3-(5-Nitrofuran-2-yl)prop-2-en-1-one Derivatives, with Potent Antituberculosis Activity, Inhibit A Novel Therapeutic Target, Arylamine N-acetyltransferase, in Mycobacteria

 , , and
, , and 
Abstract
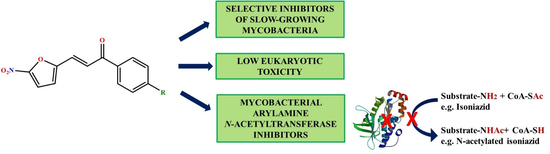
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Bacterial Strains and Cell Lines
3.2. Compounds
3.3. Evaluation of Antibacterial Potential using High-throughput Spot Culture Growth Inhibition (HT-SPOTi) Assay
3.4. Cytotoxicity Assay
3.5. Intracellular Survival Assay
3.6. Molecular Docking and MM-GBSA of the Molecules
3.7. Estimation of Inhibition against Recombinant Mycobacterium Marinum N-acetyl Transferase (MMNAT)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2019. 2019. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 15 April 2020).
- Marrakchi, H.; Lanéelle, M.-A.; Daffé, M. Mycolic Acids: Structures, Biosynthesis, and Beyond. Chem. Biol. 2014, 21, 67–85. [Google Scholar] [CrossRef]
- Upton, A.M.; Mushtaq, A.; Victor, T.C.; Sampson, S.L.; Sandy, J.; Smith, D.; Van Helden, P.V.; Sim, E. Arylamine N-acetyltransferase of Mycobacterium tuberculosis is a polymorphic enzyme and a site of isoniazid metabolism. Mol. Microbiol. 2001, 42, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, S.; Besra, G.S.; Upton, A.M.; Parish, T.; Sholto-Douglas-Vernon, C.; Gibson, K.J.C.; Knutton, S.; Gordon, S.; daSilva, R.P.; Anderton, M.C.; et al. Arylamine N-Acetyltransferase Is Required for Synthesis of Mycolic Acids and Complex Lipids in Mycobacterium bovis BCG and Represents a Novel Drug Target. J. Exp. Med. 2004, 199, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Anderton, M.C.; Bhakta, S.; Besra, G.S.; Jeavons, P.; Eltis, L.D.; Sim, E. Characterization of the putative operon containing arylamine N-acetyltransferase (nat) in Mycobacterium bovis BCG. Mol. Microbiol. 2005, 59, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Van der Geize, R.; Yam, K.; Heuser, T.; Wilbrink, M.H.; Hara, H.; Anderton, M.C.; Sim, E.; Dijkhuizen, L.; Davies, J.E.; Mohn, W.W.; et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Westwood, I.M.; Bhakta, S.; Russell, A.J.; Fullam, E.; Anderton, M.C.; Kawamura, A.; Mulvaney, A.W.; Vickers, R.J.; Bhowruth, V.; Besra, G.S.; et al. Identification of arylamine N-acetyltransferase inhibitors as an approach towards novel anti-tuberculars. Protein Cell 2010, 1, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Abuhammad, A.; Fullam, E.; Bhakta, S.; Russell, A.J.; Morris, G.M.; Finn, P.W.; Sim, E. Exploration of piperidinols as potential antitubercular agents. Molecules 2014, 19, 16274–16290. [Google Scholar] [CrossRef]
- Libardo, M.D.J.; Boshoff, H.I.M.; Barry, C.E., III. The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol. 2018, 42, 81–94. [Google Scholar] [CrossRef]
- Tawari, N.R.; Bairwa, R.; Ray, M.K.; Rajan, M.G.R.; Degani, M.S. Design, synthesis, and biological evaluation of 4-(5-nitrofuran-2-yl)prop-2-en-1-one derivatives as potent antitubercular agents. Bioorg. Med. Chem. Lett. 2010, 20, 6175–6178. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Zuccotto, F.; Hsinpin, H.; Sandberg, L.; Via, L.E.; Marriner, G.A.; Masquelin, T.; Wyatt, P.; Ray, P.; Dartois, V. Prediction of Drug Penetration in Tuberculosis Lesions. ACS Infect. Dis. 2016, 2, 552–563. [Google Scholar] [CrossRef]
- Edith, S.; Nicola, L. Arylamine N-acetyltransferases in Health And Disease: From Pharmacogenetics To Drug Discovery and Diagnostics; World Scientific: Singapore, 2018. [Google Scholar]
- Fullam, E.; Westwood, I.M.; Anderton, M.C.; Lowe, E.D.; Sim, E.; Noble, M.E.M. Divergence of Cofactor Recognition across Evolution: Coenzyme A Binding in a Prokaryotic Arylamine N-Acetyltransferase. J. Mol. Biol. 2008, 375, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Fullam, E.; Kawamura, A.; Wilkinson, H.; Abuhammad, A.; Westwood, I.; Sim, E. Comparison of the Arylamine N-Acetyltransferase from Mycobacterium marinum and Mycobacterium tuberculosis. Protein J. 2009, 28, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Abuhammad, A.M.; Lowe, E.D.; Fullam, E.; Noble, M.; Garman, E.F.; Sim, E. Probing the architecture of the Mycobacterium marinum arylamine N-acetyltransferase active site. Protein Cell 2010, 1, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Abuhammad, A.; Lowe, E.D.; McDonough, M.A.; Shaw Stewart, P.D.; Kolek, S.A.; Sim, E.; Garman, E.F. Structure of arylamine N-acetyltransferase from Mycobacterium tuberculosis determined by cross-seeding with the homologous protein from M. marinum: Triumph over adversity. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1433–1446. [Google Scholar] [CrossRef]
- Grandjean, L.; Martin, L.; Gilman, R.H.; Valencia, T.; Herrera, B.; Quino, W.; Ramos, E.; Rivero, M.; Montoya, R.; Escombe, A.R. Tuberculosis diagnosis and multidrug resistance testing by direct sputum culture in selective broth without decontamination or centrifugation. J. Clin. Microbiol. 2008, 46, 2339–2344. [Google Scholar] [CrossRef][Green Version]
- Agre, N.; Khambete, M.; Maitra, A.; Gupta, A.; Munshi, T.; Bhakta, S.; Degani, M. Exploration of 5-(5-nitrothiophen-2-yl)-4,5-dihydro-1H-pyrazoles as selective, multitargeted antimycobacterial agents. Chem. Biol. Drug Des. 2020, 95, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, S.; McHugh, T. Antibiotics Resistance Protocols, 2nd ed.; Springer: New York, NY, USA, 2010. [Google Scholar]
- Gupta, A.; Bhakta, S. An integrated surrogate model for screening of drugs against Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2012, 67, 1380–1391. [Google Scholar] [CrossRef]
- Guzman, J.D.; Evangelopoulos, D.; Gupta, A.; Birchall, K.; Mwaigwisya, S.; Saxty, B.; McHugh, T.D.; Gibbons, S.; Malkinson, J.; Bhakta, S. Antitubercular specific activity of ibuprofen and the other 2-arylpropanoic acids using the HT-SPOTi whole-cell phenotypic assay. BMJ Open 2013, 3, e002672. [Google Scholar] [CrossRef]
- Bhakta, S.; Scalacci, N.; Maitra, A.; Brown, A.K.; Dasugari, S.; Evangelopoulos, D.; McHugh, T.D.; Mortazavi, P.N.; Twist, A.; Petricci, E. Design and Synthesis of 1-((1, 5-Bis (4-chlorophenyl)-2-methyl-1 H-pyrrol-3-yl) methyl)-4-methylpiperazine (BM212) and N-Adamantan-2-yl-N′-((E)-3, 7-dimethylocta-2, 6-dienyl) ethane-1, 2-diamine (SQ109) Pyrrole Hybrid Derivatives: Discovery of Potent Antitubercular Agents Effective Against Multidrug-resistant Mycobacteria. J. Med. Chem. 2016, 59, 2780–2793. [Google Scholar]
- Johnson, B.K.; Abramovitch, R.B. Macrophage Infection Models for Mycobacterium tuberculosis. In Mycobacteria Protocols; Parish, T., Roberts, D.M., Eds.; Springer: New York, NY, USA, 2015; pp. 329–341. [Google Scholar]
- Jena, P.; Mohanty, S.; Mohanty, T.; Kallert, S.; Morgelin, M.; Lindstrøm, T.; Borregaard, N.; Stenger, S.; Sonawane, A.; Sørensen, O.E. Azurophil Granule Proteins Constitute the Major Mycobactericidal Proteins in Human Neutrophils and Enhance the Killing of Mycobacteria in Macrophages. PLoS ONE 2012, 7, e50345. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinforma. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Fukano, H.; Miyamoto, Y.; Shibayama, K.; Suzuki, M.; Hoshino, Y. Complete Genome Sequence of Mycobacterium marinum ATCC 927(T), Obtained Using Nanopore and Illumina Sequencing Technologies. Genome Announc. 2018, 6, e00397-18. [Google Scholar] [CrossRef] [PubMed]
- Abuhammad, A.; Fullam, E.; Lowe, E.D.; Staunton, D.; Kawamura, A.; Westwood, I.M.; Bhakta, S.; Garner, A.C.; Wilson, D.L.; Seden, P.T. Piperidinols that show anti-tubercular activity as inhibitors of arylamine N-acetyltransferase: An essential enzyme for mycobacterial survival inside macrophages. PLoS ONE 2012, 7, e52790. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Compound | MIC (mg/L) | GIC (mg/L) | IC50 (µM) (mg/L) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mtb H37Rv | MDR-TB | Mycobacterium bovis BCG | Mycobacterium aurum | Escherichia coli | Staphylococcus aureus | THP-1 Cell Line | RAW 264.7 Cell Line | MMNAT | |
| 1 | 0.244 | 0.976 | 0.06 | NAa | NAa | NAa | 62.5 | 125 | 0.207 (0.068) |
| 2 | 0.031 | 1.953 | 0.0075 | NAa | NAa | NAa | 62.5 | 125 | 0.269 (0.088) |
| 3 | 0.488 | 0.488 | 0.03 | NAa | 250 | 62.5 | 15.6 | 31.25 | 0.134 (0.042) |
| Isoniazid | 0.80 | NAa | 0.625 | 1.25 | ND | ND | NAa | NAa | NAb |
| Rifampicin | 0.125 | NAa | 0.50 # | 0.40 # | ND | ND | ND | ND | NAb |
| Ethambutol | 2.00 | 8.00 | 2.00 # | 0.10 # | ND | ND | ND | ND | NAb |
| Pretomanid | 0.061 | 0.488 | ND | ND | ND | ND | ND | ND | NAb |
| Ampicillin | ND | ND | NAa | ND | 2.00 | 0.125 | ND | ND | ND |
| Methotrexate | NAa | 250 | 500 | ND | ND | ND | 0.975 | 0.122 | ND |
| Compound | SI | |||||
|---|---|---|---|---|---|---|
| THP-1 | RAW 264.7 | |||||
| Mtb H37Rv | MDR-TB | M. bovis BCG | Mtb H37Rv | MDR-TB | M. bovis BCG | |
| 1 | 256 | 64 | 1042 | 512 | 128 | 2083 |
| 2 | 2016 | 32 | 8333 | 4032 | 64 | 16997 |
| 3 | 32 | 32 | 520 | 64 | 64 | 1042 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agre, N.; Tawari, N.; Maitra, A.; Gupta, A.; Munshi, T.; Degani, M.; Bhakta, S. 3-(5-Nitrofuran-2-yl)prop-2-en-1-one Derivatives, with Potent Antituberculosis Activity, Inhibit A Novel Therapeutic Target, Arylamine N-acetyltransferase, in Mycobacteria. Antibiotics 2020, 9, 368. https://doi.org/10.3390/antibiotics9070368
Agre N, Tawari N, Maitra A, Gupta A, Munshi T, Degani M, Bhakta S. 3-(5-Nitrofuran-2-yl)prop-2-en-1-one Derivatives, with Potent Antituberculosis Activity, Inhibit A Novel Therapeutic Target, Arylamine N-acetyltransferase, in Mycobacteria. Antibiotics. 2020; 9(7):368. https://doi.org/10.3390/antibiotics9070368
Chicago/Turabian StyleAgre, Neha, Nilesh Tawari, Arundhati Maitra, Antima Gupta, Tulika Munshi, Mariam Degani, and Sanjib Bhakta. 2020. "3-(5-Nitrofuran-2-yl)prop-2-en-1-one Derivatives, with Potent Antituberculosis Activity, Inhibit A Novel Therapeutic Target, Arylamine N-acetyltransferase, in Mycobacteria" Antibiotics 9, no. 7: 368. https://doi.org/10.3390/antibiotics9070368
APA StyleAgre, N., Tawari, N., Maitra, A., Gupta, A., Munshi, T., Degani, M., & Bhakta, S. (2020). 3-(5-Nitrofuran-2-yl)prop-2-en-1-one Derivatives, with Potent Antituberculosis Activity, Inhibit A Novel Therapeutic Target, Arylamine N-acetyltransferase, in Mycobacteria. Antibiotics, 9(7), 368. https://doi.org/10.3390/antibiotics9070368