Critical Parameters for the Development of Novel Therapies for Severe and Resistant Infections—A Case Study on CAL02, a Non-Traditional Broad-Spectrum Anti-Virulence Drug

Abstract
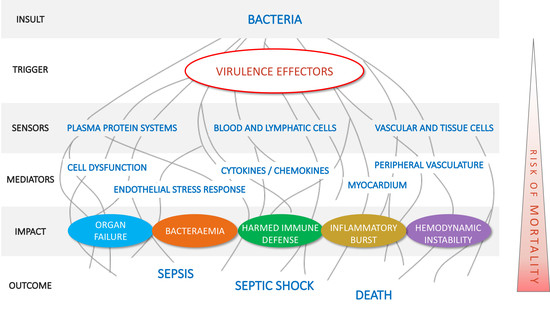
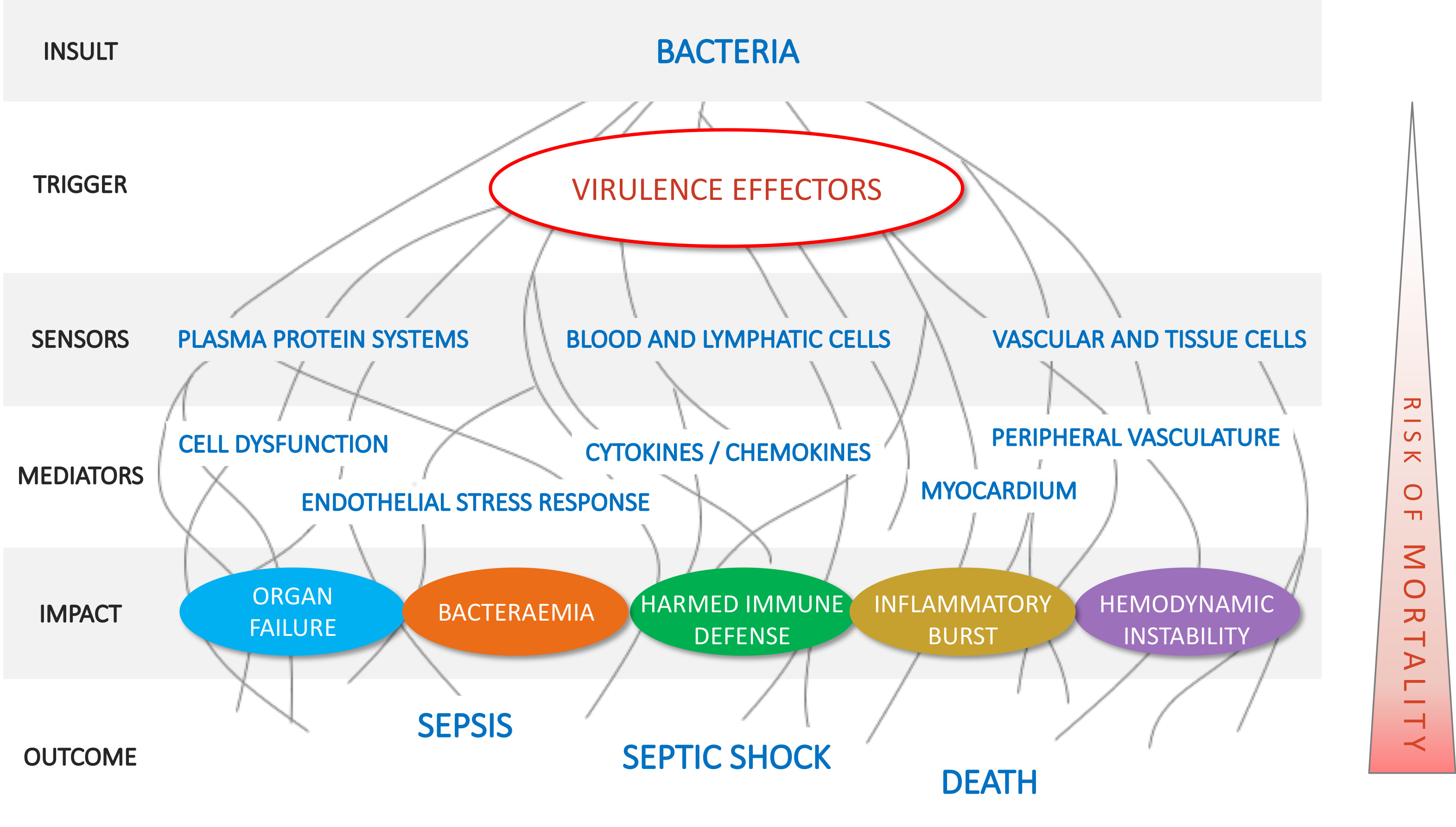
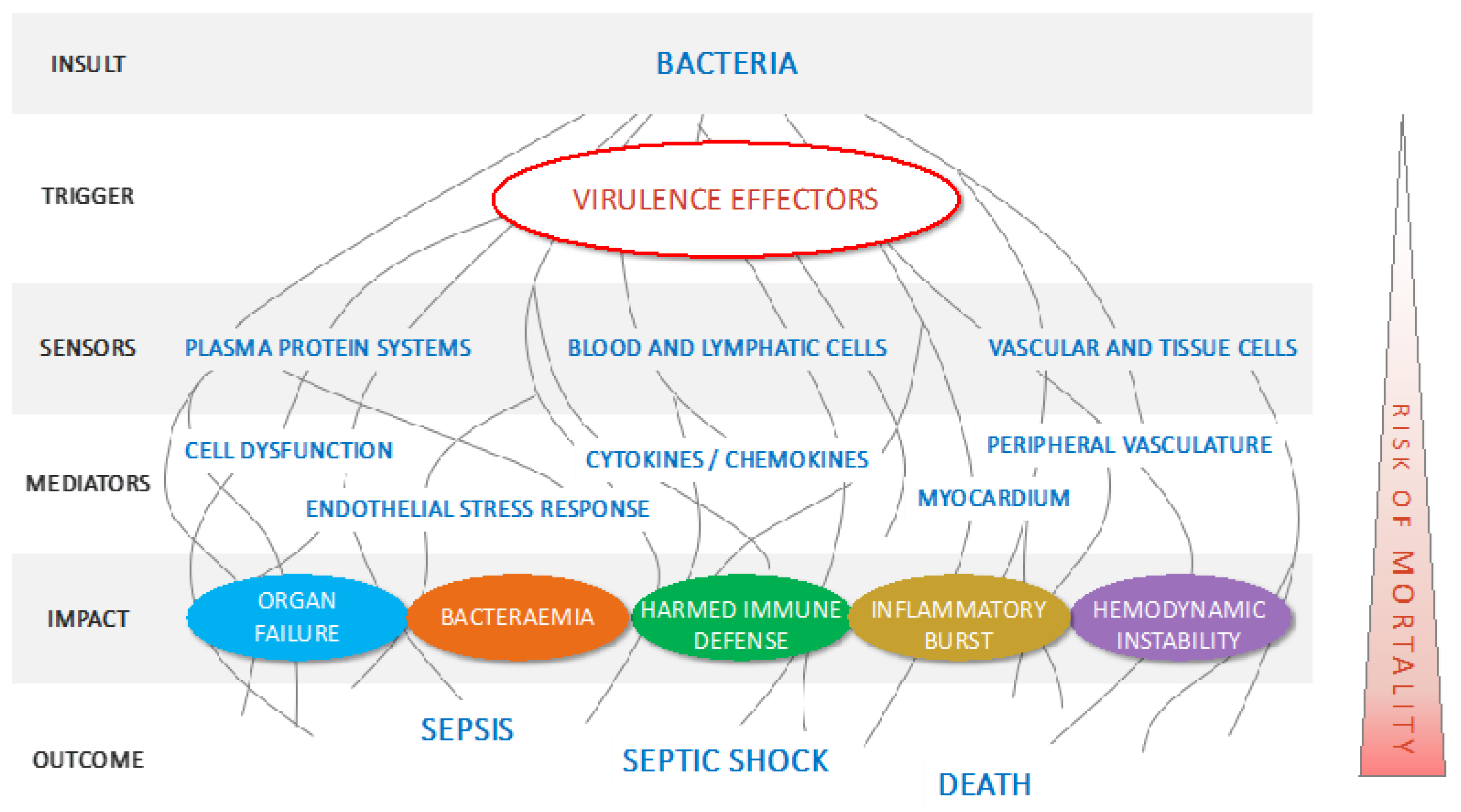
1. Introduction
2. Developing a Novel Anti-Virulence Drug: The Foundations
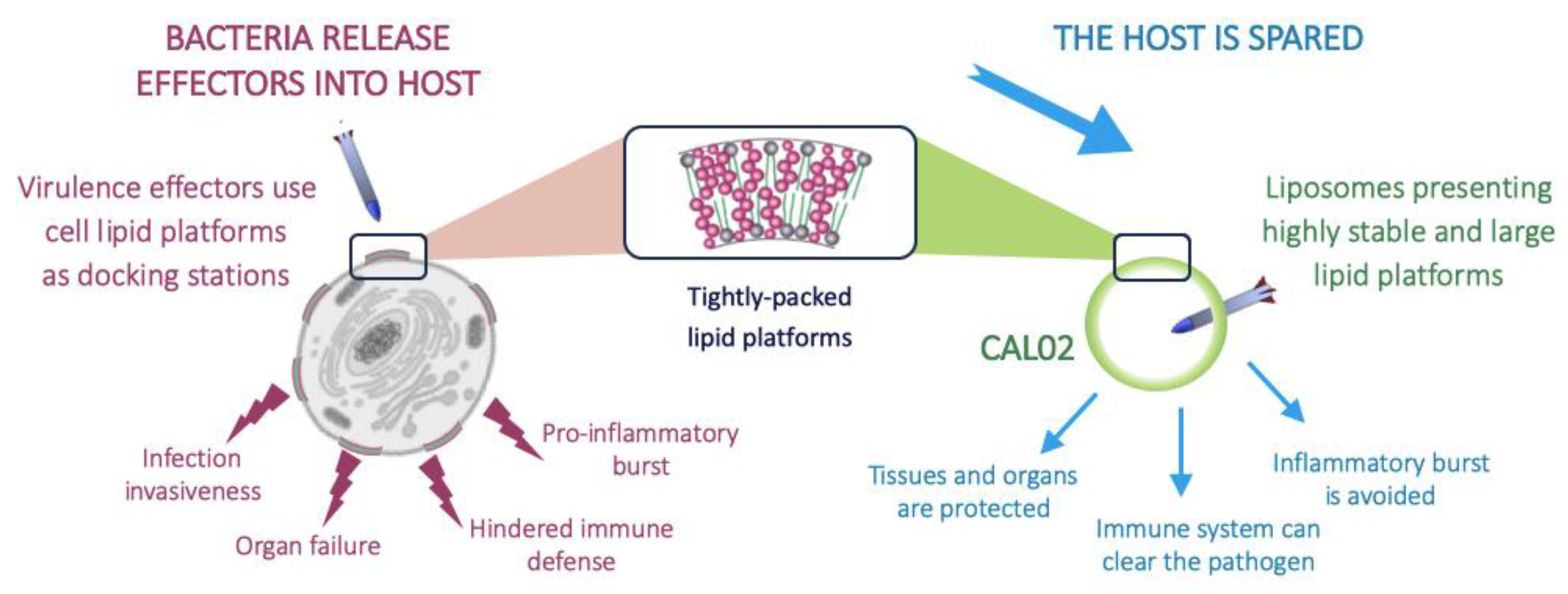
2.1. Pillar 1: Target Pathogenic Triggers
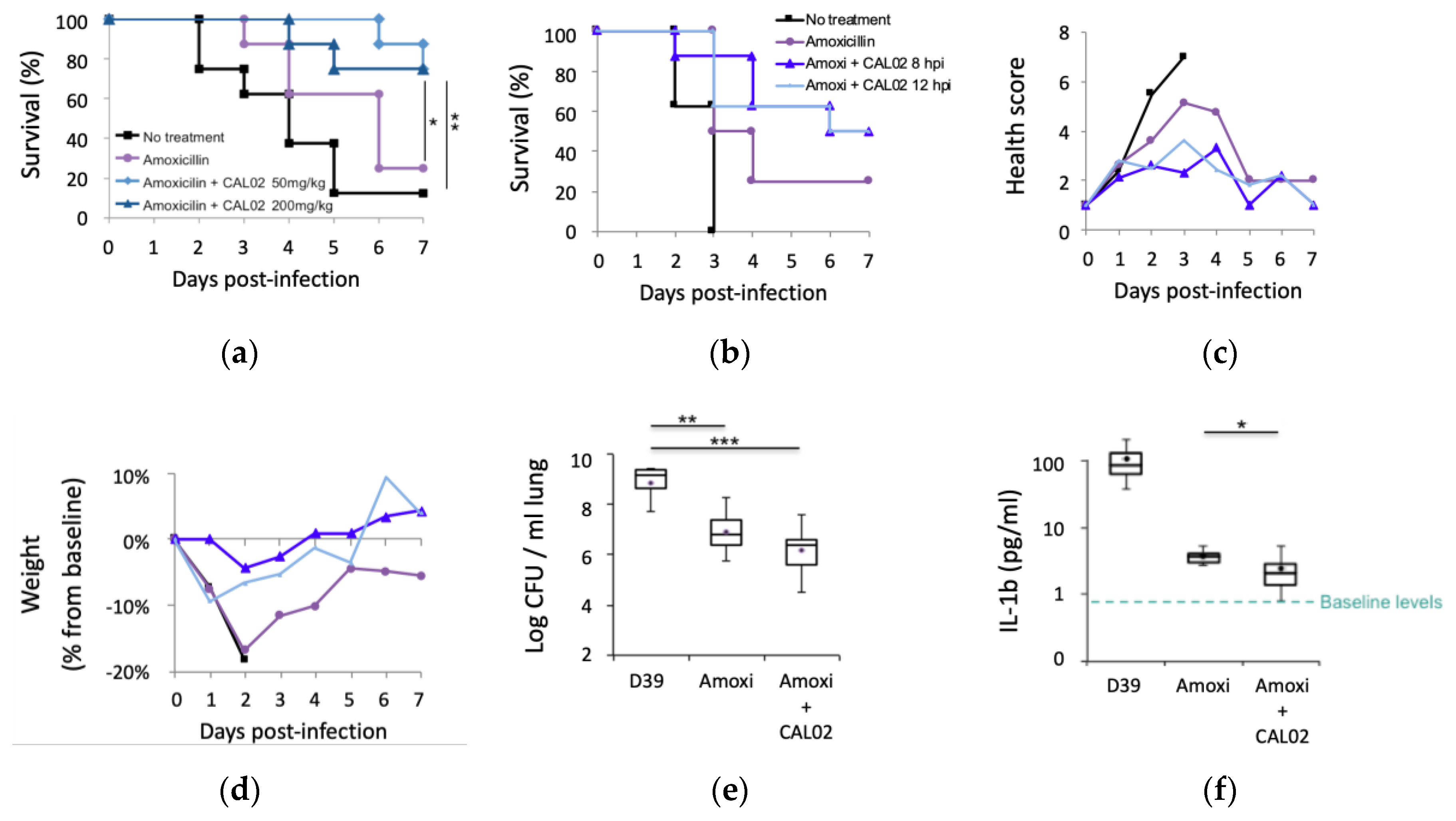
2.2. Pillar 2: Timely Intervention
2.3. Pillar 3: Target for Comprehensive and Hard Efficacy Endpoints
2.4. Pillar 4: Aiming at Economic Sustainability
3. Conclusions and Implications of Key Findings
Author Contributions
Funding
Conflicts of Interest
References
- CDC Antibiotic Resistance Threats in the United States. 2019. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 19 February 2020).
- Los, F.C.O.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of Pore-Forming Toxins in Bacterial Infectious Diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef] [PubMed]
- Rello, J.; Parisella, F.R.; Perez, A. Alternatives to antibiotics in an era of difficult-to-treat resistance: New insights. Expert Rev. Clin. Pharmacol. 2019, 12, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Friedman, N.D.; Temkin, E.; Carmeli, Y. The negative impact of antibiotic resistance. Clin. Microbiol. Infect. 2016, 22, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Zaas, D.W.; Duncan, M.; Rae Wright, J.; Abraham, S.N. The role of lipid rafts in the pathogenesis of bacterial infections. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2005, 1746, 305–313. [Google Scholar] [CrossRef]
- Henry, B.D.; Neill, D.R.; Becker, K.A.; Gore, S.; Bricio-Moreno, L.; Ziobro, R.; Edwards, M.J.; Mühlemann, K.; Steinmann, J.; Kleuser, B.; et al. Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat. Biotechnol. 2015, 33, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.J.C. Pore-forming toxins. Cell. Mol. Life Sci. CMLS 2002, 59, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Tam, K.; Torres, V.J. Staphylococcus aureus Secreted Toxins and Extracellular Enzymes. Microbiol. Spectr. 2019, 7, 640–668. [Google Scholar] [CrossRef]
- Wolfmeier, H.; Mansour, S.C.; Liu, L.T.; Pletzer, D.; Draeger, A.; Babiychuk, E.B.; Hancock, R.E.W. Liposomal Therapy Attenuates Dermonecrosis Induced by Community-Associated Methicillin-Resistant Staphylococcus aureus by Targeting α-Type Phenol-Soluble Modulins and α-Hemolysin. EBioMedicine 2018, 33, 211–217. [Google Scholar] [CrossRef]
- Azeredo da Silveira, S.; Lajaunias, F.; Perez, A. CAL02 anti-toxin agent against Pseudomonas aeruginosa: First demonstrations of efficacy. In Proceedings of the 27th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Vienna, Austria, 22–25 April 2017; Available online: https://www.escmid.org/escmid_publications/escmid_elibrary/material/?mid=50933 (accessed on 19 February 2020).
- Oliveira, D.; Borges, A.; Simões, M. Staphylococcus aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins 2018, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- DiGiandomenico, A.; Keller, A.E.; Gao, C.; Rainey, G.J.; Warrener, P.; Camara, M.M.; Bonnell, J.; Fleming, R.; Bezabeh, B.; Dimasi, N.; et al. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci. Transl. Med. 2014, 6, 262ra155. [Google Scholar] [CrossRef]
- Hua, L.; Hilliard, J.J.; Shi, Y.; Tkaczyk, C.; Cheng, L.I.; Yu, X.; Datta, V.; Ren, S.; Feng, H.; Zinsou, R.; et al. Assessment of an anti-alpha-toxin monoclonal antibody for prevention and treatment of Staphylococcus aureus-induced pneumonia. Antimicrob. Agents Chemother. 2014, 58, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Peetermans, W.E.; Viegi, G.; Blasi, F. Risk factors for community-acquired pneumonia in adults in Europe: A literature review. Thorax 2013, 68, 1057–1065. [Google Scholar] [CrossRef]
- GBD 2016 Lower Respiratory Infections Collaborators Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1191–1210. [CrossRef]
- Torres, A. Community-acquired pneumonia: Changing paradigms about mortality. Community Acquir. Infect. 2014, 1, 1. [Google Scholar] [CrossRef]
- Rello, J.; Perez, A. Precision medicine for the treatment of severe pneumonia in intensive care. Expert Rev. Respir. Med. 2016, 10, 297–316. [Google Scholar] [CrossRef]
- Ibn Saied, W.; Mourvillier, B.; Cohen, Y.; Ruckly, S.; Reignier, J.; Marcotte, G.; Siami, S.; Bouadma, L.; Darmon, M.; de Montmollin, E.; et al. A Comparison of the Mortality Risk Associated With Ventilator-Acquired Bacterial Pneumonia and Nonventilator ICU-Acquired Bacterial Pneumonia. Crit. Care Med. 2019, 47, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Kalil, A.C.; Metersky, M.L.; Klompas, M.; Muscedere, J.; Sweeney, D.A.; Palmer, L.B.; Napolitano, L.M.; O’Grady, N.P.; Bartlett, J.G.; Carratalà, J.; et al. Management of Adults with Hospital-acquired and Ventilator-associated Pneumonia: 2016 Clinical Practice Guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin. Infect. Dis. 2016, 63, e61–e111. [Google Scholar] [CrossRef]
- Welte, T. Managing CAP patients at risk of clinical failure. Respir. Med. 2015, 109, 157–169. [Google Scholar] [CrossRef]
- Woodhead, M.; Blasi, F.; Ewig, S.; Garau, J.; Huchon, G.; Ieven, M.; Ortqvist, A.; Schaberg, T.; Torres, A.; van der Heijden, G.; et al. Guidelines for the management of adult lower respiratory tract infections—Full version. Clin. Microbiol. Infect. 2011, 17, E1–E59. [Google Scholar] [CrossRef]
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Crit. Care Med. 2017, 45, 486–552. [Google Scholar] [CrossRef] [PubMed]
- Mouton, J.W.; Brown, D.F.J.; Apfalter, P.; Cantón, R.; Giske, C.G.; Ivanova, M.; MacGowan, A.P.; Rodloff, A.; Soussy, C.-J.; Steinbakk, M.; et al. The role of pharmacokinetics/pharmacodynamics in setting clinical MIC breakpoints: The EUCAST approach. Clin. Microbiol. Infect. 2012, 18, E37–E45. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, D.; Aebi, S.; Grandgirard, D.; Leib, S.L.; Draeger, A.; Babiychuk, E.; Hathaway, L.J. Clinical Streptococcus pneumoniae isolates induce differing CXCL8 responses from human nasopharyngeal epithelial cells which are reduced by liposomes. BMC Microbiol. 2016, 16, 154–160. [Google Scholar] [CrossRef]
- Lajaunias, F.; Azeredo da Silveira, S.; Draeger, A.; Babiychuk, E. CAL02 neutralizes bacterial toxins to combat severe pneumonia. In Proceedings of the 25th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Copenhagen, Denmark, 25–28 April 2015; Available online: https://www.escmid.org/escmid_publications/escmid_elibrary/material/?mid=22277 (accessed on 19 February 2020).
- Azeredo da Silveira, S. Development of CAL02, Liposomes Engineered to Neutralize a Broad Spectrum of Virulent Effectors. In Proceedings of the 2019 ASM/ASCMID Conference on Drug Development to Meet the Challenge of Antimicrobial Resistance, Boston, MA, USA, 3–6 September 2019; Available online: https://www.dropbox.com/sh/hq9il9wj19rk16k/AABJB8ElxuKdOS0ebhPLg4ywa/Azeredo%20da%20Silveira%20Lajaunias%2C%20Samareh%20The%20Road%20from%20Pre-Clinical%20Fri%20AM.pdf?dl=0 (accessed on 19 February 2020).
- Jones, R.N. Microbial etiologies of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia. Clin. Infect. Dis. 2010, 51 (Suppl. 1), S81–S87. [Google Scholar] [CrossRef]
- Kieninger, A.N.; Lipsett, P.A. Hospital-acquired pneumonia: Pathophysiology, diagnosis, and treatment. Surg. Clin. North Am. 2009, 89, 439–461. [Google Scholar] [CrossRef]
- NICE Pneumonia (Hospital-Acquired): Antimicrobial Prescribing. 2019. Available online: https://www.nice.org.uk/guidance/ng139 (accessed on 19 February 2020).
- Koulenti, D.; Tsigou, E.; Rello, J. Nosocomial pneumonia in 27 ICUs in Europe: Perspectives from the EU-VAP/CAP study. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Sabrià, M.; Sopena, N. HAP in nonventilated patients. In Nosocomial and Ventilator-Associated Pneumonia; European Respiratory Monograph; European Respiratory Society: Sheffield, UK, 2011; Volume 53, pp. 138–150. [Google Scholar] [CrossRef]
- Welte, T.; Dellinger, R.P.; Ebelt, H.; Ferrer, M.; Opal, S.M.; Singer, M.; Vincent, J.-L.; Werdan, K.; Martin-Loeches, I.; Almirall, J.; et al. Efficacy and safety of trimodulin, a novel polyclonal antibody preparation, in patients with severe community-acquired pneumonia: A randomized, placebo-controlled, double-blind, multicenter, phase II trial (CIGMA study). Intensive Care Med. 2018, 44, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Liapikou, A.; Cillóniz, C.; Torres, A. Ceftobiprole for the treatment of pneumonia: A European perspective. Drug Des. Dev. Ther. 2015, 9, 4565–4572. [Google Scholar] [CrossRef]
- Torres, A.; Chalmers, J.D.; Dela Cruz, C.S.; Dominedò, C.; Kollef, M.; Martin-Loeches, I.; Niederman, M.; Wunderink, R.G. Challenges in severe community-acquired pneumonia: A point-of-view review. Intensive Care Med. 2019, 45, 159–171. [Google Scholar] [CrossRef]
- Laterre, P.-F.; Colin, G.; Dequin, P.-F.; Dugernier, T.; Boulain, T.; Azeredo da Silveira, S.; Lajaunias, F.; Perez, A.; François, B. CAL02, a novel antitoxin liposomal agent, in severe pneumococcal pneumonia: A first-in-human, double-blind, placebo-controlled, randomised trial. Lancet Infect. Dis. 2019, 19, 620–630. [Google Scholar] [CrossRef]
- Von Seth, M.; Hillered, L.; Otterbeck, A.; Hanslin, K.; Larsson, A.; Sjölin, J.; Lipcsey, M.; Cove, M.; Chew, N.S.; Vu, L.H.; et al. 37th International Symposium on Intensive Care and Emergency Medicine (part 3 of 3). Crit. Care 2017, 21, 39–40. [Google Scholar] [CrossRef]
- Pletz, M.W.; Bauer, M.; Brakhage, A.A. One step closer to precision medicine for infectious diseases. Lancet Infect. Dis. 2019, 19, 564–565. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L.; Moreno, R.; Takala, J.; Willatts, S.; De Mendonça, A.; Bruining, H.; Reinhart, C.K.; Suter, P.M.; Thijs, L.G. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996, 22, 707–710. [Google Scholar] [CrossRef]
- EMA. Addendum to the guideline on the evaluation of medicinal products indicated for treatment of Bacterial Infections. EMA/CHMP/351889/2013. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/addendum-guideline-evaluation-medicinal-products-indicated-treatment-bacterial-infections_en.pdf (accessed on 19 February 2020).
- FDA. Antibacterial Therapies for Patients with an Unmet Medical Need for the Treatment of Serious Bacterial Diseases-Guidance for Industry. 2017. Available online: https://www.fda.gov/files/drugs/published/Antibacterial-Therapies-for-Patients-With-an-Unmet-Medical-Need-for-the-Treatment-of-Serious-Bacterial-Diseases.pdf (accessed on 19 February 2020).
- Blasi, F.; Mantero, M.; Santus, P.; Tarsia, P. Understanding the burden of pneumococcal disease in adults. Clin. Microbiol. Infect. 2012, 18, 7–14. [Google Scholar] [CrossRef] [PubMed]
- File, T.M.; Marrie, T.J. Burden of Community-Acquired Pneumonia in North American Adults. Postgrad. Med. 2010, 122, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Jencks, S.F.; Williams, M.V.; Coleman, E.A. Rehospitalizations among Patients in the Medicare Fee-for-Service Program. New Engl. J. Med. 2009, 360, 1418–1428. [Google Scholar] [CrossRef]
- Welte, T.; Torres, A.; Nathwani, D. Clinical and economic burden of community-acquired pneumonia among adults in Europe. Thorax 2012, 67, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Study National Clinical Trial (NCT) Identifier Phase—Status | Indication Study Drug | Primary Efficacy Endpoint(s) |
|---|---|---|
| Antibiotics | ||
| Comparison of Two Antibiotic Regimens in the Treatment of Severe Sepsis and Septic Shock (MaxSep) NCT00534287Phase 3—Completed (2010) | Severe sepsis/Septic shock Meropenem vs. Meropenem plus Moxifloxacin | Mean total SOFA score (study duration or up to Day 14) |
| Clinical Outcome Study of High-Dose Meropenem in Sepsis and Septic Shock PatientsNCT03344627 N/A—Completed (2018) | Sepsis/Septic shock Meropenem | Change of total SOFA score from Baseline to Day 4 |
| Immuno-Modulator Approaches | ||
| Cx611-0204 SEPCELL Study (SEPCELL) NCT03158727 Phase 1/2—ONGOING | Severe CABP Cx611 (allogeneic adipose-derived stem cells) | Composite: Reduction of the duration of mechanical ventilation and/or vasopressors needed and/or improved survival, and/or clinical cure of the CABP, and other infection-related endpoints |
| Esomeprazole to Reduce Organ Failure in Sepsis (PPI-SEPSIS) NCT03452865 Phase 3—Not Yet Recruiting | Sepsis/Septic shock Esomeprazole | SOFA score reduction (Days 1–28) |
| Efficacy, Safety and Tolerability of Nangibotide in Patients with Septic Shock (ASTONISH) NCT04055909 Phase 2—ONGOING | Septic shock Nangibotide (formerly LR12, TREM-1 inhibitor) | Change of total SOFA score from baseline to Day 3 (in the subgroup defined by patients with elevated sTREM-1 baseline levels and in the overall population) |
| Approaches Targeting Hemodynamic Instability and Shock | ||
| Selepressin Evaluation Programme for Sepsis-Induced Shock—Adaptive Clinical Trial (SEPSIS-ACT) NCT02508649Phase 2/3—Completed (2018) | Septic shock Selepressin | Vasopressor- and mechanical ventilator-free days: Defined as number of days from start of treatment to 30 days thereafter during which the patient is 1) alive; 2) free of treatment with vasopressors; 3) free of any mechanical ventilation |
| Rapid Administration of Carnitine in sEpsis (RACE) NCT01665092 Phase 2—Completed (2019) | Septic shock Levo-Carnitine | Delta SOFA Score (48 h) |
| Treatment of Patients with Early Septic Shock and Bio- ADM Concentration > 70 pg/mL With ADRECIZUMAB (AdrenOSS-2) NCT03085758 Phase 2—ONGOING | Septic shock & ADM > 70 pg/mL ADRECIZUMAB (monoclonal antibody targeting adrenomedullin) | SSI within 14 day follow-up defined as follows: Each day on vasopressor, and/or mechanical ventilation, and/or renal failure (defined as renal SOFA = 4), or not alive, is counted 1; the sum over the follow up period is defined as SSI. Among secondary outcomes: SOFA score and its changes over time (composite) |
| Remote Ischemic Conditioning in Septic Shock (RECO-Sepsis) NCT03201575 N/A—ONGOING | Septic shock Remote ischemic conditioning (inflations and deflations of a brachial cuff) | Average SOFA score (96 h) |
| Efficacy and Safety of Rheosorbilact® Solution for Infusion, in a Complex Therapy of Pneumonia NCT03824457 Phase 4—ONGOING | CAP with PSI/PORT index score ≥ IV and SOFA ≥ 2 points and < 48 h since beginning of antibacterial therapy Rheosorbilact® | A change in the total SOFA score (while at ICU) vs. baseline score upon admission |
| Efficacy and Safety of Rheosorbilact® Solution for Infusion, in a Complex Therapy of Sepsis NCT03764085 Phase 4—Completed (2020) | Sepsis Rheosorbilact® | A change in the total SOFA score (while at ICU) vs. baseline score upon admission |
| Ilomedin in Septic Shock with Persistent Microperfusion Defects (I-MICRO) (I-MICRO) NCT03788837 Phase 3—Not yet recruiting | Septic Shock Hyperdynamic Ilomedin (prostaglandin analog) | Delta SOFA score between infusion onset and Day 7 and patients deceased before Day 7 will be attributed a maximum SOFA score. |
| Guided Fluid-Balance Optimization with Mini-Fluid Challenge During Septic Shock (GOAL) NCT03461900 N/A—Not Yet Recruiting | Septic shock Minifluid challenge | Delta SOFA score (between Day 0 and 5) |
| Hemoadsorbers | ||
| Adsorbtion of Cytokines Early in Septic Shock: The ACESS Study NCT02288975 Medical device—Completed (2017) | Septic shock CytoSorb 300 mL device (3804606CE01) | Cytokine response AND Organ dysfunctions (incl. SOFA) In the first 48 h of septic shock |
| A Double-Blind, Randomized Placebo-Controlled Clinical Investigation With Alteco® LPS Adsorber (ASSET) NCT02335723 Medical device—Completed (2017) | Septic shock Alteco® LPS Adsorber | Relative change from baseline in SOFA score (6–28 days) |
| Hemoadsorption for Prevention of Vasodilatory Shock in Cardiac Surgery Patients with Infective Endocarditis (REMOVE) NCT03266302 Medical device—ONGOING | Infective Endocarditis Hemoadsorber for removal of cytokines | Mean SOFA score (between 24 h before until day 9 post-surgery) |
| Use of Extracorporeal Treatment with the Cytosorb-Adsorber for the Reduction of SIRS in Heart Surgery Patients (CASHSP) NCT02265419 Medical device—ONGOING | Heart surgery with SIRS criterions and postoperative central venous oxygen saturation >75% and need of vasopressors within 6 h postoperative Extracorporeal treatment with the Cytosorb adsorber | Mean SOFA score (to Day 7) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azeredo da Silveira, S.; Shorr, A.F. Critical Parameters for the Development of Novel Therapies for Severe and Resistant Infections—A Case Study on CAL02, a Non-Traditional Broad-Spectrum Anti-Virulence Drug. Antibiotics 2020, 9, 94. https://doi.org/10.3390/antibiotics9020094
Azeredo da Silveira S, Shorr AF. Critical Parameters for the Development of Novel Therapies for Severe and Resistant Infections—A Case Study on CAL02, a Non-Traditional Broad-Spectrum Anti-Virulence Drug. Antibiotics. 2020; 9(2):94. https://doi.org/10.3390/antibiotics9020094
Chicago/Turabian StyleAzeredo da Silveira, Samareh, and Andrew F. Shorr. 2020. "Critical Parameters for the Development of Novel Therapies for Severe and Resistant Infections—A Case Study on CAL02, a Non-Traditional Broad-Spectrum Anti-Virulence Drug" Antibiotics 9, no. 2: 94. https://doi.org/10.3390/antibiotics9020094
APA StyleAzeredo da Silveira, S., & Shorr, A. F. (2020). Critical Parameters for the Development of Novel Therapies for Severe and Resistant Infections—A Case Study on CAL02, a Non-Traditional Broad-Spectrum Anti-Virulence Drug. Antibiotics, 9(2), 94. https://doi.org/10.3390/antibiotics9020094