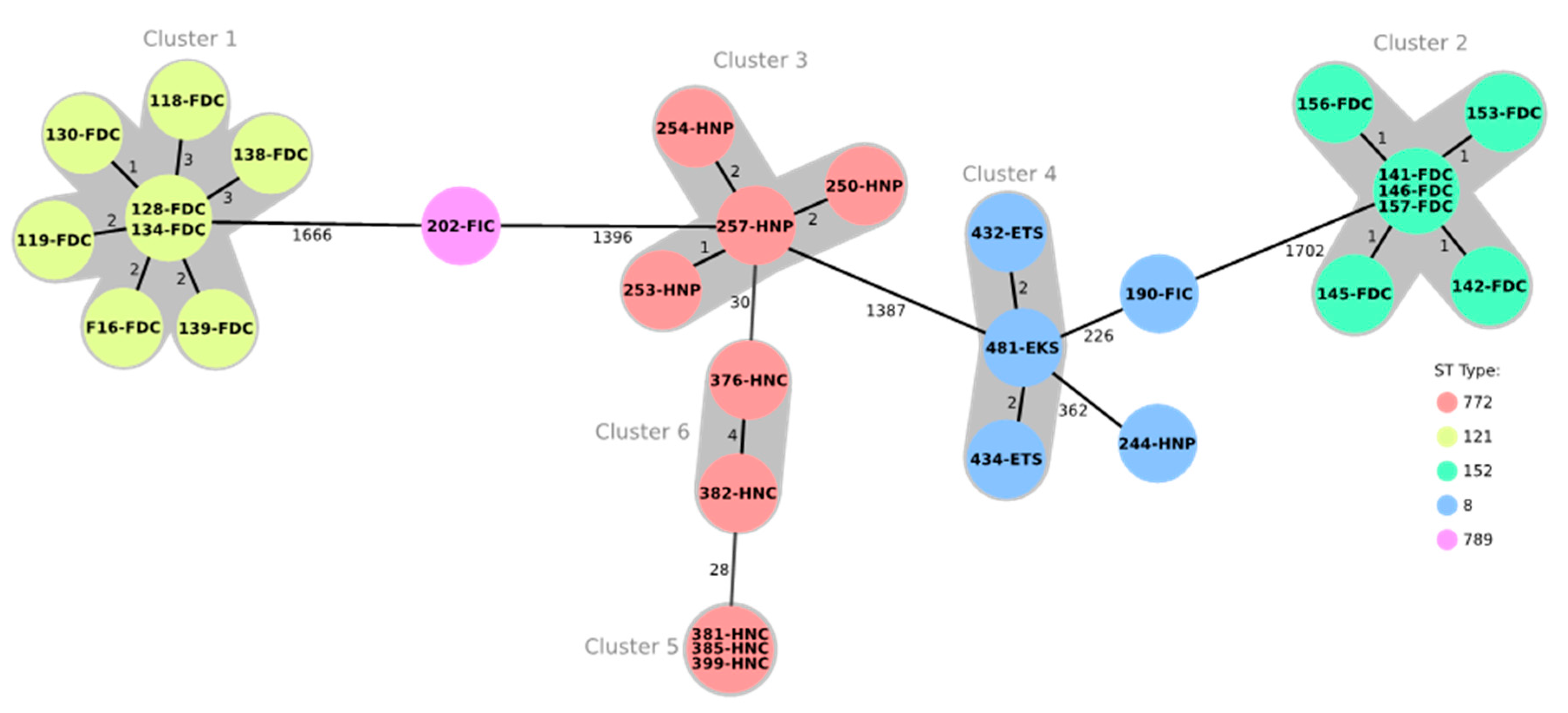
There was diversity in the STs, the cgMLST clusters,
spa and
dru types among the 30 MRSA isolates subjected to WGS. All STs identified in this study are associated with the community-associated MRSA (CA-MRSA) involved in human infections. LA-MRSA clones were not detected in this study. Earlier studies have identified LA-MRSA ST398 in poultry meat [
22,
23], diseased poultry [
24] or chickens at slaughter [
25]. MRSA ST8 is a CA-MRSA with high diversity and has been reported in hospital and community settings in several African countries, including Nigeria [
26,
27]. MRSA ST8 with
spa type t1476 identified in frozen chicken meat in this study had been reported in clinical samples from humans in Ghana [
28]. ST121 has been described as highly disseminated CA-MRSA, and has been reported in Nigeria [
7,
29], as well as in other African countries [
28,
30]. All the ST121 isolates detected in the present study had
spa type t314, similar to those reported in human healthcare institutions in Ghana [
28]. In the present study, all the MRSA isolates from freshly dressed chicken in Ibadan belonged to ST152 with
spa type t4690, which has also been reported in human clinical samples in Ghana [
28], and from human cases of osteomyelitis in Ile-Ife, southwestern Nigeria [
6]. MRSA ST772 has emerged as a CA-MRSA with a global spread [
31,
32]. All isolated MRSA belonging to ST772 in the present study were from human samples. ST772 has been reported in human infections in a tertiary care hospital in Southwest Nigeria [
7]. The only isolate belonging to ST789 had
spa type t091 and was recovered from frozen chicken in Abeokuta. A similar MRSA ST789 with
spa type t091 was reported in hospital settings in Angola [
33].
MRSA showed varying degrees of resistance to the tested antimicrobial agents. All MRSA isolates but one from humans were multidrug resistant. Multidrug resistance could be due to the co-selection of multiple resistance genes co-located on the same mobile genetic element following selective pressure induced by antimicrobial usage. An earlier study on the epidemiology of MRSA in Nigeria reported a high percentage of gentamicin, ciprofloxacin, and erythromycin resistance in MRSA of human origin [
26], similar to the findings in the present study. However, in that study [
26], MRSA isolates from humans also showed a high percentage of resistance to tetracycline, clindamycin and trimethoprim/sulfamethoxazole, contrary to the findings of the present study. In our study, the
mecA gene was detected in all selected MRSA isolates. This gene was located in SCC
mec cassettes of the types IVa, V or Vc. SCC
mec elements of types IV and V are the most commonly encountered SCC
mec types in CA-MRSA [
27,
34,
35]. SCC
mec elements of types IVa and V have been reported in hospital-associated MRSA (HA-MRSA) in Nigeria [
29]. In addition to
mecA, which confers resistance to a very broad range of β-lactams, a β-lactamase-encoding gene (
blaZ) was detected among MRSA isolates from this study. Phenotypic resistance to tetracycline, gentamicin and erythromycin was supported by the detection of
tet(K),
aphA3 and
msr(A)/
mph(C). All the tested MRSA harbored the gene
dfrG, which confers resistance to trimethoprim. Moreover, most MRSA isolates in this study possessed
fosB (resistance to fosfomycin) and one also possessed
sat4 (resistance to streptothricin), but our isolates were not investigated for phenotypic susceptibility to fosfomycin and streptothricin. The presence of these genes is likely to be explained by co-selection, as both antimicrobial agents are neither approved for nor used in chickens. The
sat4 gene has been described to be present in a resistance gene cluster consisting of the genes
aadE–sat4–aphA3 in
S. pseudintermedius [
36,
37]. A further analysis showed that this cluster was not completely available in any of the sequenced MRSA isolates. Instead, a truncated cluster comprising
aadE–sat4–Δ
aphA3 was detected in the single ST789/t091/dt11a isolate, whereas a different truncated cluster comprising
aadE–Δ
sat4–Δ
aphA3 was found in the seven ST772/t657/dt10ao isolates and in the two ST772/t657/dt10cj isolates.
The expression of virulence genes influences the pathogenicity of the respective isolates and the severity of staphylococcal infections [
3]. All the MRSA isolates in this study possessed genes for virulence factors that play critical roles in adhesion, tissue destruction, invasion, immune modulation, erythrocytes and leucocyte lysis, and toxin production. The expression of these genes determines the pathogenesis and prognosis of MRSA infections [
38]. Enterotoxins are responsible for staphylococcal food poisoning. All ST152/t4690 isolates had no enterotoxin genes, while MRSA of other STs possessed one or more of the enterotoxin genes
sea,
seb and
sec. These enterotoxin genes are located on mobile genetic elements (MGEs), including bacteriophages (
sea), plasmids (
seb), or pathogenicity islands (
sec) [
39], which explains their presence or absence in individual isolates by the acquisition or loss of the respective MGEs. Many of the virulence genes detected in this study have been reported in CA-MRSA isolates from humans in hospital settings [
40]. The diversity in virulence genes among the STs in the present study signifies variability in virulence-carrying plasmid and bacteriophages in the isolates [
41,
42]. The frequent occurrence of virulence genes in the MRSA isolates suggests that they may cause severe infections in susceptible hosts including humans. Thus, the presence of these potential pathogens in chicken meat, environmental sources and apparently healthy human carriers portends their role as a threat to public health.
4. Materials and Method
4.1. Sampling Locations and Sample Collection
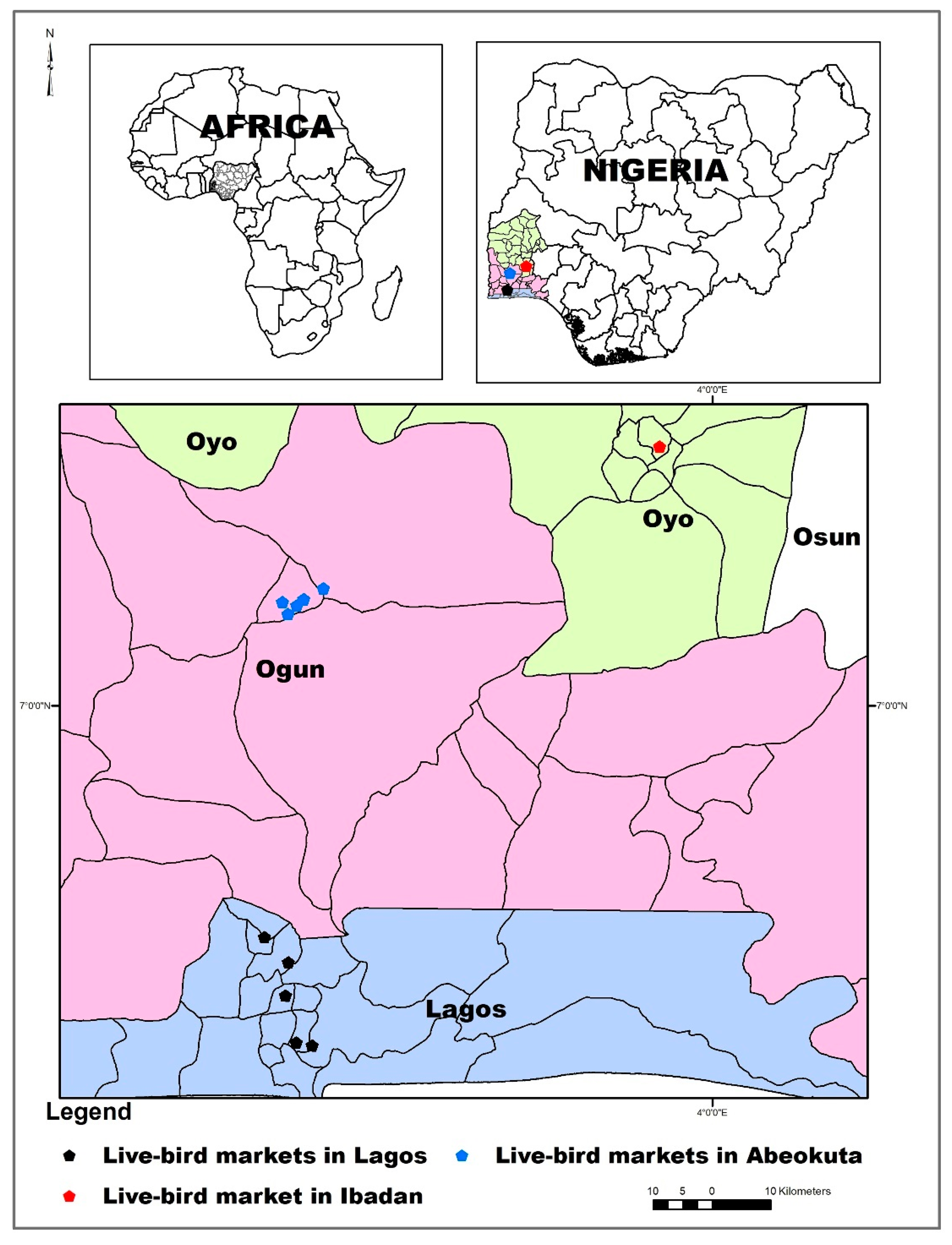
Samples were collected from human, chicken, chicken meat, and environmental sources within live bird markets and chicken slaughter centers in Lagos, Abeokuta and Ibadan, all located in the southwestern part of Nigeria (
Figure 2).
Both Lagos and Ibadan have large markets for chicken products, including chicken meat. Ogun State with its capital Abeokuta is the largest producer of chickens in Nigeria. Farmers and middle men bring live chickens in large numbers from farms and supply these to vendors/retailers in the live bird markets. These vendors sell the chickens to individual consumers. It is very common for people to purchase chickens from vendors in live bird markets for household consumption. The chickens are slaughtered and processed (dressed) by processors within the market, either at the point of purchase or in nearby vicinity. Samples comprised nasal swabs from humans, meat of freshly dressed chicken carcasses, as well as a swab of the surface of utensils, such as knives and tables, used by chicken meat processors in slaughterhouses of purchase, or in the nearby vicinity. Samples comprised nasal swabs from humans and the meat of freshly dressed chicken carcasses, as well as swabs of the surface of utensils, such as knives and tables used by chicken meat processors in slaughterhouses (
Table 6). Human samples were from chicken meat processors/vendors and consumers/buyers that purchased freshly dressed chicken meat from the slaughter centers. In addition, samples were collected from imported frozen chicken meat being sold within the premises of the live bird market. As a trace-back strategy, oropharyngeal swabs were collected from live chickens in three poultry farms that supplied live chickens to vendors at the live bird markets in Lagos and Abeokuta. Sampling was done within a five-month period from December 2016 to April 2017. Samples were labeled, preserved in icepacks and transported to the laboratory for microbiological analysis.
4.2. Isolation and Phenotypic Identification of Staphylococci Including S. aureus
Sample swabs were inoculated into 10 mL of Mueller–Hinton broth (MHB) containing 6.5% sodium chloride (NaCl) for pre-enrichment. Meat samples (25 g each) were thoroughly homogenized and inoculated into 225 mL of MHB for pre-enrichment. Pre-enrichment cultures were incubated at 35 ± 2 °C for 24 h. After the pre-enrichment stage, MHB cultures were streaked onto mannitol salt agar (MSA) with graded oxacillin supplementation; first at 2 mg/L and later at 4 mg/L. The MSA plates were incubated at 35 ± 2 °C for 24 to 48 h. The MSA plates were inspected for the presence of oxacillin-resistant, mannitol-fermenting colonies of bacteria. Oxacillin-resistant isolates were further sub-cultured on MSA supplemented with cefoxitin at 4 μg/mL. Cefoxitin-resistant isolates were noted. All oxacillin-resistant isolates (regardless of their cefoxitin resistance status) were selected and streaked onto blood agar (5% sheep blood) for the phenotypic identification of staphylococci. Identification tests included colony morphology and type of hemolysis on blood agar, Gram staining and microscopy, catalase, and oxidase, as well as coagulase and clumping factor tests. All oxacillin-resistant, catalase-positive, oxidase-negative Gram-positive cocci with cluster-like arrangement irrespective of coagulase production were selected for confirmatory tests to identify S. aureus. In total, 636 isolates, suspected to be staphylococci, were identified from 734 samples. Some samples yielded more than one suspected isolate, while others yielded none. Identified isolates were preserved on Tryptic Soy agar (supplemented with oxacillin) slants and refrigerated. For species identification and molecular characterization of isolates, preserved cultures were shipped to the laboratories at the Institute of Microbiology and Epizootics, Freie Universität (FU) Berlin, and the National Reference Laboratory for coagulase-positive staphylococci including S. aureus (NRL-Staph), Federal Institute for Risk Assessment (BfR), both located in Berlin, Germany.
Fresh staphylococcal colonies on blood agar were tested by MALDI-TOF mass spectrometry after an initial screening on Baird–Parker agar (BPA) for species identification. Isolates with inconclusive identification results after MALDI-TOF were further subjected to confirmation as
S. aureus by an in-house multiplex real-time polymerase chain reaction (RT-PCR) assay, as described elsewhere [
43] targeting the genus-specific gene
tuf and the species-specific gene
nuc. In parallel, and based on the presence/absence of the
mecA gene coding for methicillin resistance, isolates were further identified as MRSA (
mecA+) and MSSA (
mecA−) genotypes, respectively.
4.3. Antimicrobial Susceptibility Testing
Of the 56 MRSA isolates, minimum inhibitory concentrations (MICs) were determined for 16 non-β-lactam antimicrobial agents or combinations of agents by broth microdilution, according to the recommendations of the CLSI [
10]. For this, the microtitre plate layouts (Sensititre™), that were used in the national resistance monitoring program GE
RM-Vet, were also used in this study. The antimicrobial agents tested and the test ranges in mg/L were as follows: ciprofloxacin (0.08–16), clindamycin (0.03–64), doxycycline (0.06–128), enrofloxacin (0.08–16), erythromycin (0.015–32), florfenicol (0.12–256), gentamicin (0.12–256), linezolid (0.03–64), nalidixic acid (0.06–128), neomycin (0.12–64), quinupristin/dalfopristin (0.015–8), streptomycin (0.25–512), tetracycline (0.12–256), tiamulin (0.03–64), trimethoprim/sulfamethoxazole (0.015/0.3–32/608), and vancomycin (0.015–32).
S. aureus ATCC
®29213 was included in the tests for quality control purposes.
4.4. DNA Extraction and Whole Genome Sequencing
Out of the 56 isolates confirmed as MRSA, 30 isolates were selected for WGS based on phenotypic resistance profiles, sample source and sampling locations. Representative isolates were selected among groups of isolates with similar phenotypic resistance profiles from different sample categories in each geographical location. Hence, all the identified phenotypic resistance profiles observed among isolates from different sample types within each geographical location were represented in the test collection submitted to WGS. All selected isolates had oxacillin MICs of ≥16 mg/L, suggesting that they were phenotypic MRSA.
S. aureus isolates grown in brain heart infusion (BHI) broth at 37 ± 2 °C for 24 h were harvested and DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). The quality of the DNA was evaluated by spectral analysis (NanoDrop Spectrophotometer, Thermo Fisher Scientific, Waltham, MA, USA), and the concentration was fluorometrically quantified by a Qubit 3.0 Fluorometer (dsDNA HS Assay Kit 0.2–100 ng; Thermo Fisher Scientific, Waltham, MA, USA). DNA libraries were prepared using the Nextera DNA Flex Library Prep Kit, according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Library quality was assessed using the fragment analyzer 3408 (Advanced Analytical Technologies Inc., Ames, IA, USA). Paired-end sequencing was performed on the Illumina NextSeq platform (2 × 151 cycles) using the NextSeq 500/550 Mid Output kit v2.5 (300 cycles).
Raw Illumina reads were trimmed and de novo assembled with our in-house developed AQUAMIS pipeline (
https://gitlab.com/bfr_bioinformatics/AQUAMIS/), which implements fastp [
44] for trimming, and shovill (based on SPAdes) (
https://github.com/tseemann/shovill) for assembly. Furthermore, this pipeline performs Mash v 2.1 for reference search (
http://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0997-x), as well as QUAST v 5.0.2 for assembly quality control (
https://github.com/ablab/quast). The Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession numbers JABUZL000000000-JABVAO000000000. To determine the phylogenetic relationships, sequences were analyzed by Ridom Seqsphere+ v. 6.0.0 (April 2019) (Ridom, Muenster, Germany), using the proposed cgMLST scheme of 1783 gene targets [
45], with 98% required identity and 98% required percentage of coverage to one of the known alleles (allele library status December 2019). Bacterial characterization was conducted with our in-house developed BakCharak pipeline (
https://gitlab.com/bfr_bioinformatics/bakcharak), which implements ABRicate (
https://github.com/tsmeeann/abricate) for the screening of antimicrobial resistance genes (using the NCBI AMRFinder database [
46]), plasmid markers (using the PlasmidFinder database [
47]) and virulence factors (using the VFDB database (
https://dx.doi.org/10.1093%2Fnar%2Fgki008)). In addition, the ResFinder 4.0 database (
https://cge.cbs.dtu.dk/services/ResFinder/) was used for identification of antimicrobial resistance genes and chromosomal mutations conferring antimicrobial resistance. The
spa (
https://spaserver.ridom.de/),
dru (
http://dru-typing.org/), and MLST (
https://pubmlst.org/bigsdb?db=pubmlst_saureus_seqdef) types were deduced from the whole genome sequences.


 ,
, 

{kind=link}
{kind=link}