Candidiasis and Mechanisms of Antifungal Resistance



Abstract
1. Introduction
2. Antifungals and Their Targets
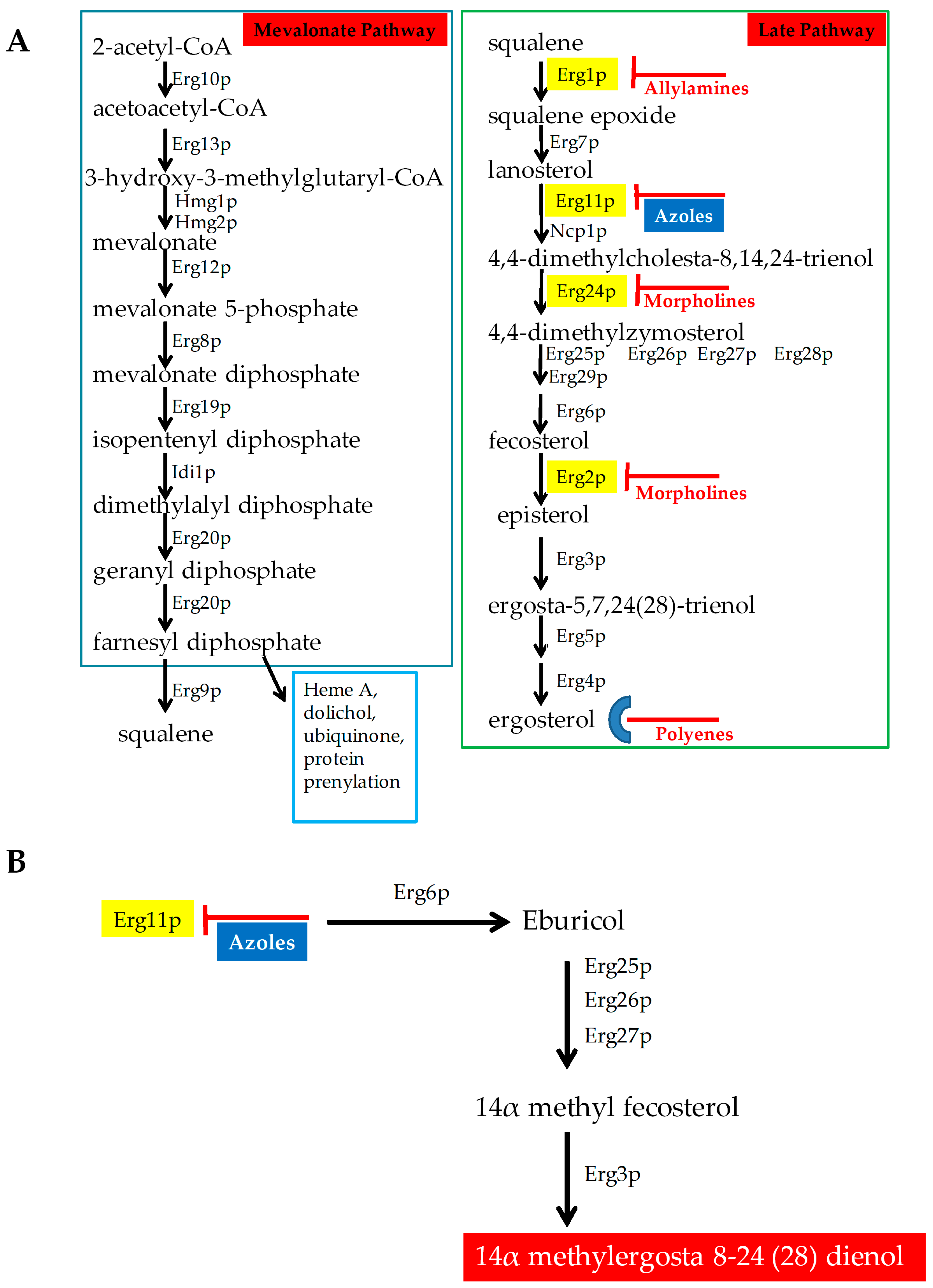
2.1. Antifungals that Target Ergosterol and Its Biosynthesis
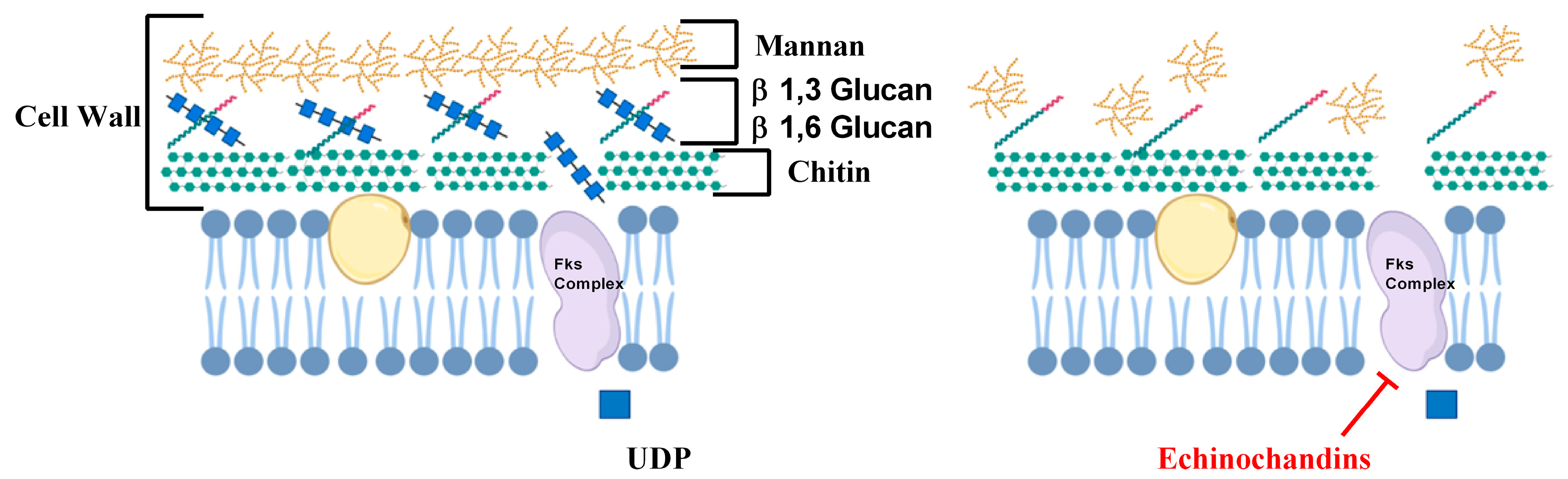
2.2. Inhibitors of Cell Wall Biosynthesis
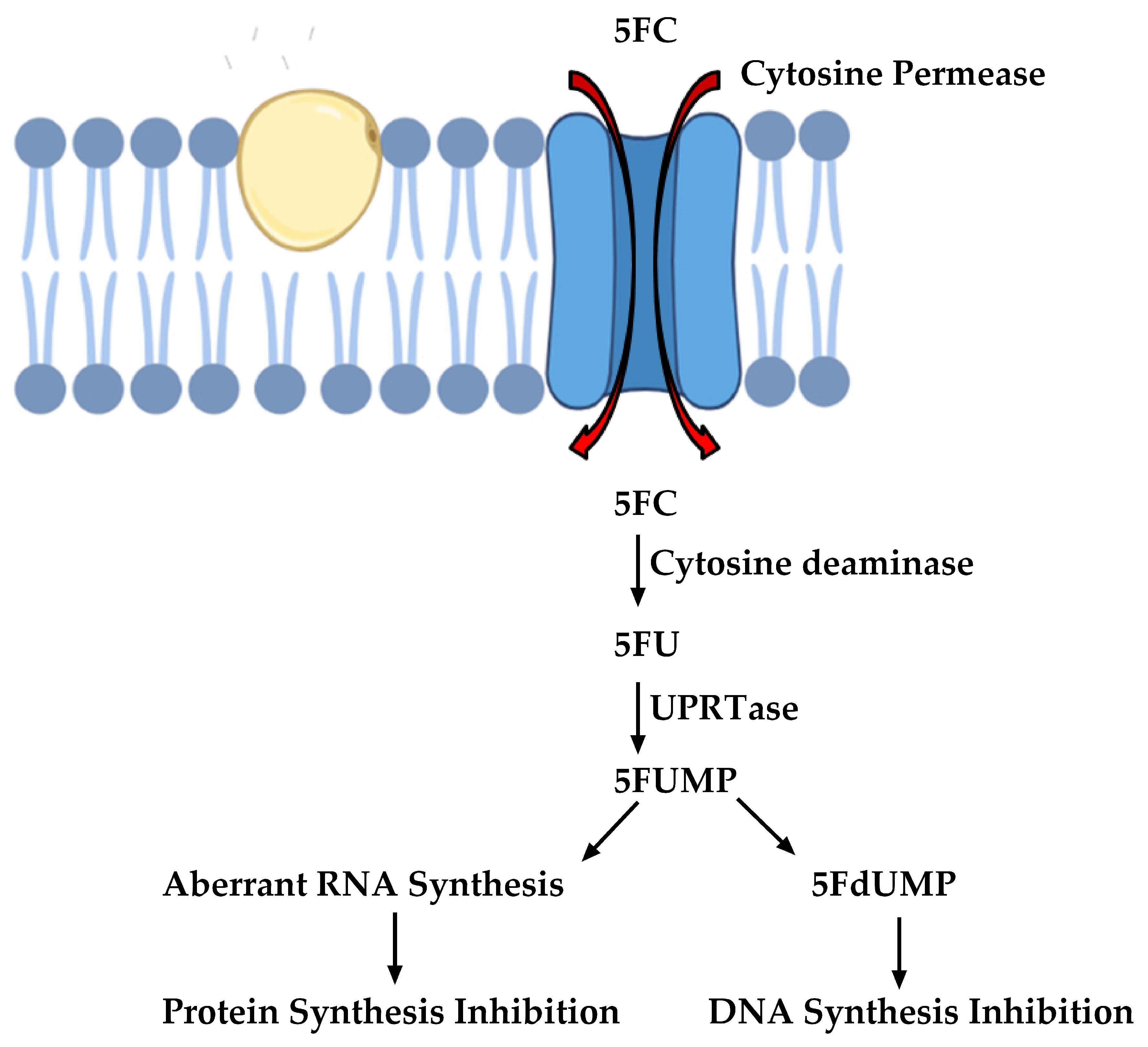
2.3. Inhibitors of Nucleic Acid Biosynthesis
3. Drug Efficacies and Therapeutic Failure
4. Antifungal Drug Resistance
4.1. Azole Drug Resistance
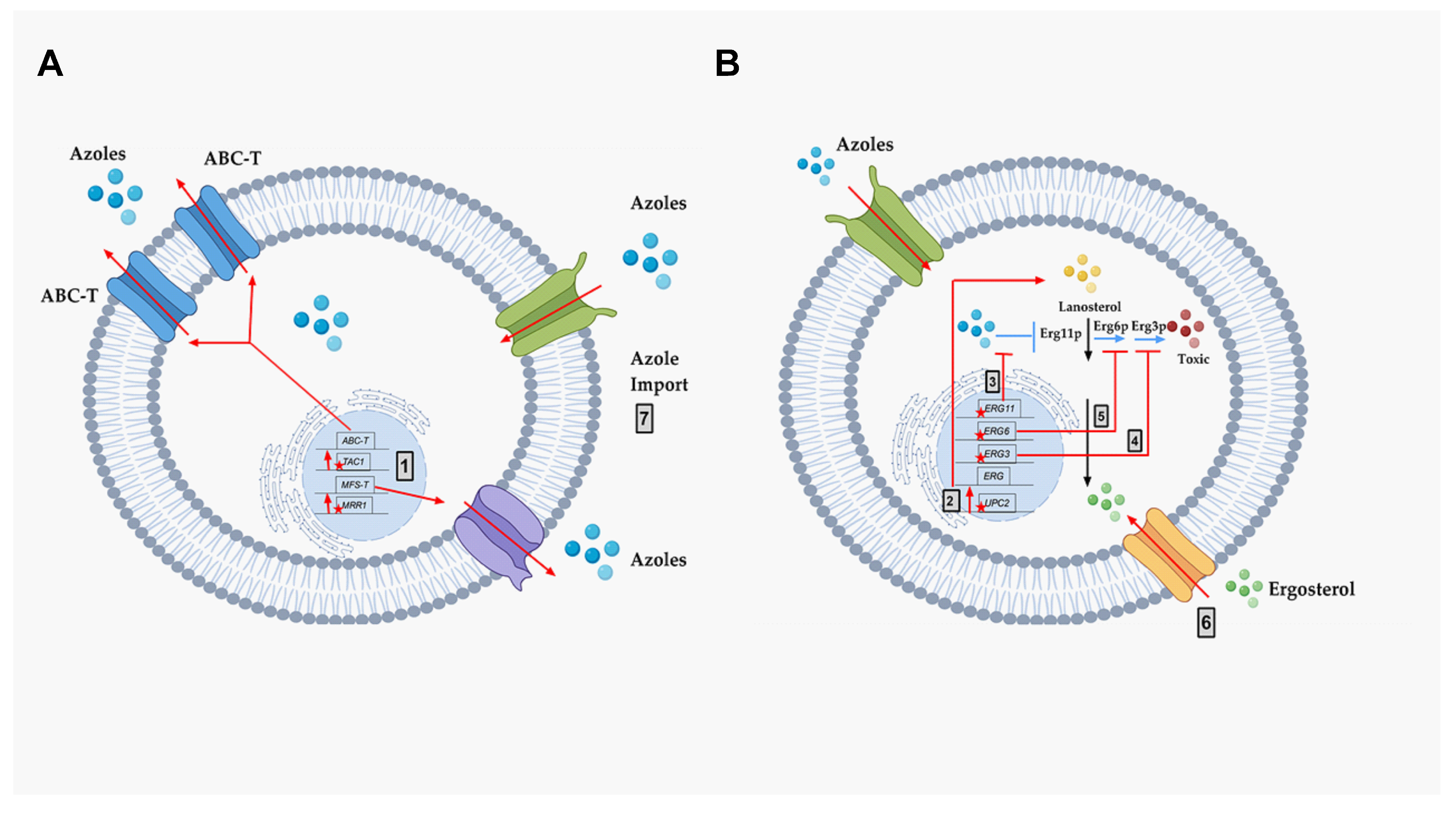
4.1.1. Over-Expression of Membrane Transporters
- a)
- ABC-transporters. Adenosine Triphosphate (ATP)-binding cassette transporters (ABC-T) are active transporters requiring ATP as an energy source. Each ABC-T consists of two membrane-spanning domains (MSD), each containing six transmembrane segments, and two nucleotide binding domains (NBD). Each NBD consists of an ATP-binding cassette (ABC) that binds ATP [39]. Azoles are the substrates for the ABC-Ts that are listed in Table 1 [40].
- b)
- MFS-transporters. Major facilitator transporters (MFS-T) require a proton gradient of the plasma membrane as an energy source to transport xenobiotics. MFS-Ts do not have the NBDs that are characteristic of ABC-Ts, and they have 12 to 14 transmembrane segments [41]. Azoles are also the substrates of the MFS-Ts listed in Table 1.
4.1.2. Altered Ergosterol Biosynthesis
- Mutation and/or overexpression of ergosterol pathway genes (Figure 4; Grey box 3)
- ERG11/CYP51: Erg11p is a cytochrome p450 enzyme that regulates a rate-limiting step in the ergosterol biosynthetic pathway [12]. It is an essential enzyme in the pathway, and cellular ergosterol levels decrease when Erg11p is inhibited by azoles [75]. ERG11 overexpression is linked with azole resistance in many fungi. Many azole-resistant C. albicans clinical isolates show increased CaERG11 expression [25,42,76]. CgERG11 overexpression is observed in azole-resistant C. glabrata isolates [77]. Overexpression of ERG11 was also recently observed in generationally aging C. auris that showed increased resilience to azoles [72]. Several point mutations in ERG11 have been identified in resistant clinical isolates. For example, CaErg11p mutations A61V, A114S, Y132F, Y132H, K143Q, K143R, Y257H, S405F, G448E, F449S, G464S, R467K, and I471T contribute to azole resistance in C. albicans [78]. Many of these point mutations lower azole binding in the active site of the enzyme. Point mutations in ERG11 associated with resistance have been identified in other azole-resistant Candida sp. These include, C. glabrata mutations C108G, C423T, and A1581G, and C. krusei mutations A497C and G1570A [79].
- ERG3: Besides ERG11, ERG3 is also linked with azole resistance. Erg3p is a C5 sterol desaturase enzyme, and converts episterol to ergosta-5,7,24 (28)- trienol during ergosterol biosynthesis (Figure 4; Grey box 4). When Erg11p is inhibited, Erg3p and Erg6p synthesize a toxic sterol, 14α methyl ergosta 8,24 (28)-dien-3β, 6α-diol, in both C. albicans and C. glabrata, [80,81,82]. Disruption of ERG3 results in increased azole resistance in C. albicans [83]. Q139A in Erg3p is reported in azole-resistant C. glabrata isolates [84].
- ERG6: In the presence of azoles, ERG6 contributes to the formation of the toxic diol from lanosterol [80,81,82] (Figure 4; Grey box 5). Erg6p is a Δ24 sterol C-methyl transferase, a non-essential enzyme in the ergosterol biosynthetic pathway. Significant azole resistance is observed in the heterozygous ERG6 deletion in C. albicans [85]. Similarly, Δerg6 in S. cerevisiae showed increased azole resistance [86], increased membrane permeability, and low Pdr5p efflux activity [87]. This suggests that ERG6-dependent azole resistance is the result of toxic sterol formation, and not due to efflux pump overexpression.
- Gain of Function Mutation (GOF) in UPC2 (Figure 4; Grey box 2)
4.1.3. Altered Sterol Import
4.1.4. Genome Plasticity
4.1.5. Other Hypothetical Azole Resistance Mechanism—Altered Azole Import
4.2. Resistance to Other Drugs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pfaller, M.A.; Diekema, D. Epidemiology of Invasive Candidiasis: A Persistent Public Health Problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef]
- Preventions, C.f.D.C.a. DRUG-RESISTANT CANDIDA SPECIES. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/candida-508.pdf (accessed on 9 June 2020).
- Pfaller, M.; Neofytos, D.; Diekema, D.; Azie, N.; Meier-Kriesche, H.-U.; Quan, S.-P.; Horn, D. Epidemiology and outcomes of candidemia in 3648 patients: Data from the Prospective Antifungal Therapy (PATH Alliance®) registry, 2004–2008. Diagn. Microbiol. Infect. Dis. 2012, 74, 323–331. [Google Scholar] [CrossRef]
- Kojic, E.M.; Darouiche, R.O. Candida Infections of Medical Devices. Clin. Microbiol. Rev. 2004, 17, 255–267. [Google Scholar] [CrossRef]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.R.; Clancy, C.J.; A Marr, K.; Ostrosky-Zeichner, L.L.; Reboli, A.; Schuster, M.G.; A Vazquez, J.; Walsh, T.J.; et al. Executive Summary: Clinical Practice Guideline for the Management of Candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2016, 62, 409–417. [Google Scholar] [CrossRef]
- Kodedová, M.; Sychrová, H. Changes in the Sterol Composition of the Plasma Membrane Affect Membrane Potential, Salt Tolerance and the Activity of Multidrug Resistance Pumps in Saccharomyces cerevisiae. PLoS ONE 2015, 10, e0139306. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Esquivel, B.D.; White, T.C. Overexpression or Deletion of Ergosterol Biosynthesis Genes Alters Doubling Time, Response to Stress Agents, and Drug Susceptibility inSaccharomyces cerevisiae. mBio 2018, 9, e01291-18. [Google Scholar] [CrossRef]
- Yasu, T.; Konuma, T.; Kuroda, S.; Takahashi, S.; Tojo, A. Effect of Cumulative Intravenous Voriconazole Dose on Renal Function in Hematological Patients. Antimicrob. Agents Chemother. 2018, 62, e00507-18. [Google Scholar] [CrossRef]
- Jenks, J.D.; Salzer, H.J.; Prattes, J.; Krause, R.; Buchheidt, D.; Hoenigl, M. Spotlight on isavuconazole in the treatment of invasive aspergillosis and mucormycosis: Design, development, and place in therapy. Drug Des. Dev. Ther. 2018, 12, 1033–1044. [Google Scholar] [CrossRef]
- Wilson, D.; Dimondi, V.P.; Johnson, S.W.; Jones, T.M.; Drew, R.H. Role of isavuconazole in the treatment of invasive fungal infections. Ther. Clin. Risk Manag. 2016, 12, 1197–1206. [Google Scholar] [CrossRef]
- Pasqualotto, A.C.; Falci, D.R. Profile of isavuconazole and its potential in the treatment of severe invasive fungal infections. Infect. Drug Resist. 2013, 6, 163–174. [Google Scholar] [CrossRef][Green Version]
- Veen, M.; Stahl, U.; Lang, C. Combined overexpression of genes of the ergosterol biosynthetic pathway leads to accumulation of sterols in Saccharomyces cerevisiae. FEMS Yeast Res. 2003, 4, 87–95. [Google Scholar] [CrossRef]
- Delattin, N.; Cammue, B.P.; Thevissen, K. Reactive oxygen species-inducing antifungal agents and their activity against fungal biofilms. Future Med. Chem. 2014, 6, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Efimova, S.S.; Schagina, L.V.; Ostroumova, O.S. Investigation of Channel-Forming Activity of Polyene Macrolide Antibiotics in Planar Lipid Bilayers in the Presence of Dipole Modifiers. Acta Nat. 2014, 6, 67–79. [Google Scholar] [CrossRef]
- Hamill, R.J. Amphotericin B Formulations: A Comparative Review of Efficacy and Toxicity. Drugs 2013, 73, 919–934. [Google Scholar] [CrossRef]
- Vogelsinger, H.; Weiler, S.; Djanani, A.; Kountchev, J.; Bellmann-Weiler, R.; Wiedermann, C.J.; Bellmann, R. Amphotericin B tissue distribution in autopsy material after treatment with liposomal amphotericin B and amphotericin B colloidal dispersion. J. Antimicrob. Chemother. 2006, 57, 1153–1160. [Google Scholar] [CrossRef]
- Sanglard, D.; Coste, A.T.; Ferrari, S. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res. 2009, 9, 1029–1050. [Google Scholar] [CrossRef]
- Shirwaikar, A.A.; Thomas, T.; Shirwaikar, A.; Lobo, R.; Prabhu, K.S. Treatment of Onychomycosis: An Update. Indian J. Pharm. Sci. 2008, 70, 710–714. [Google Scholar] [CrossRef]
- Popolo, L.; Gualtieri, T.; Ragni, E. The yeast cell-wall salvage pathway. Med. Mycol. 2001, 39, 111–121. [Google Scholar] [CrossRef]
- Perlin, D.S. Current perspectives on echinocandin class drugs. Future Microbiol. 2011, 6, 441–457. [Google Scholar] [CrossRef]
- Douglas, C.M. Fungal beta(1,3)-D-glucan synthesis. Med. Mycol. 2001, 39, 55–66. [Google Scholar] [CrossRef]
- Munro, C. Fungal echinocandin resistance. F1000 Boil. Rep. 2010, 2, 66. [Google Scholar] [CrossRef]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.; Benjamin, D.K.; Calandra, T.; Edwards, J.J.E.; Filler, S.G.; Fisher, J.F.; Kullberg, B.-J.; Ostrosky-Zeichner, L.L.; et al. Clinical Practice Guidelines for the Management of Candidiasis: 2009 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 503–535. [Google Scholar] [CrossRef]
- Vermes, A.; Guchelaar, H.-J.; Dankert, J. Flucytosine: A review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J. Antimicrob. Chemother. 2000, 46, 171–179. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Sobel, J.D.; White, T.C. A Combination Fluorescence Assay Demonstrates Increased Efflux Pump Activity as a Resistance Mechanism in Azole-Resistant Vaginal Candida albicans Isolates. Antimicrob. Agents Chemother. 2016, 60, 5858–5866. [Google Scholar] [CrossRef]
- White, T.C.; Marr, K.A.; Bowden, R.A. Clinical, Cellular, and Molecular Factors That Contribute to Antifungal Drug Resistance. Clin. Microbiol. Rev. 1998, 11, 382–402. [Google Scholar] [CrossRef]
- Rex, J.H.; Rinaldi, M.G.; Pfaller, M.A. Resistance of Candida species to fluconazole. Antimicrob. Agents Chemother. 1995, 39, 1–8. [Google Scholar] [CrossRef]
- White, T.C. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 1997, 41, 1482–1487. [Google Scholar] [CrossRef]
- Rodrigues, C.F.; Henriques, M. Oral mucositis caused byCandida glabratabiofilms: Failure of the concomitant use of fluconazole and ascorbic acid. Ther. Adv. Infect. Dis. 2017, 4, 10–17. [Google Scholar] [CrossRef]
- Sabra, R.; Branch, R.A. Amphotericin B Nephrotoxicity. Drug Saf. 1990, 5, 94–108. [Google Scholar] [CrossRef]
- Faustino, C.; Pinheiro, L. Lipid Systems for the Delivery of Amphotericin B in Antifungal Therapy. Pharmaceutics 2020, 12, 29. [Google Scholar] [CrossRef]
- Sau, K.; Mambula, S.S.; Latz, E.; Henneke, P.; Golenbock, D.T.; Levitz, S.M. The Antifungal Drug Amphotericin B Promotes Inflammatory Cytokine Release by a Toll-like Receptor- and CD14-dependent Mechanism. J. Boil. Chem. 2003, 278, 37561–37568. [Google Scholar] [CrossRef] [PubMed]
- Re, V.L.; Carbonari, D.; Goldberg, D.; Forde, K.A.; Goldberg, D.S.; Reddy, K.R.; Haynes, K.; Roy, J.A.; Sha, D.; Marks, A.R.; et al. Oral Azole Antifungal Medications and Risk of Acute Liver Injury, Overall and by Chronic Liver Disease Status. Am. J. Med. 2015, 129, 283–291.e5. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Zapata, D.; Petraitiene, R.; Petraitis, V. Echinocandins: The Expanding Antifungal Armamentarium. Clin. Infect. Dis. 2015, 61, S604–S611. [Google Scholar] [CrossRef] [PubMed]
- Lamping, E.; Monk, B.C.; Niimi, K.; Holmes, A.; Tsao, S.; Tanabe, K.; Niimi, M.; Uehara, Y.; Cannon, R.D. Characterization of Three Classes of Membrane Proteins Involved in Fungal Azole Resistance by Functional Hyperexpression in Saccharomyces cerevisiae. Eukaryot. Cell 2007, 6, 1150–1165. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, N.K.; Wasi, M.; Nair, R.; Gupta, M.; Kumar, M.; Mondal, A.K.; Gaur, N.A.; Prasad, R. Vacuolar Sequestration of Azoles, a Novel Strategy of Azole Antifungal Resistance Conserved across Pathogenic and Nonpathogenic Yeast. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Leighton, J.; Schatz, G. An ABC transporter in the mitochondrial inner membrane is required for normal growth of yeast. EMBO J. 1995, 14, 188–195. [Google Scholar] [CrossRef]
- Kispal, G.; Csere, P.; Guiard, B.; Lill, R. The ABC transporter Atm1p is required for mitochondrial iron homeostasis. FEBS Lett. 1997, 418, 346–350. [Google Scholar] [CrossRef]
- Michealis, S.; Berkower, C. Sequence Comparison of Yeast ATP-binding Cassette Proteins. Cold Spring Harb. Symp. Quant. Boil. 1995, 60, 291–307. [Google Scholar] [CrossRef]
- Cannon, R.D.; Lamping, E.; Holmes, A.; Niimi, K.; Baret, P.V.; Keniya, M.V.; Tanabe, K.; Niimi, M.; Goffeau, A.; Monk, B.C. Efflux-Mediated Antifungal Drug Resistance. Clin. Microbiol. Rev. 2009, 22, 291–321. [Google Scholar] [CrossRef]
- Marger, M.D.; Saier, M.H. A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biochem. Sci. 1993, 18, 13–20. [Google Scholar] [CrossRef]
- White, T.C.; Holleman, S.; Dy, F.; Mirels, L.F.; Stevens, D.A. Resistance Mechanisms in Clinical Isolates of Candida albicans. Antimicrob. Agents Chemother. 2002, 46, 1704–1713. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.; Rahkhoodaee, F.; Raymond, M. Relative Contributions of the Candida albicans ABC Transporters Cdr1p and Cdr2p to Clinical Azole Resistance. Antimicrob. Agents Chemother. 2009, 53, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, D.; Bille, J.; Sanglard, D. A novel multidrug efflux transporter gene of the major facilitator superfamily from Candida albicans (FLU1) conferring resistance to fluconazole. Microbiology 2000, 146, 2743–2754. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Yeater, K.M.; Hoyer, L.L. Cellular and Molecular Biology of Candida albicans Estrogen Response. Eukaryot. Cell 2006, 5, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Panwar, S.L.; Pasrija, R.; Prasad, R. Membrane homoeostasis and multidrug resistance in yeast. Biosci. Rep. 2008, 28, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Saidane, S.; Weber, S.; De Deken, X.; St-Germain, G.; Raymond, M. PDR16-mediated azole resistance in Candida albicans. Mol. Microbiol. 2006, 60, 1546–1562. [Google Scholar] [CrossRef]
- Lohberger, A.; Coste, A.T.; Sanglard, D. Distinct Roles of Candida albicans Drug Resistance Transcription FactorsTAC1,MRR1, andUPC2in Virulence. Eukaryot. Cell 2013, 13, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Morschhäuser, J.; Barker, K.S.; Liu, T.T.; Blaß-Warmuth, J.; Homayouni, R.; Rogers, P.D. The Transcription Factor Mrr1p Controls Expression of the MDR1 Efflux Pump and Mediates Multidrug Resistance in Candida albicans. PLoS Pathog. 2007, 3, e164. [Google Scholar] [CrossRef]
- Coste, A.T.; Turner, V.; Ischer, F.; Morschhäuser, J.; Forche, A.; Selmecki, A.; Berman, J.; Bille, J.; Sanglard, D. A Mutation in Tac1p, a Transcription Factor Regulating CDR1 and CDR2, Is Coupled With Loss of Heterozygosity at Chromosome 5 to Mediate Antifungal Resistance in Candida albicans. Genetics 2006, 172, 2139–2156. [Google Scholar] [CrossRef]
- Dunkel, N.; Blass, J.; Rogers, P.D.; Morschhäuser, J. Mutations in the multi-drug resistance regulatorMRR1, followed by loss of heterozygosity, are the main cause ofMDR1overexpression in fluconazole-resistantCandida albicansstrains. Mol. Microbiol. 2008, 69, 827–840. [Google Scholar] [CrossRef]
- Kalkandelen, K.T.; Dereli, M.D. [Investigation of mutations in transcription factors of efflux pump genes in fluconazole-resistant Candida albicans strains overexpressing the efflux pumps]. Mikrobiyoloji Bulteni 2015, 49, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Rogers, P.D.; Morschhäuser, J. Gain-of-Function Mutations in the Transcription Factor MRR1 Are Responsible for Overexpression of the MDR1 Efflux Pump in Fluconazole-Resistant Candida dubliniensis Strains. Antimicrob. Agents Chemother. 2008, 52, 4274–4280. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-G.; Yang, Y.-L.; Shih, H.-I.; Su, C.-L.; Lo, H.-J. CaNdt80 Is Involved in Drug Resistance in Candida albicans by Regulating CDR1. Antimicrob. Agents Chemother. 2004, 48, 4505–4512. [Google Scholar] [CrossRef]
- Talibi, D.; Raymond, M. Isolation of a Putative Candida albicansTranscriptional Regulator Involved in Pleiotropic Drug Resistance by Functional Complementation of a pdr1 pdr3 Mutation inSaccharomyces cerevisiae. J. Bacteriol. 1999, 181, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Mogavero, S.; Tavanti, A.; Senesi, S.; Rogers, P.D.; Morschhäuser, J. Differential Requirement of the Transcription Factor Mcm1 for Activation of the Candida albicans Multidrug Efflux PumpMDR1by Its Regulators Mrr1 and Cap1. Antimicrob. Agents Chemother. 2011, 55, 2061–2066. [Google Scholar] [CrossRef] [PubMed]
- Alarco, A.-M.; Raymond, M. The bZip Transcription Factor Cap1p Is Involved in Multidrug Resistance and Oxidative Stress Response inCandida albicans. J. Bacteriol. 1999, 181, 700–708. [Google Scholar] [CrossRef]
- Sanguinetti, M.; Posteraro, B.; Fiori, B.; Ranno, S.; Torelli, R.; Fadda, G. Mechanisms of Azole Resistance in Clinical Isolates of Candida glabrata Collected during a Hospital Survey of Antifungal Resistance. Antimicrob. Agents Chemother. 2005, 49, 668–679. [Google Scholar] [CrossRef]
- Vermitsky, J.-P.; Edlind, T.D. Azole Resistance in Candida glabrata: Coordinate Upregulation of Multidrug Transporters and Evidence for a Pdr1-Like Transcription Factor. Antimicrob. Agents Chemother. 2004, 48, 3773–3781. [Google Scholar] [CrossRef]
- Culakova, H.; Dzugasova, V.; Valencikova, R.; Gbelska, Y.; Šubík, J. Stress response and expression of fluconazole resistance associated genes in the pathogenic yeast Candida glabrata deleted in the CgPDR16 gene. Microbiol. Res. 2015, 174, 17–23. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Fries, B.C. Enhanced Efflux Pump Activity in OldCandida glabrataCells. Antimicrob. Agents Chemother. 2018, 62, e02227-17. [Google Scholar] [CrossRef]
- Costa, C.; Dias, P.; Sá-Correia, I.; Teixeira, M. MFS multidrug transporters in pathogenic fungi: Do they have real clinical impact? Front. Physiol. 2014, 5, 197. [Google Scholar] [CrossRef]
- Costa, C.; Ribeiro, J.; Miranda, I.M.; Silva-Dias, A.; Cavalheiro, M.; Costa-De-Oliveira, S.; Rodrigues, A.G.; Teixeira, M. Clotrimazole Drug Resistance in Candida glabrata Clinical Isolates Correlates with Increased Expression of the Drug:H+ Antiporters CgAqr1, CgTpo1_1, CgTpo3, and CgQdr2. Front. Microbiol. 2016, 7, 1773. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Ischer, F.; Calabrese, D.; Posteraro, B.; Sanguinetti, M.; Fadda, G.; Rohde, B.; Bauser, C.; Bader, O.; Sanglard, D. Gain of Function Mutations in CgPDR1 of Candida glabrata Not Only Mediate Antifungal Resistance but Also Enhance Virulence. PLoS Pathog. 2009, 5, e1000268. [Google Scholar] [CrossRef] [PubMed]
- Ni, Q.; Wang, C.; Tian, Y.; Dong, D.; Jiang, C.; Mao, E.; Peng, Y. CgPDR1 gain-of-function mutations lead to azole-resistance and increased adhesion in clinical Candida glabrata strains. Mycoses 2018, 61, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Vale-Silva, L.A.; Moeckli, B.; Torelli, R.; Posteraro, B.; Sanguinetti, M.; Sanglard, D. Upregulation of the Adhesin Gene EPA1 Mediated by PDR1 in Candida glabrata Leads to Enhanced Host Colonization. mSphere 2016, 1, e00065-15. [Google Scholar] [CrossRef]
- Costa, C.; Pires, C.; Cabrito, T.R.; Renaudin, A.; Ohno, M.; Chibana, H.; Sá-Correia, I.; Teixeira, M. Candida glabrata Drug:H+Antiporter CgQdr2 Confers Imidazole Drug Resistance, Being Activated by Transcription Factor CgPdr1. Antimicrob. Agents Chemother. 2013, 57, 3159–3167. [Google Scholar] [CrossRef]
- Lamping, E.; Ranchod, A.; Nakamura, K.; Tyndall, J.D.A.; Niimi, K.; Holmes, A.; Niimi, M.; Cannon, R.D. Abc1p is a Multidrug Efflux Transporter That Tips the Balance in Favor of Innate Azole Resistance in Candida krusei. Antimicrob. Agents Chemother. 2008, 53, 354–369. [Google Scholar] [CrossRef]
- Guinea, J.; Sánchez-Somolinos, M.; Cuevas, O.; Peláez, T.; Bouza, E. Fluconazole resistance mechanisms inCandida krusei: The contribution of efflux-pumps. Med. Mycol. 2006, 44, 575–578. [Google Scholar] [CrossRef]
- Zhang, L.; Xiao, M.; Watts, M.R.; Wang, H.; Fan, X.; Kong, F.; Xu, Y.-C. Development of fluconazole resistance in a series of Candida parapsilosis isolates from a persistent candidemia patient with prolonged antifungal therapy. BMC Infect. Dis. 2015, 15, 340. [Google Scholar] [CrossRef]
- Moran, G.P.; Pinjon, E.; Coleman, D.; Sullivan, D. Azole susceptibility and resistance in Candida dubliniensis. Biochem. Soc. Trans. 2005, 33, 1210. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Holowka, T.; Orner, E.P.; Fries, B.C. Gene Duplication Associated with Increased Fluconazole Tolerance in Candida auris cells of Advanced Generational Age. Sci. Rep. 2019, 9, 5052. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, F.; Graf, A.; Jequier, L.; Coste, A.T. Review on Antifungal Resistance Mechanisms in the Emerging Pathogen Candida auris. Front. Microbiol. 2019, 10, 2788. [Google Scholar] [CrossRef] [PubMed]
- Rybak, J.M.; Muñoz, J.F.; Barker, K.S.; Parker, J.E.; Esquivel, B.D.; Berkow, E.L.; Lockhart, S.R.; Gade, L.; Palmer, G.E.; White, T.C.; et al. Mutations in TAC1B: A Novel Genetic Determinant of Clinical Fluconazole Resistance in Candida auris. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Arthington-Skaggs, B.A.; Jradi, H.; Desai, T.; Morrison, C.J. Quantitation of Ergosterol Content: Novel Method for Determination of Fluconazole Susceptibility of Candida albicans. J. Clin. Microbiol. 1999, 37, 3332–3337. [Google Scholar] [CrossRef] [PubMed]
- Flowers, S.A.; Barker, K.S.; Berkow, E.L.; Toner, G.; Chadwick, S.G.; Gygax, S.E.; Morschhäuser, J.; Rogers, P.D. Gain-of-Function Mutations inUPC2Are a Frequent Cause ofERG11Upregulation in Azole-Resistant Clinical Isolates of Candida albicans. Eukaryot. Cell 2012, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Pam, V.K.; Akpan, J.U.; O Oduyebo, O.; O Nwaokorie, F.; A Fowora, M.; O Oladele, R.; Ogunsola, F.T.; I Smith, S. Fluconazole susceptibility and ERG11 gene expression in vaginal candida species isolated from lagos Nigeria. Int. J. Mol. Epidemiol. Genet. 2012, 3, 84–90. [Google Scholar]
- Xiang, M.-J.; Liu, J.-Y.; Ni, P.-H.; Wang, S.; Shi, C.; Wei, B.; Ni, Y.-X.; Ge, H.-L. Erg11mutations associated with azole resistance in clinical isolates ofCandida albicans. FEMS Yeast Res. 2013, 13, 386–393. [Google Scholar] [CrossRef]
- Silva, D.B.D.S.; Rodrigues, L.M.C.; De Almeida, A.A.; De Oliveira, K.M.P.; Grisolia/, A.B. Novel point mutations in the ERG11 gene in clinical isolates of azole resistant Candida species. Memórias do Instituto Oswaldo Cruz 2016, 111, 192–199. [Google Scholar] [CrossRef]
- Hull, C.M.; Parker, J.E.; Bader, O.; Weig, M.; Gross, U.; Warrilow, A.; Kelly, D.E.; Kelly, S.L. Facultative Sterol Uptake in an Ergosterol-Deficient Clinical Isolate of Candida glabrata Harboring a Missense Mutation inERG11and Exhibiting Cross-Resistance to Azoles and Amphotericin B. Antimicrob. Agents Chemother. 2012, 56, 4223–4232. [Google Scholar] [CrossRef]
- Chau, A.S.; Gurnani, M.; Hawkinson, R.; Laverdiere, M.; Cacciapuoti, A.; McNicholas, P.M. Inactivation of Sterol Δ5,6-Desaturase Attenuates Virulence in Candida albicans. Antimicrob. Agents Chemother. 2005, 49, 3646–3651. [Google Scholar] [CrossRef]
- Kelly, S.L.; Lamb, D.C.; Kelly, D.E.; Manning, N.J.; Loeffler, J.; Hebart, H.; Schumacher, U.; Einsele, H. Resistance to fluconazole and cross-resistance to amphotericin B in Candida albicans from AIDS patients caused by defective sterol delta5,6-desaturation. FEBS Lett. 1997, 400, 80–82. [Google Scholar] [CrossRef]
- Sanglard, D.; Ischer, F.; Parkinson, T.; Falconer, D.; Bille, J. Candida albicans Mutations in the Ergosterol Biosynthetic Pathway and Resistance to Several Antifungal Agents. Antimicrob. Agents Chemother. 2003, 47, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.I.; Choi, C.W.; Lee, K.M.; Lee, Y.S. Gene Expression and Identification Related to Fluconazole Resistance of Candida glabrata Strains. Osong Public Heal. Res. Perspect. 2010, 1, 36–41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, D.; Jiang, B.; Ketela, T.; Lemieux, S.; Veillette, K.; Martel, N.; Davison, J.; Sillaots, S.; Trosok, S.; Bachewich, C.; et al. Genome-Wide Fitness Test and Mechanism-of-Action Studies of Inhibitory Compounds in Candida albicans. PLoS Pathog. 2007, 3, e92. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.B.; Sirjusingh, C.; Parsons, A.B.; Boone, C.; Wickens, C.; E Cowen, L.; Kohn, L.M. Mode of selection and experimental evolution of antifungal drug resistance in Saccharomyces cerevisiae. Genetics 2003, 163, 1287–1298. [Google Scholar]
- Akins, R. An update on antifungal targets and mechanisms of resistance inCandidaalbicans. Med Mycol. 2005, 43, 285–318. [Google Scholar] [CrossRef]
- Silver, P.M.; Oliver, B.G.; White, T.C. Role of Candida albicans Transcription Factor Upc2p in Drug Resistance and Sterol Metabolism. Eukaryot. Cell 2004, 3, 1391–1397. [Google Scholar] [CrossRef]
- Yang, H.; Tong, J.; Lee, C.W.; Ha, S.; Eom, S.H.; Im, Y.J. Structural mechanism of ergosterol regulation by fungal sterol transcription factor Upc2. Nat. Commun. 2015, 6, 6129. [Google Scholar] [CrossRef]
- Hoot, S.J.; Oliver, B.G.; White, T.C. Candida albicans UPC2 is transcriptionally induced in response to antifungal drugs and anaerobicity through Upc2p-dependent and -independent mechanisms. Microbiology 2008, 154, 2748–2756. [Google Scholar] [CrossRef]
- Davies, B.S.; Wang, H.S.; Rine, J. Dual Activators of the Sterol Biosynthetic Pathway of Saccharomyces cerevisiae: Similar Activation/Regulatory Domains but Different Response Mechanisms. Mol. Cell. Boil. 2005, 25, 7375–7385. [Google Scholar] [CrossRef]
- Hoot, S.J.; Smith, A.R.; Brown, R.P.; White, T.C. An A643V Amino Acid Substitution in Upc2p Contributes to Azole Resistance in Well-Characterized Clinical Isolates ofCandida albicans. Antimicrob. Agents Chemother. 2010, 55, 940–942. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, C.J.; Schneider, S.; Barker, K.S.; Rogers, P.D.; Morschhäuser, J. An A643T Mutation in the Transcription Factor Upc2p Causes Constitutive ERG11 Upregulation and Increased Fluconazole Resistance in Candida albicans. Antimicrob. Agents Chemother. 2009, 54, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Nagi, M.; Nakayama, H.; Tanabe, K.; Bard, M.; Aoyama, T.; Okano, M.; Higashi, S.; Ueno, K.; Chibana, H.; Niimi, M.; et al. Transcription factors CgUPC2A and CgUPC2B regulate ergosterol biosynthetic genes in Candida glabrata. Genes Cells 2010, 16, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Zavrel, M.; Hoot, S.J.; White, T.C. Comparison of Sterol Import under Aerobic and Anaerobic Conditions in Three Fungal Species, Candida albicans, Candida glabrata, and Saccharomyces cerevisiae. Eukaryot. Cell 2013, 12, 725–738. [Google Scholar] [CrossRef]
- Kuo, D.; Tan, K.; Zinman, G.; Ravasi, T.; Bar-Joseph, Z.; Ideker, T. Evolutionary divergence in the fungal response to fluconazole revealed by soft clustering. Genome Boil. 2010, 11, R77. [Google Scholar] [CrossRef]
- Wilcox, L.J.; Balderes, D.A.; Wharton, B.; Tinkelenberg, A.H.; Rao, G.; Sturley, S.L. Transcriptional Profiling Identifies Two Members of the ATP-binding Cassette Transporter Superfamily Required for Sterol Uptake in Yeast. J. Boil. Chem. 2002, 277, 32466–32472. [Google Scholar] [CrossRef]
- Nakayama, H.; Tanabe, K.; Bard, M.; Hodgson, W.; Wu, S.; Takemori, D.; Aoyama, T.; Kumaraswami, N.S.; Metzler, L.; Takano, Y.; et al. The Candida glabrata putative sterol transporter gene CgAUS1 protects cells against azoles in the presence of serum. J. Antimicrob. Chemother. 2007, 60, 1264–1272. [Google Scholar] [CrossRef]
- A Lewis, T.; Taylor, F.R.; Parks, L.W. Involvement of heme biosynthesis in control of sterol uptake by Saccharomyces cerevisiae. J. Bacteriol. 1985, 163, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Crowley, J.H.; Leak, F.W.; Shianna, K.V.; Tove, S.; Parks, L.W. A Mutation in a Purported Regulatory Gene Affects Control of Sterol Uptake in Saccharomyces cerevisiae. J. Bacteriol. 1998, 180, 4177–4183. [Google Scholar] [CrossRef] [PubMed]
- Ness, F.; Achstetter, T.; Duport, C.; Karst, F.; Spagnoli, R.; Degryse, E. Sterol Uptake in Saccharomyces cerevisiae Heme Auxotrophic Mutants is Affected by Ergosterol and Oleate but Not by Palmitoleate or by Sterol Esterification. J. Bacteriol. 1998, 180, 1913–1919. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.B.; Funt, J.M.; Abbey, D.; Issi, L.; Guiducci, C.; A Martinez, D.; DeLorey, T.; Li, B.Y.; White, T.C.; Cuomo, C.A.; et al. The evolution of drug resistance in clinical isolates of Candida albicans. eLife 2015, 4, 00662. [Google Scholar] [CrossRef]
- Selmecki, A.; Forche, A.; Berman, J. Aneuploidy and Isochromosome Formation in Drug-Resistant Candida albicans. Science 2006, 313, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.Z.; Saha, A.; Haseeb, A.; Bennett, R.J. A chromosome 4 trisomy contributes to increased fluconazole resistance in a clinical isolate of Candida albicans. Microbiology 2017, 163, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Perepnikhatka, V.; Fischer, F.J.; Niimi, M.; Baker, R.A.; Cannon, R.D.; Wang, Y.-K.; Sherman, F.; Rustchenko, E. Specific Chromosome Alterations in Fluconazole-Resistant Mutants of Candida albicans. J. Bacteriol. 1999, 181, 4041–4049. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, F.; Li, D.; Zhou, M.; Wang, X.; Xu, Q.; Zhang, Y.; Yan, L.; Jiang, Y. Trisomy of chromosome R confers resistance to triazoles in Candida albicans. Med. Mycol. 2015, 53, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Poláková, S.; Blume, C.; Zárate, J.; Mentel, M.; Jørck-Ramberg, D.; Stenderup, J.; Piškur, J. Formation of new chromosomes as a virulence mechanism in yeastCandida glabrata. Proc. Natl. Acad. Sci. USA 2009, 106, 2688–2693. [Google Scholar] [CrossRef]
- Marichal, P.; Bossche, H.V.; Odds, F.C.; Nobels, G.; Warnock, D.W.; Timmerman, V.; Van Broeckhoven, C.; Fay, S.; Mose-Larsen, P. Molecular biological characterization of an azole-resistant Candida glabrata isolate. Antimicrob. Agents Chemother. 1997, 41, 2229–2237. [Google Scholar] [CrossRef]
- Mansfield, B.E.; Oltean, H.N.; Oliver, B.G.; Hoot, S.J.; Leyde, S.E.; Hedstrom, L.; White, T.C. Azole Drugs Are Imported By Facilitated Diffusion in Candida albicans and Other Pathogenic Fungi. PLoS Pathog. 2010, 6, e1001126. [Google Scholar] [CrossRef]
- Vandeputte, P.; Ferrari, S.; Coste, A.T. Antifungal Resistance and New Strategies to Control Fungal Infections. Int. J. Microbiol. 2011, 2012, 1–26. [Google Scholar] [CrossRef]
- Kahn, J.N.; Garcia-Effron, G.; Hsu, M.-J.; Park, S.; Marr, K.A.; Perlin, D.S. Acquired Echinocandin Resistance in a Candida krusei Isolate Due to Modification of Glucan Synthase. Antimicrob. Agents Chemother. 2007, 51, 1876–1878. [Google Scholar] [CrossRef]
- Walker, L.A.; Gow, N.A.; Munro, C.A. Fungal echinocandin resistance. Fungal Genet. Biol. 2010, 2, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K.; Alastruey-Izquierdo, A.; Healey, K.; Johnson, M.E.; Perlin, D.S.; Edlind, T.D. Fks1 and Fks2 Are Functionally Redundant but Differentially Regulated in Candida glabrata: Implications for Echinocandin Resistance. Antimicrob. Agents Chemother. 2012, 56, 6304–6309. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.K.; Kline, E.G.; Healey, K.; Kordalewska, M.; Perlin, D.S.; Nguyen, M.H.; Clancy, C.J. Spontaneous Mutational Frequency and FKS Mutation Rates Vary by Echinocandin Agent against Candida glabrata. Antimicrob. Agents Chemother. 2018, 63, e01692-18. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-J.; Nelliat, A. A Double-Edged Sword: Aneuploidy is a Prevalent Strategy in Fungal Adaptation. Genes 2019, 10, 787. [Google Scholar] [CrossRef]
- Hope, W.; Tabernero, L.; Denning, D.; Anderson, M.J. Molecular Mechanisms of Primary Resistance to Flucytosine in Candida albicans. Antimicrob. Agents Chemother. 2004, 48, 4377–4386. [Google Scholar] [CrossRef]
- Chapeland-Leclerc, F.; Bouchoux, J.; Goumar, A.; Chastin, C.; Villard, J.; Noël, T. Inactivation of the FCY2 Gene Encoding Purine-Cytosine Permease Promotes Cross-Resistance to Flucytosine and Fluconazole in Candida lusitaniae. Antimicrob. Agents Chemother. 2005, 49, 3101–3108. [Google Scholar] [CrossRef]
- Chapeland-Leclerc, F.; Hennequin, C.; Papon, N.; Girard, A.; Socié, G.; Ribaud, P.; Lacroix, C. Acquisition of Flucytosine, Azole, and Caspofungin Resistance in Candida glabrata Bloodstream Isolates Serially Obtained from a Hematopoietic Stem Cell Transplant Recipient. Antimicrob. Agents Chemother. 2009, 54, 1360–1362. [Google Scholar] [CrossRef]
- Arendrup, M.C.; Patterson, T.F. Multidrug-Resistant Candida: Epidemiology, Molecular Mechanisms, and Treatment. J. Infect. Dis. 2017, 216, S445–S451. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J.; Chang, Y.C. Aneuploidy and Drug Resistance in Pathogenic Fungi. PLoS Pathog. 2012, 8, e1003022. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Efflux Pump | Pump Type | Organism |
|---|---|---|
| Cdr1p, Cdr2p | ABC-T | Candida albicans |
| CgCdr1p | ABC-T | Candida glabrata |
| CgFlr1p | MFS-T | Candida glabrata |
| CgPdh1p | ABC-T | Candida glabrata |
| CgQdr2p | MFS-T | Candida glabrata |
| CgSnq2p | ABC-T | Candida glabrata |
| CkAbc1p | ABC-T | Candida krusei |
| CkAbc2p | ABC-T | Candida krusei |
| Mdr1p | MFS-T | Candida albicans |
| Cdr1p | ABC-T | Candida auris |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharya, S.; Sae-Tia, S.; Fries, B.C. Candidiasis and Mechanisms of Antifungal Resistance. Antibiotics 2020, 9, 312. https://doi.org/10.3390/antibiotics9060312
Bhattacharya S, Sae-Tia S, Fries BC. Candidiasis and Mechanisms of Antifungal Resistance. Antibiotics. 2020; 9(6):312. https://doi.org/10.3390/antibiotics9060312
Chicago/Turabian StyleBhattacharya, Somanon, Sutthichai Sae-Tia, and Bettina C. Fries. 2020. "Candidiasis and Mechanisms of Antifungal Resistance" Antibiotics 9, no. 6: 312. https://doi.org/10.3390/antibiotics9060312
APA StyleBhattacharya, S., Sae-Tia, S., & Fries, B. C. (2020). Candidiasis and Mechanisms of Antifungal Resistance. Antibiotics, 9(6), 312. https://doi.org/10.3390/antibiotics9060312