
Iron Chelation in Murine Models of Systemic Inflammation Induced by Gram-Positive and Gram-Negative Toxins



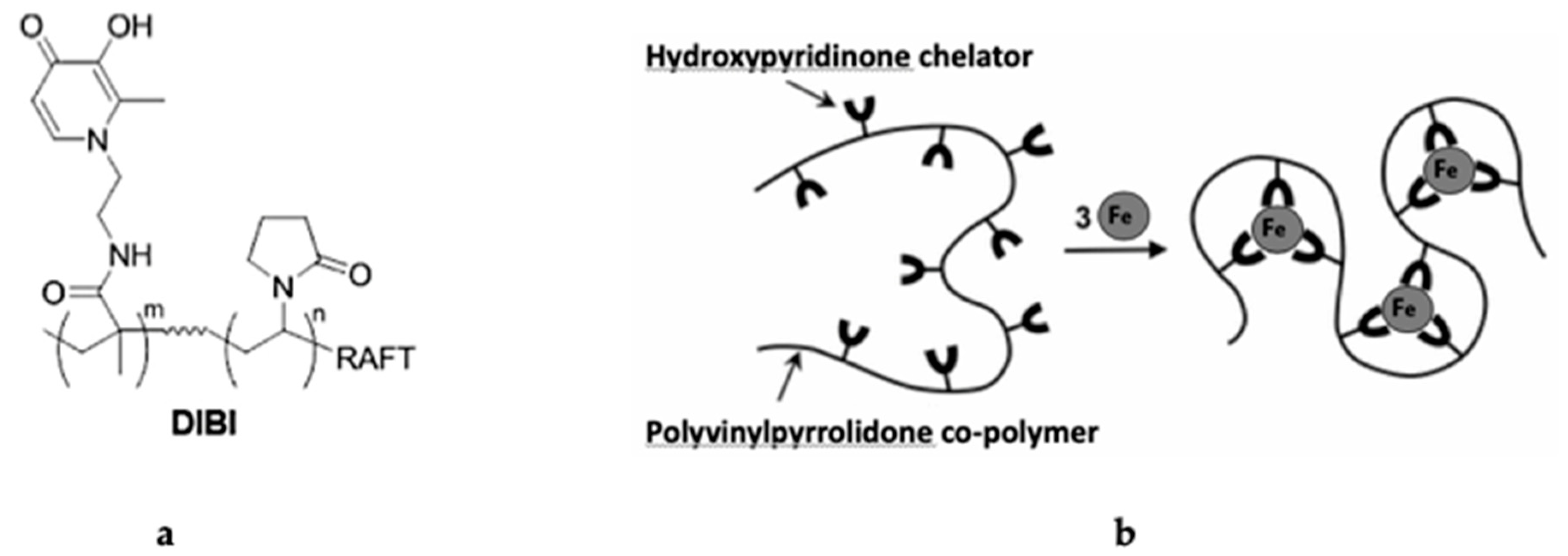{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. IVM
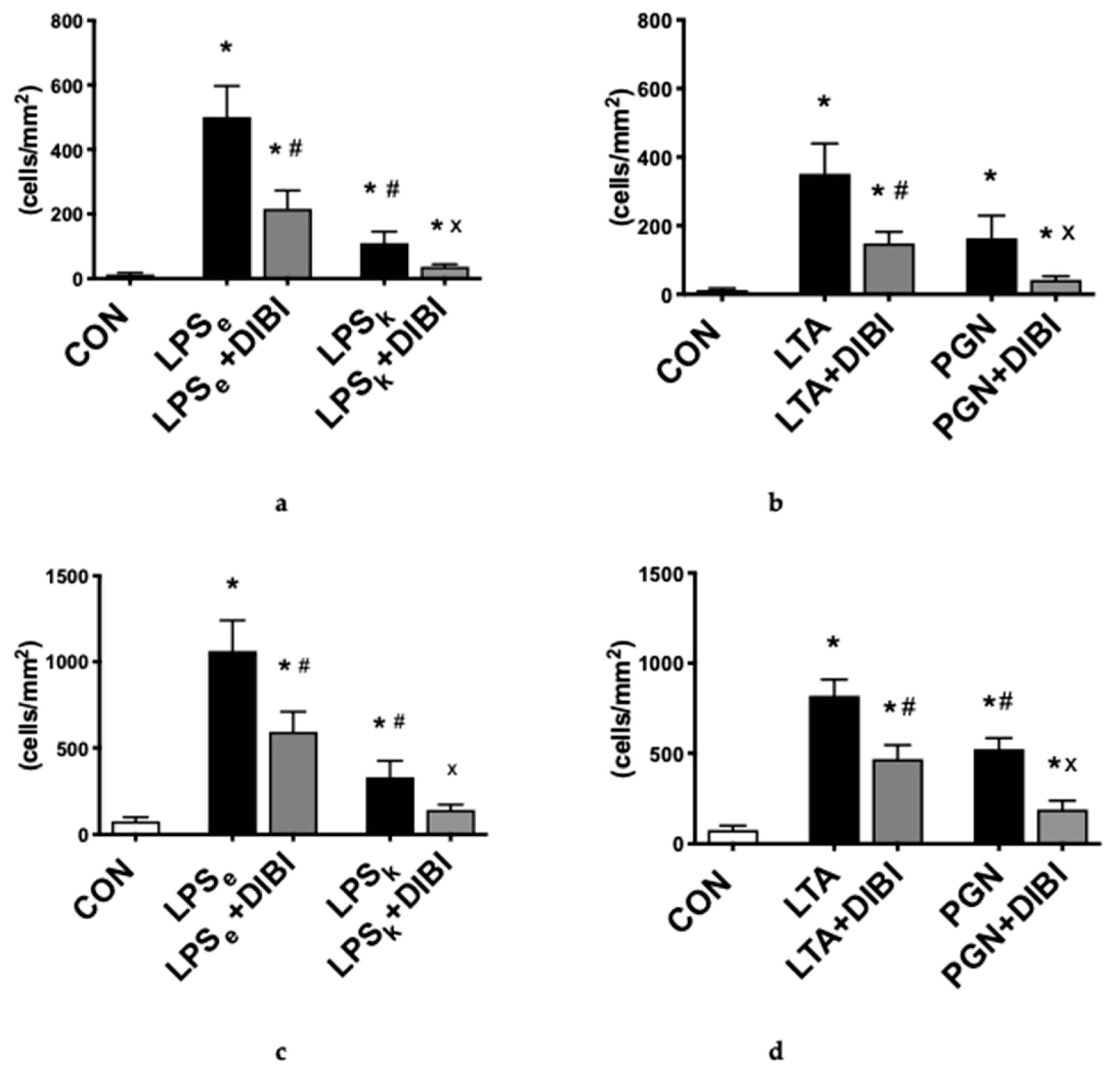
2.1.1. Leukocyte Adhesion
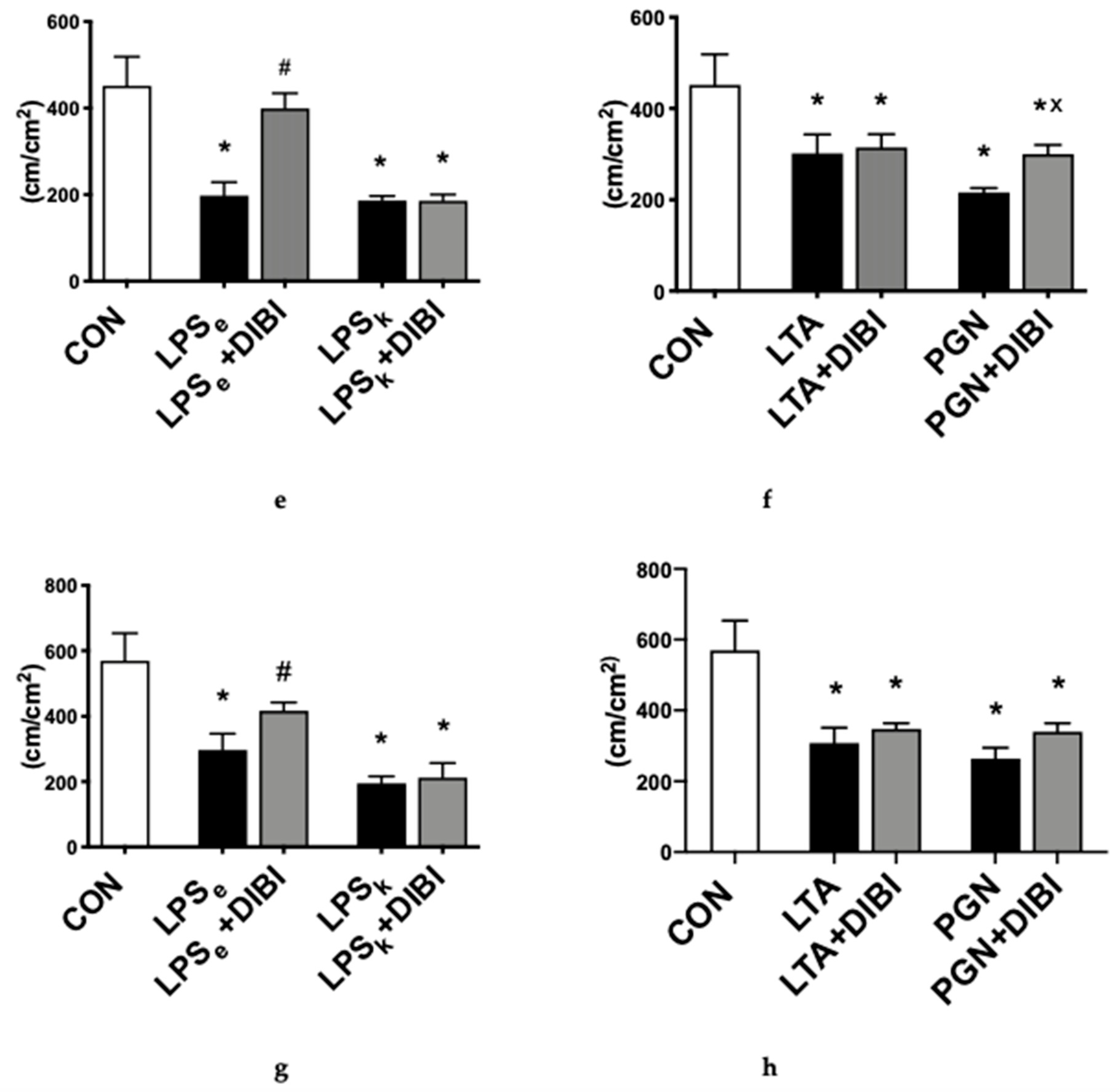
2.1.2. Capillary Perfusion
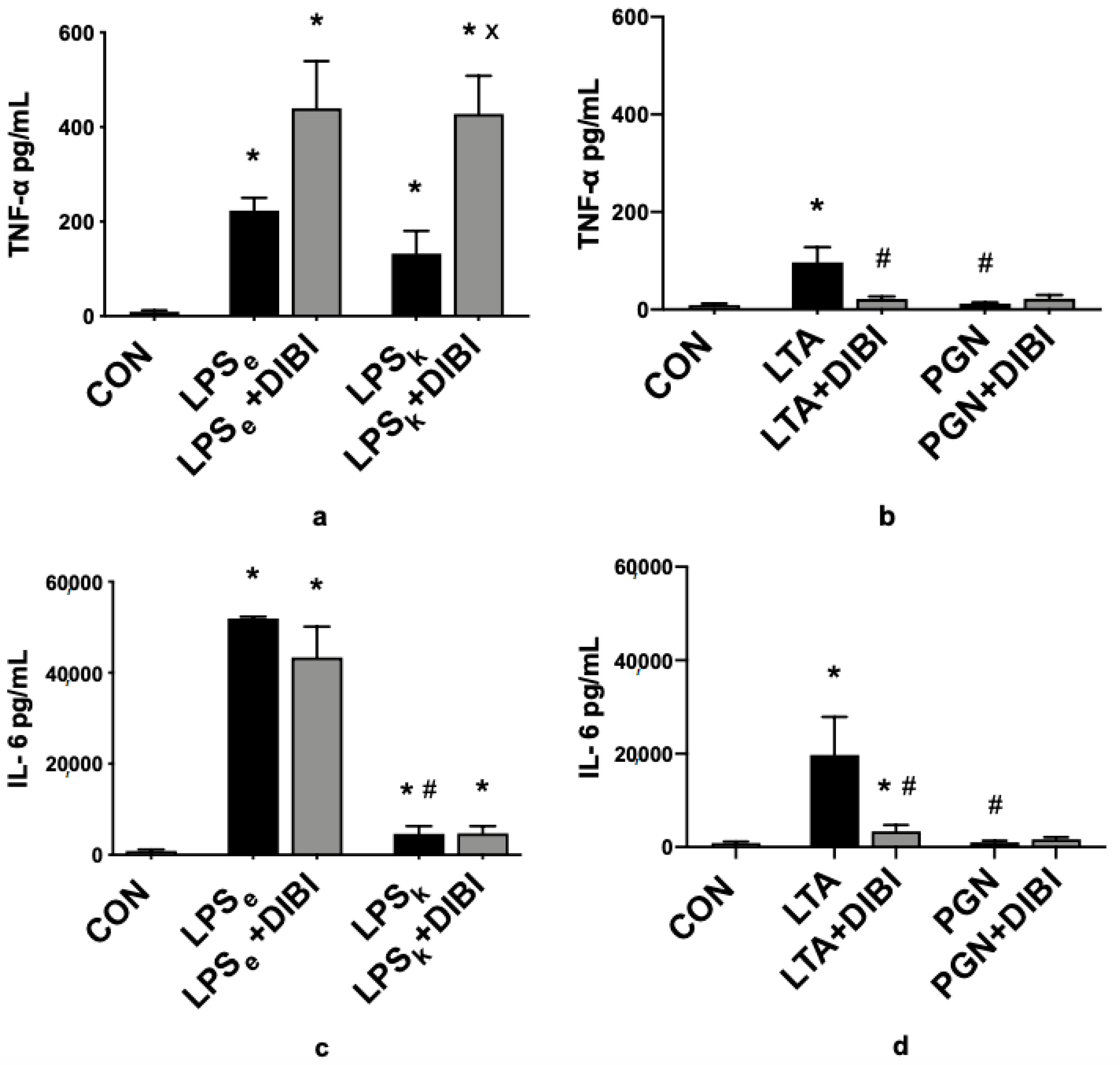
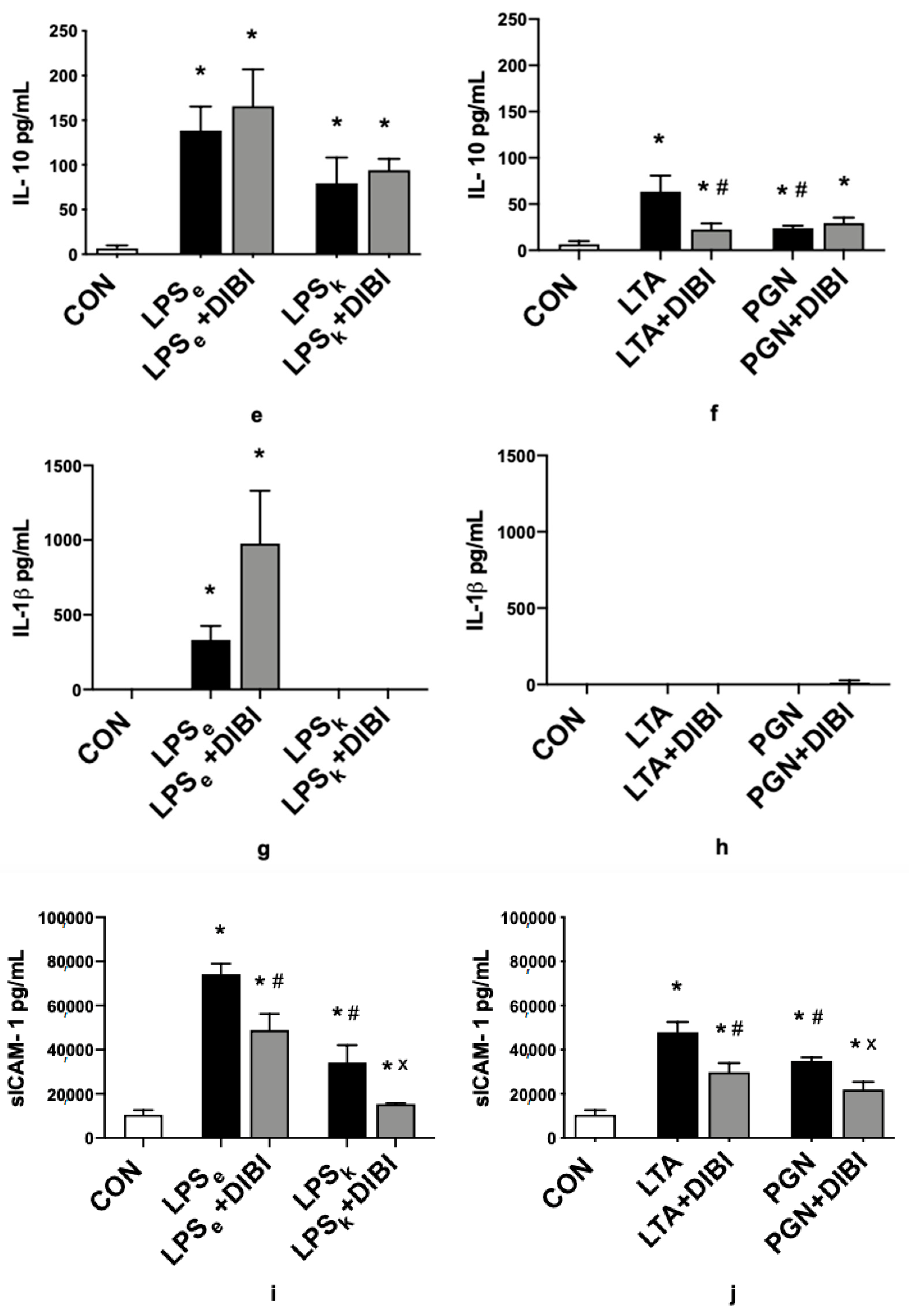
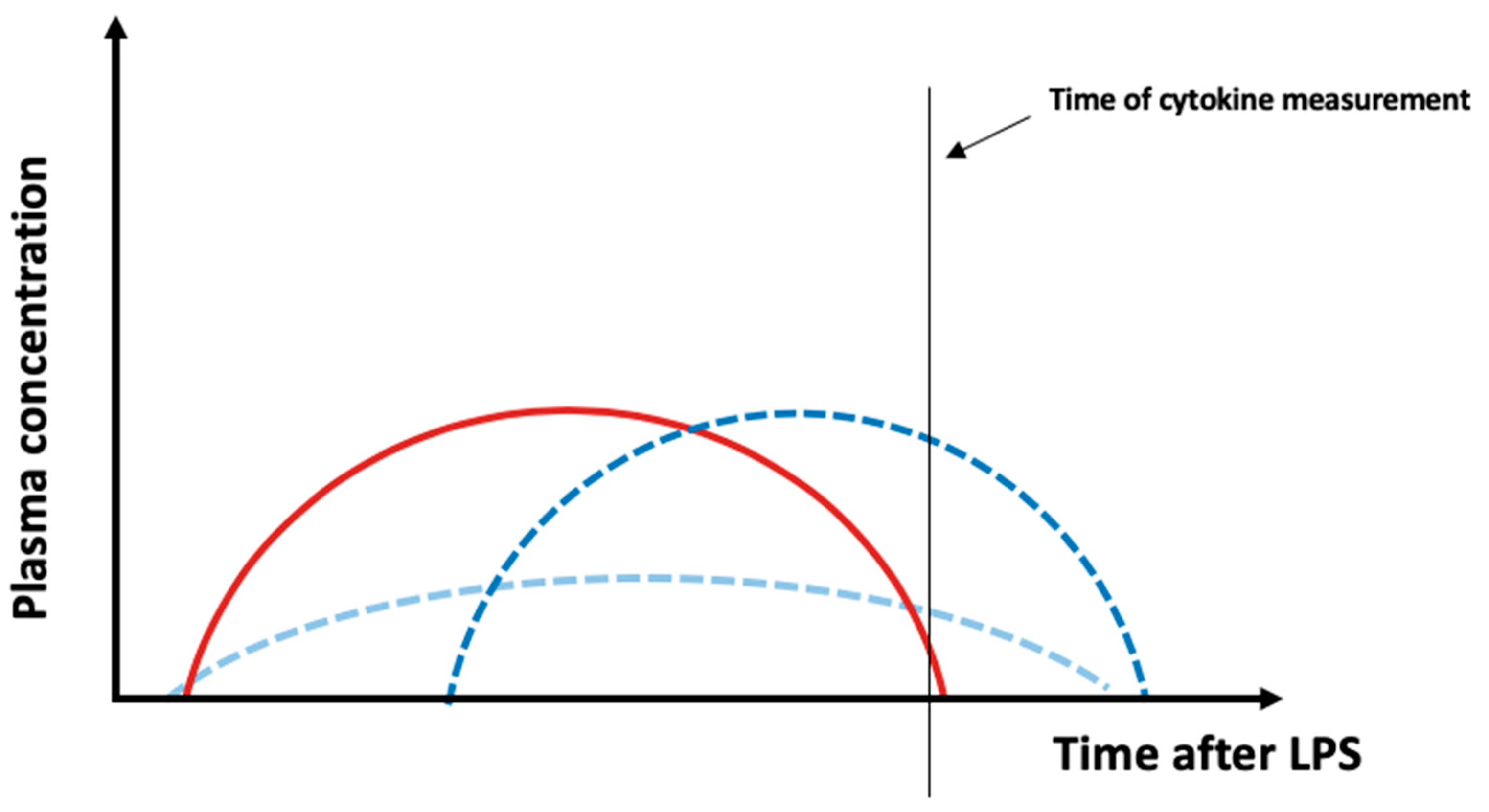
2.2. Cytokines
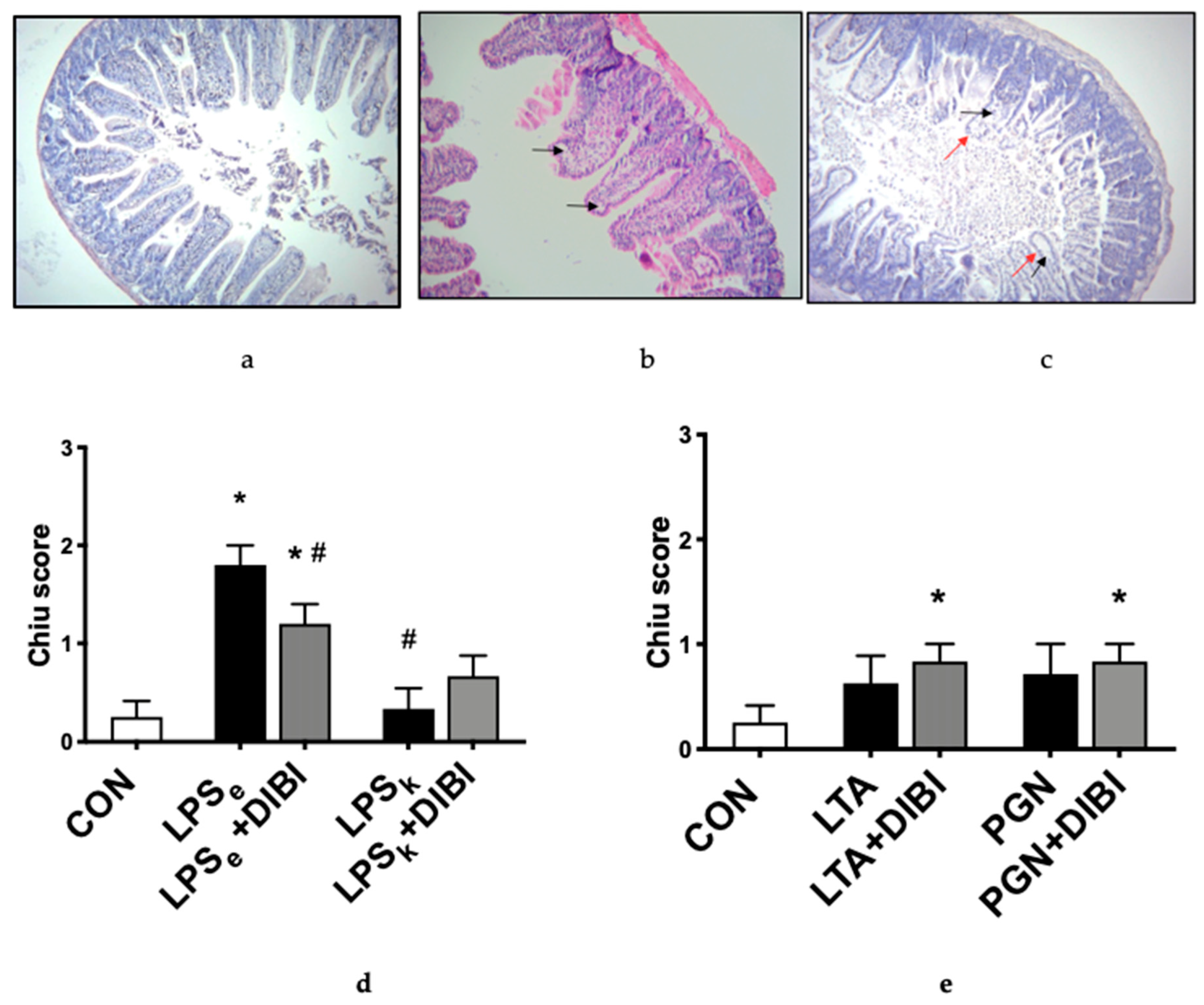
2.3. Histology
3. Discussion
4. Materials and Methods
4.1. Bacterial Toxins
4.2. Animals
4.3. Experimental Groups
4.4. Experimental Model
4.5. Intravital Microscopy
4.6. Plasma Cytokine Measurements
4.7. Histology
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sheftel, A.D.; Mason, A.B.; Ponka, P. The long history of iron in the Universe and in health and disease. Biochim. Biophys. ActaGen. Subj. 2012, 1820, 161–187. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Luhr, R.; Cao, Y.; Söderquist, B.; Cajander, S. Trends in sepsis mortality over time in randomised sepsis trials: A systematic literature review and meta-analysis of mortality in the control arm, 2002–2016. Crit. Care 2019, 23, 241. [Google Scholar] [CrossRef]
- Kwizera, A.; Baelani, I.; Mer, M.; Kissoon, N.; Schultz, M.J.; Patterson, A.J.; Musa, N.; Farmer, J.C.; Dünser, M.W.; Adhikari, N.; et al. The long sepsis journey in low- and middle-income countries begins with a first step but on which road? Crit. Care 2018, 22, 64. [Google Scholar] [CrossRef]
- Gyawali, B.; Ramakrishna, K.; Dhamoon, A.S. Sepsis: The evolution in definition, pathophysiology, and management. Sage Open Med. 2019, 7, 1–13. [Google Scholar] [CrossRef]
- Ramsey, H.; Wu, M.X. Mitochondrial anti-oxidant protects IEX-1 deficient mice from organ damage during endotoxemia. Int. Immunopharmacol. 2014, 23, 658–663. [Google Scholar] [CrossRef]
- Li, G.; Wu, J.; Li, R.; Yuan, D.; Fan, Y.; Yang, J.; Ji, M.; Zhu, S. Protective Effects of Antioxidant Peptide SS-31 Against Multiple Organ Dysfunctions During Endotoxemia. Inflammation 2016, 39, 54–64. [Google Scholar] [CrossRef]
- Lehmann, C.; Islam, S.; Jarosch, S.; Zhou, J.; Hoskin, D.; Greenshields, A. The Utility of Iron Chelators in the Management of Inflammatory Disorders. Mediat. Inflamm. 2015, 2015, 516740. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Mobarra, N.; Shanaki, M.; Ehteram, H.; Nasiri, H.; Sahmani, M.; Saeidi, M. A review on iron chelators in treatment of iron overload syndromes. Int. J. Hematol. Oncol. Stem Cell Res. 2016, 10, 239–247. [Google Scholar]
- Power, C.M.R.; Grant, T.; Greenshields, A.L.; Arsenault, D.J.; Holbein, B.E.; Hoskin, D.W. Inhibitory effect of iron withdrawal by chelation on the growth of human and murine mammary carcinoma and fibrosarcoma cells. Exp. Mol. Pathol. 2015, 99, 262–270. [Google Scholar] [CrossRef]
- Savage, K.A.; Parquet, M.C.; Allan, D.S.; Davidson, R.J.; Holbein, B.E.; Lilly, E.A. Iron restriction to clinical isolates of Candida albicans by the novel chelator DIBI inhibits growth and increases sensitivity to azoles in vitro and in vivo in a murine model of experimental vaginitis. Antimicrob. Agents Chemother. 2018, 62, e02576-17. [Google Scholar] [CrossRef]
- Ang, M.T.C.; Gumbau-Brisa, R.; Allan, D.S.; McDonald, R.; Ferguson, M.J.; Holbein, B.E.; Bierenstiel, M. DIBI, a 3-hydroxypyridin-4-one chelator iron-binding polymer with enhanced antimicrobial activity. Medchemcomm 2018, 9, 1206–1212. [Google Scholar] [CrossRef]
- Li, L.; Frei, B. Iron chelation inhibits NF-κB-mediated adhesion molecule expression by inhibiting p22phox protein expression and NADPH oxidase activity. Arter. Thromb. Vasc. Biol. 2006, 26, 2638–2643. [Google Scholar] [CrossRef]
- Van, E.L.T.; Heemskerk, S.; Van, D.P.R.W.; Van, W.S.M.; Peters, W.H.M.; Van, D.H.J.G.; Kox, M.; Swinkels, D.W.; Pickkers, P. The effect of iron loading and iron chelation on the innate immune response and subclinical organ injury during human endotoxemia: A randomized trial. Haematologica 2014, 99, 579–587. [Google Scholar]
- Vulcano, M.; Meiss, R.P.; Isturiz, M.A. Deferoxamine reduces tissue injury and lethality in LPS-treated mice. Int. J. Immunopharmacol. 2000, 22, 635–644. [Google Scholar] [CrossRef]
- Messaris, E.; Antonakis, P.T.; Memos, N.; Chatzigianni, E.; Leandros, E.; Konstadoulakis, M.M. Deferoxamine administration in septic animals: Improved survival and altered apoptotic gene expression. Int. Immunopharmacol. 2004, 4, 455–459. [Google Scholar] [CrossRef]
- Thorburn, V.T.; Aali, M.; Kostek, L.; LeTourneau-Paci, C.; Colp, P.; Zhou, J.; Holbein, B.E.; Hoskin, D.; Lehmann, C. Anti-inflammatory effects of a novel iron chelator, DIBI, in experimental sepsis. Clin. Hemorheol. Microcirc. 2017, 67, 241–250. [Google Scholar] [CrossRef]
- Arora, N.; Caldwell, A.; Wafa, K.; Szczesniak, A.; Caldwell, M.; Al-Banna, N.; Sharawy, N.; Islam, S.; Zhou, J.; Holbein, B.E.; et al. DIBI, a polymeric hydroxypyridinone iron chelator, reduces ocular inflammation in local and systemic endotoxin-induced uveitis. Clin. Hemorheol. Microcirc. 2018, 69, 153–164. [Google Scholar] [CrossRef]
- Dickson, K.; Lehmann, C. Inflammatory response to different toxins in experimental sepsis models. Int. J. Mol. Sci. 2019, 20, 4341. [Google Scholar] [CrossRef]
- Chiu, C.J.; McArdle, A.H.; Brown, R.; Scott, H.J.; Gurd, F.N. Intestinal Mucosal Lesion in Low-Flow States: I. A Morphological, Hemodynamic, and Metabolic Reappraisal. Arch. Surg. 1970, 101, 478–483. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Q.; Wang, C.; Liu, X.; Li, N.; Li, J. Disruption of tight junctions during polymicrobial sepsis in vivo. J. Pathol. 2009, 218, 210–221. [Google Scholar] [CrossRef]
- Gatt, M. The Role of the Gut in Sepsis. Surgery 2015, 33, 534–541. [Google Scholar] [CrossRef]
- Fay, K.T.; Ford, M.L.; Coopersmith, C.M. The intestinal microenvironment in sepsis. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 2574–2583. [Google Scholar] [CrossRef]
- Schnoor, M.; Alcaide, P.; Voisin, M.B.; Van, B.J.D. Crossing the Vascular Wall: Common and Unique Mechanisms Exploited by Different Leukocyte Subsets during Extravasation. Mediat. Inflamm. 2015, 2015, 946509. [Google Scholar] [CrossRef]
- Triantafilou, K.; Triantafilou, M.; Dedrick, R.L. Interactions of bacterial lipopolysaccharide and peptidoglycan with a 70 kDa and an 80 kDa protein on the cell surface of CD14+ and CD14- cells. Hum. Immunol. 2001, 62, 50–63. [Google Scholar] [CrossRef]
- Finney, S.J.; Leaver, S.K.; Evans, T.W.; Burke-Gaffney, A. Differences in lipopolysaccharide- and lipoteichoic acid-induced cytokine/chemokine expression. Intensive Care Med. 2012, 38, 324–332. [Google Scholar] [CrossRef]
- Yipp, B.G.; Andonegui, G.; Howlett, C.J.; Robbins, S.M.; Hartung, T.; Ho, M.; Kubes, P. Profound Differences in Leukocyte-Endothelial Cell Responses to Lipopolysaccharide Versus Lipoteichoic Acid. J. Immunol. 2002, 168, 4650–4658. [Google Scholar] [CrossRef]
- Finney, S.J.; Anning, P.B.; Cao, T.V.; Perretti, M.; Evans, T.W.; Burke-Gaffney, A. Butanol-extracted lipoteichoic acid induces in vivo leukocyte adhesion. Biochem. Biophys. Res. Commun. 2007, 364, 831–837. [Google Scholar] [CrossRef]
- Leistner, R.; Bloch, A.; Gastmeier, P.; Schwab, F. E. coli bacteremia in comparison to K. pneumoniae bacteremia: Influence of pathogen species and ESBL production on 7-day mortality. Antimicrob. Resist. Infect. Control 2016, 5, 5–8. [Google Scholar] [CrossRef]
- De Backer, D.; Orbegozo, C.D.; Donadello, K.; Vincent, J.L. Pathophysiology of microcirculatory dysfunction and the pathogenesis of septic shock. Virulence 2014, 5, 73–79. [Google Scholar] [CrossRef]
- De Backer, D.; Donadello, K.; Taccone, F.S.; Ospina-Tascon, G.; Salgado, D.; Vincent, J.L. Microcirculatory alterations: Potential mechanisms and implications for therapy. Ann. Intensive Care 2011, 1, 27. [Google Scholar] [CrossRef]
- Kvietys, P.R. Intramural blood and lymphatic vessels. In the Gastrointestinal Circulation; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. Available online: https://www.ncbi.nlm.nih.gov/books/NBK53099/ (accessed on 11 December 2019).
- Thorburn, T.V. Iron-Related Immune Cell Function in Sepsis. Ph.D. Thesis, Dalhousie University, Halifax, NS, Canada, July 2018. [Google Scholar]
- Massey, M.J.; Hou, P.C.; Filbin, M.; Wang, H.; Ngo, L.; Huang, D.T.; Aird, W.C.; Novack, V.; Trzeciak, S.; Yealy, D.M.; et al. Microcirculatory perfusion disturbances in septic shock: Results from the ProCESS trial. Crit. Care 2018, 22, 308. [Google Scholar] [CrossRef]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; Mcguire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of cytokines as a double-edged sword in sepsis. Vivo 2013, 27, 669–684. [Google Scholar]
- Dinarello, C.A. Proinflammatory cytokines. Chest 2000, 118, 503–508. [Google Scholar] [CrossRef]
- Mera, S.; Tatulescu, D.; Cismaru, C.; Bondor, C.; Slavcovici, A.; Zanc, V.; Carstina, D.; Oltean, M. Multiplex cytokine profiling in patients with sepsis. Apmis 2011, 119, 155–163. [Google Scholar] [CrossRef]
- Harrison, C. Sepsis: Calming the cytokine storm. Nat. Rev. Drug Discov. 2010, 9, 360–361. [Google Scholar] [CrossRef]
- Wang, J.E.; Dahle, M.K.; Yndestad, A.; Bauer, I.; McDonald, M.C.; Aukrust, P.; Foster, S.J.; Bauer, M.; Aasen, A.O.; Thiemermann, C. Peptidoglycan of Staphylococcus aureus causes inflammation and organ injury in the rat. Crit. Care Med. 2004, 32, 546–552. [Google Scholar] [CrossRef]
- Gillrie, M.R.; Zbytnuik, L.; McAvoy, E.; Kapadia, R.; Lee, K.; Waterhouse, C.C.M.; Davis, S.P.; Muruve, D.A.; Kubes, P.; Ho, M. Divergent roles of Toll-like receptor 2 in response to lipoteichoic acid and Staphylococcus aureus in vivo. Eur. J. Immunol. 2010, 40, 1639–1650. [Google Scholar] [CrossRef]
- Kim, H.Y.; Han, N.R.; Kim, H.M.; Jeong, H.J. The Iron Chelator and Anticancer Agent Dp44mT Relieves Allergic Inflammation in Mice with Allergic Rhinitis. Inflammation 2018, 4, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Cermanova, J.; Kadova, Z.; Dolezelova, E.; Zagorova, M.; Safka, V.; Hroch, M.; Laho, T.; Holeckova, M.; Mokry, J.; Kovarikova, P.; et al. Deferoxamine but not Dexrazoxane Alleviates Liver Injury Induced by Endotoxemia in Rats. Shock 2014, 42, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, C.; Pan, S.; Miao, Q.; Xue, J.; Xun, J.; Zhang, Y.; Gao, Y.; Duan, X.; Fan, Y. Deferoxamine attenuates lipopolysaccharide-induced inflammatory responses and protects against endotoxic shock in mice. Biochem. Biophys. Res. Commun. 2015, 465, 305–311. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, H.; Li, G.; Wang, D.; Zhou, Q.; Wu, J.; Zheng, J.; Huang, J.; Slade, D.A.; Wu, X.; et al. STING-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine 2019, 41, 497–508. [Google Scholar] [CrossRef]
- Islam, S.; Jarosch, S.; Zhou, J.; Parquet, M.D.C.; Toguri, J.T.; Colp, P.; Holbein, B.E.; Lehmann, C. Anti-inflammatory and anti-bacterial effects of iron chelation in experimental sepsis. J. Surg. Res. 2015, 200, 266–273. [Google Scholar] [CrossRef]
- Perlman, R.L. Mouse Models of Human Disease: An Evolutionary Perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef]
- Lehmann, C.; Kianian, M.; Zhou, J.; Kuster, I.; Kuschnereit, R.; Whynot, S.; Hung, O.; Shukla, R.; Johnston, B.; Cerny, V.; et al. Cannabinoid receptor 2 activation reduces intestinal leukocyte recruitment and systemic inflammatory mediator release in acute experimental sepsis. Crit. Care 2012, 16, 47. [Google Scholar] [CrossRef]
- Lehmann, C.; Götz, F.; Schuster, L.; Zhou, J. Improved setup for intestinal intravital microscopy in mice—The “floating table”. Minerva Anestesiol. 2013, 79, 102–103. [Google Scholar]
- Pavlovic, D.; Frieling, H.; Lauer, K.S.; Bac, V.H.; Richter, J.; Wendt, M.; Lehmann, C.; Usichenko, T.; Meissner, K.; Gruendling, M. Thermostatic tissue platform for intravital microscopy: “the hanging drop” model. J. Microsc. 2006, 224, 203–210. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fokam, D.; Dickson, K.; Kamali, K.; Holbein, B.; Colp, P.; Stueck, A.; Zhou, J.; Lehmann, C. Iron Chelation in Murine Models of Systemic Inflammation Induced by Gram-Positive and Gram-Negative Toxins. Antibiotics 2020, 9, 283. https://doi.org/10.3390/antibiotics9060283
Fokam D, Dickson K, Kamali K, Holbein B, Colp P, Stueck A, Zhou J, Lehmann C. Iron Chelation in Murine Models of Systemic Inflammation Induced by Gram-Positive and Gram-Negative Toxins. Antibiotics. 2020; 9(6):283. https://doi.org/10.3390/antibiotics9060283
Chicago/Turabian StyleFokam, Danielle, Kayle Dickson, Kiyana Kamali, Bruce Holbein, Patricia Colp, Ashley Stueck, Juan Zhou, and Christian Lehmann. 2020. "Iron Chelation in Murine Models of Systemic Inflammation Induced by Gram-Positive and Gram-Negative Toxins" Antibiotics 9, no. 6: 283. https://doi.org/10.3390/antibiotics9060283
APA StyleFokam, D., Dickson, K., Kamali, K., Holbein, B., Colp, P., Stueck, A., Zhou, J., & Lehmann, C. (2020). Iron Chelation in Murine Models of Systemic Inflammation Induced by Gram-Positive and Gram-Negative Toxins. Antibiotics, 9(6), 283. https://doi.org/10.3390/antibiotics9060283