Molecular Characterization of the Burkholderia cenocepacia dcw Operon and FtsZ Interactors as New Targets for Novel Antimicrobial Design

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Characterization of the dcw Operon in B. cenocepacia J2315
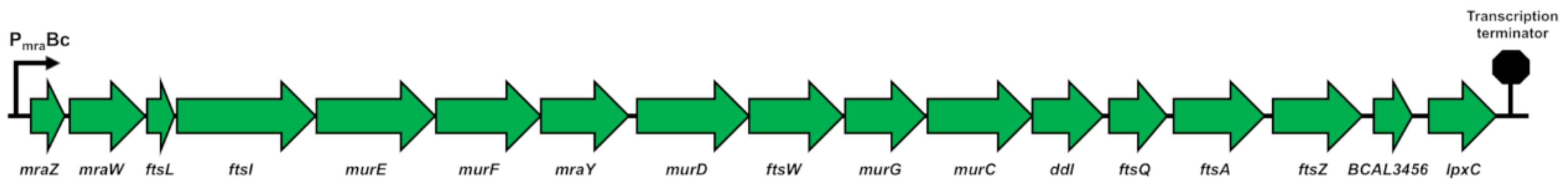
2.1.1. The dcw Cluster of B. cenocepacia J2315 Has the Conserved Gene Organization Shared by Rod-Shaped Bacteria
2.1.2. The dcw Operon is Transcribed as a Polycistronic mRNA from mraZ to ftsZ
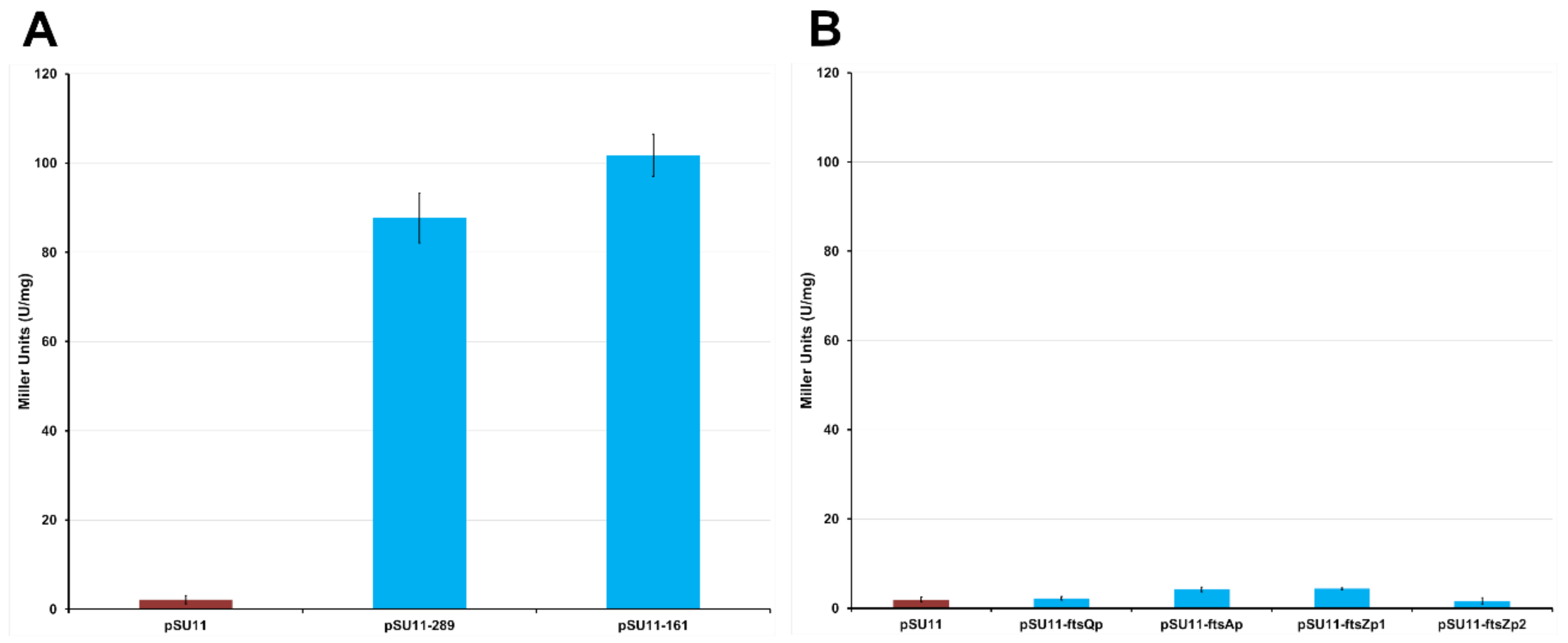
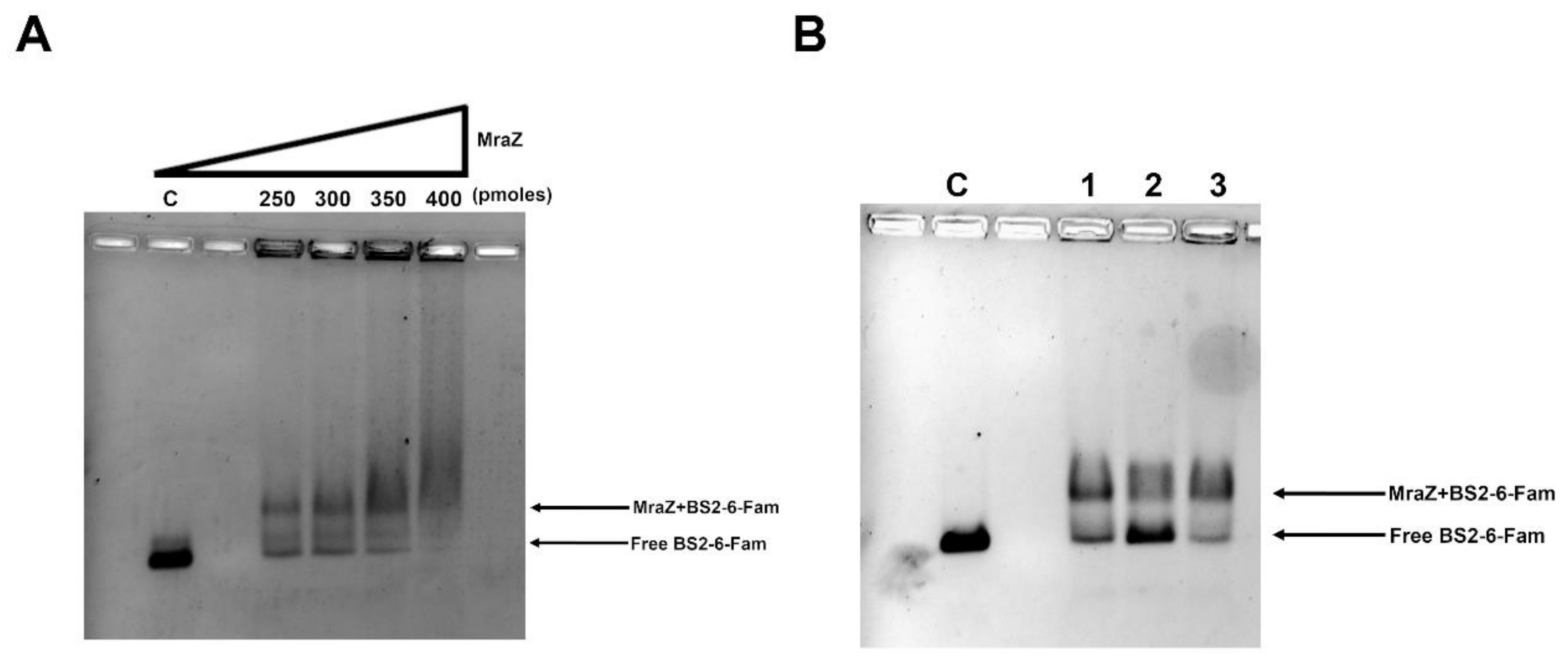
2.1.3. The Protein Mraz Is the Transcriptional Regulator of the dcw Operon
2.2. Study of the FtsZ Interactors in B. cenocepacia In Vivo and In Vitro
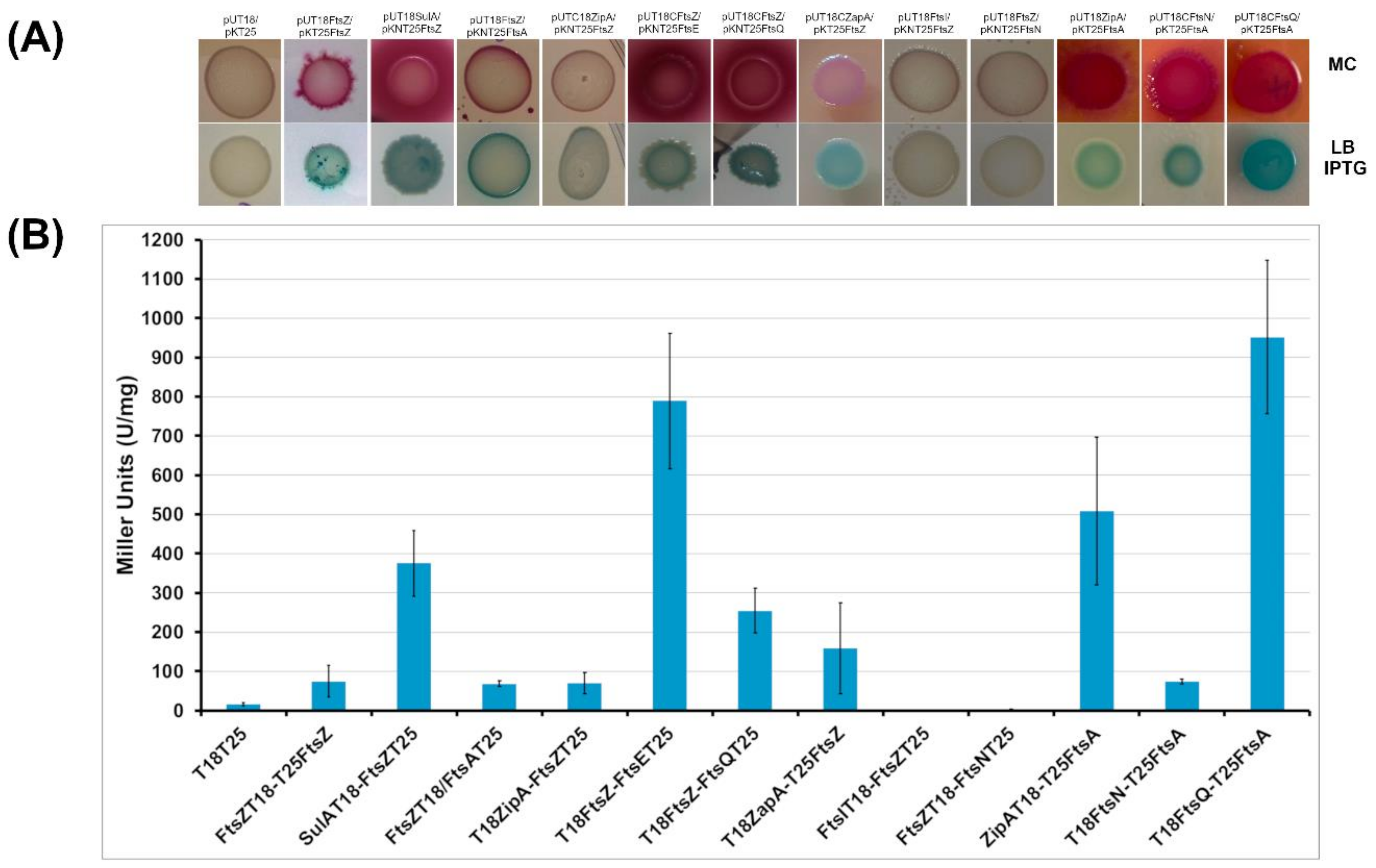
2.2.1. BACTH Analysis of B. cenocepacia Cell Division Proteins
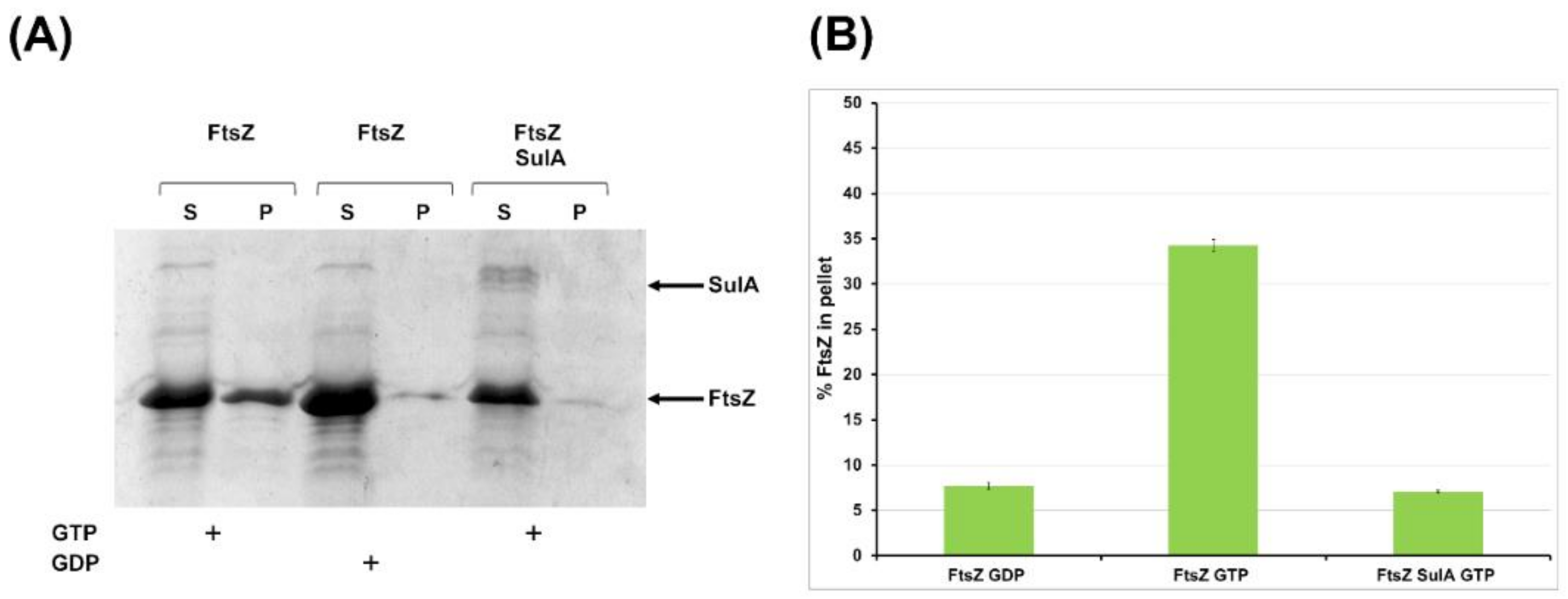
2.2.2. SulA Blocks FtsZ Polymerization In Vitro
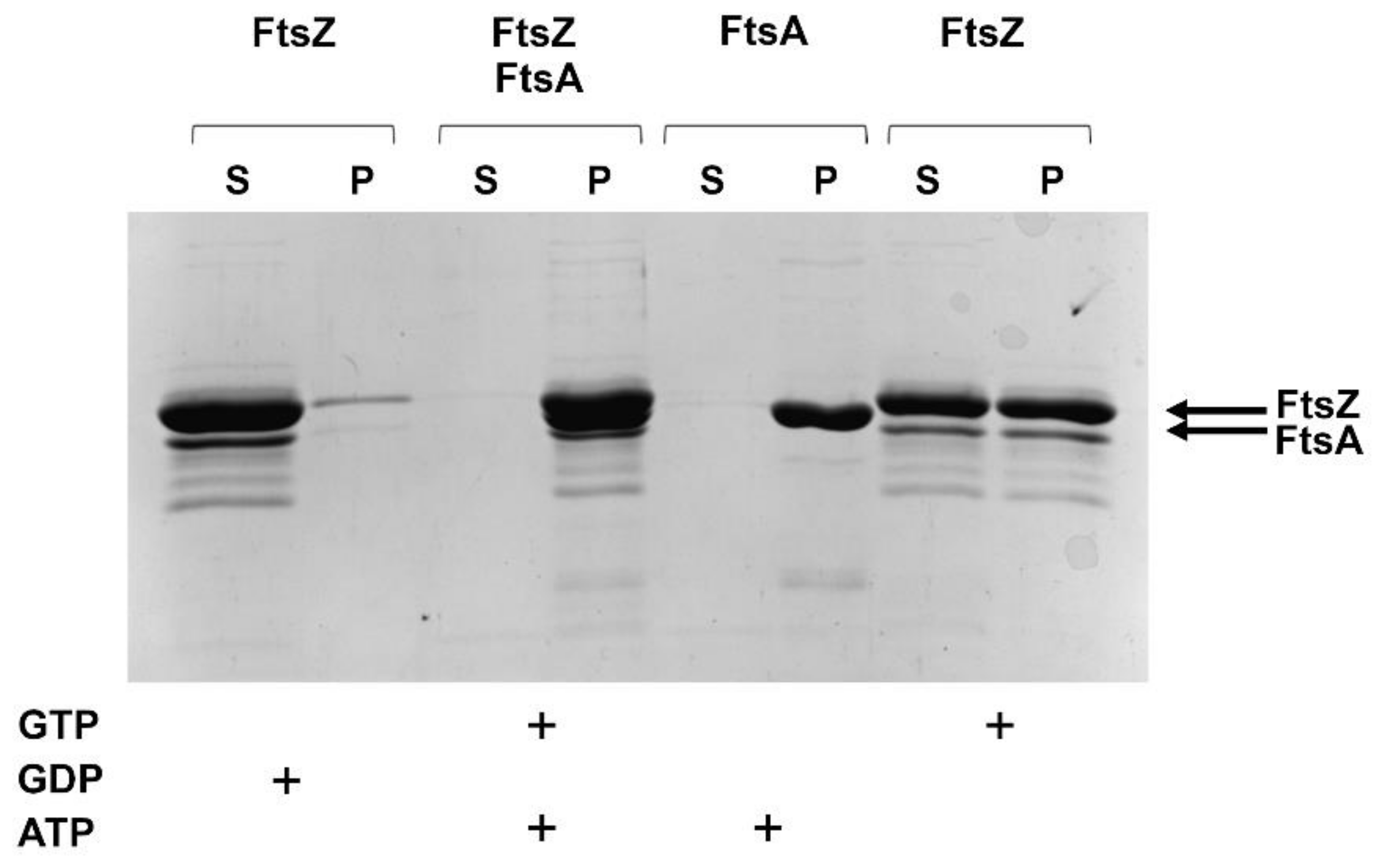
2.2.3. FtsZ and FtsA In Vitro Interaction
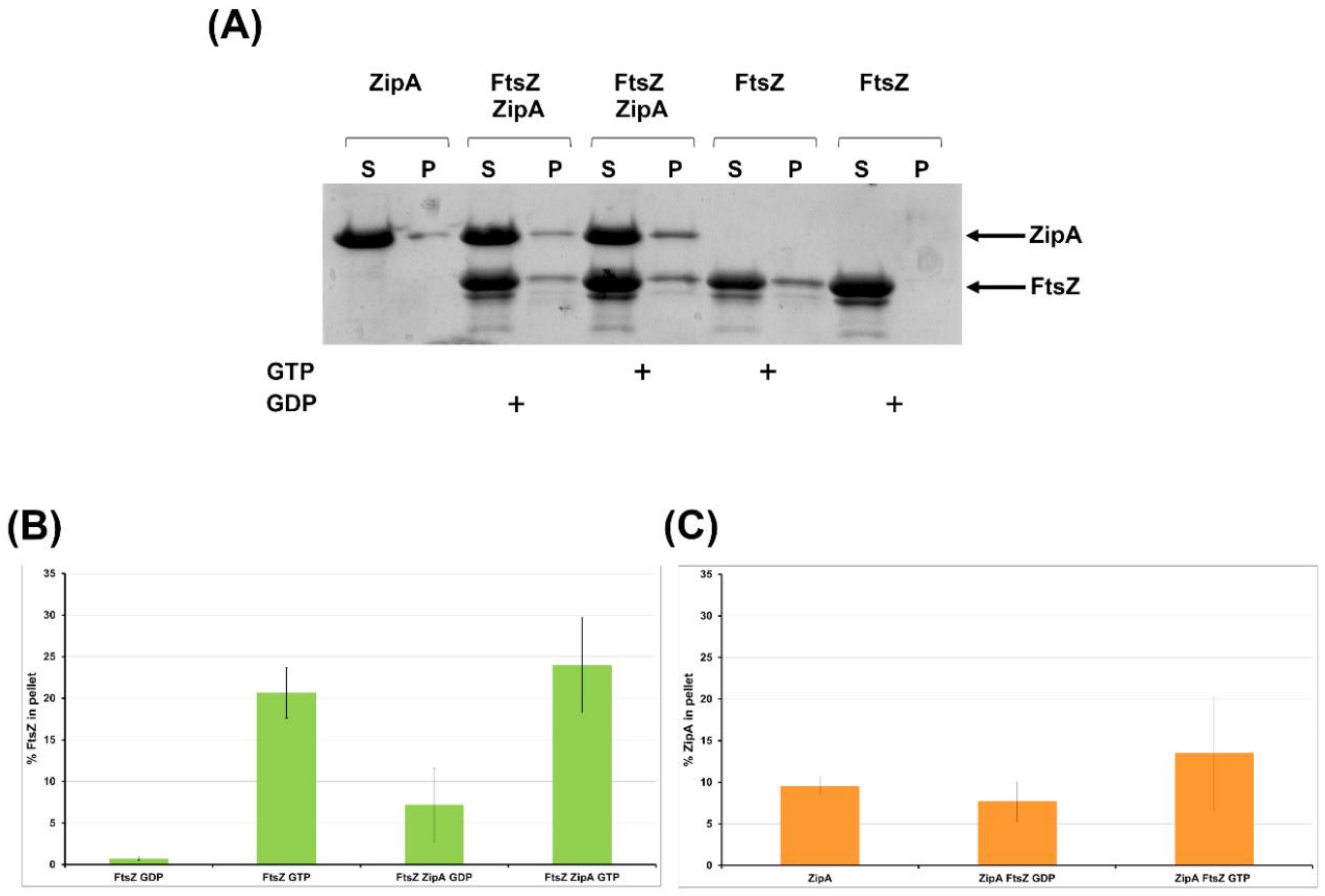
2.2.4. FtsZ Interacts with ZipA In Vitro
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, and Culture Conditions
4.2. B. cenocepacia J2315 RNA Extraction and RT-PCR
4.3. Promoter Activity Assessment by β-Galactosidase Activity Assay
4.4. 5′-Rapid Amplification of cDNA Ends (5′-RACE)
4.5. Cloning, Expression, and Purification of B. cenocepacia MraZ
4.6. Electrophoretic Mobility Shift Assay (EMSA)
4.7. Construction of the Recombinant Plasmid of the BACTH Complementation Assays
4.8. BACTH Complementation Assays
4.9. Cloning, Expression, and Purification of pBCA006, the Putative B. cenocepacia SulA
4.10. Cloning, Expression, and Purification of B. cenocepacia FtsA
4.11. Cloning, Expression, and Purification of B. cenocepacia ZipA
4.12. Co-Sedimentation Assays of FtsZ in the Presence of ZipA or SulA
4.13. Co-Sedimentation Assays of FtsZ in the Presence of FtsA and Vesicles
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thanbichler, M. Synchronization of chromosome dynamics and cell division in bacteria. Cold Spring Harb. Perspect. Biol. 2010, 2, a000331. [Google Scholar] [CrossRef] [PubMed]
- Kleckner, N.E.; Chatzi, K.; White, M.A.; Fisher, J.K.; Stouf, M. Coordination of Growth, Chromosome Replication/Segregation, and Cell Division in E. coli. Front. Microbiol. 2018, 9, 1469. [Google Scholar] [CrossRef] [PubMed]
- Osorio, A.; Camarena, L.; Cevallos, M.A.; Poggio, S. A New Essential Cell Division Protein in Caulobacter crescentus. J. Bacteriol. 2017, 199, e00811-16. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Lutkenhaus, J. Assembly and activation of the Escherichia coli divisome. Mol. Microbiol. 2017, 105, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Errington, J.; Wu, L.J. Cell Cycle Machinery in Bacillus subtilis. Subcell Biochem. 2017, 84, 67–101. [Google Scholar] [CrossRef] [PubMed]
- Vega, D.E.; Margolin, W. Direct Interaction between the Two Z Ring Membrane Anchors FtsA and ZipA. J. Bacteriol. 2019, 201, e00579-18. [Google Scholar] [CrossRef]
- Caldas, P.; López-Pelegrín, M.; Pearce, D.J.G.; Budanur, N.B.; Brugués, J.; Loose, M. Cooperative ordering of treadmilling filaments in cytoskeletal networks of FtsZ and its crosslinker ZapA. Nat. Commun. 2019, 10, 5744. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Ray, S.; Singh, D.; Dhaked, H.P.; Panda, D. ZapC promotes assembly and stability of FtsZ filaments by binding at a different site on FtsZ than ZipA. Int. J. Biol. Macromol. 2015, 81, 435–442. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Huang, K.H.; Zeng, W.; Janakiraman, A. Structure of the Z Ring-associated Protein, ZapD, Bound to the C-terminal Domain of the Tubulin-like Protein, FtsZ, Suggests Mechanism of Z Ring Stabilization through FtsZ Cross-linking. J. Biol. Chem. 2017, 292, 3740–3750. [Google Scholar] [CrossRef]
- Du, S.; Henke, W.; Pichoff, S.; Lutkenhaus, J. How FtsEX localizes to the Z ring and interacts with FtsA to regulate cell division. Mol. Microbiol. 2019, 112, 881–895. [Google Scholar] [CrossRef]
- Männik, J.; Bailey, M.W.; O’Neill, J.C.; Männik, J. Kinetics of large-scale chromosomal movement during asymmetric cell division in Escherichia coli. PLoS Genet. 2017, 13, e1006638. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Beckwith, J. FtsQ, FtsL and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Mol. Microbiol. 2001, 42, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Boes, A.; Olatunji, S.; Breukink, E.; Terrak, M. Regulation of the Peptidoglycan Polymerase Activity of PBP1b by Antagonist Actions of the Core Divisome Proteins FtsBLQ and FtsN. mBio 2019, 10, e01912-18. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Welsh, M.A.; Marmont, L.S.; Lee, W.; Sjodt, M.; Kruse, A.C.; Kahne, D.; Bernhardt, T.G.; Walker, S. FtsW is a peptidoglycan polymerase that is functional only in complex with its cognate penicillin-binding protein. Nat. Microbiol. 2019, 4, 587–594. [Google Scholar] [CrossRef]
- Wang, L.; Khattar, M.K.; Donachie, W.D.; Lutkenhaus, J. FtsI and FtsW are localized to the septum in Escherichia coli. J. Bacteriol. 1998, 180, 2810–2816. [Google Scholar] [CrossRef]
- Pichoff, S.; Du, S.; Lutkenhaus, J. Disruption of divisome assembly rescued by FtsN-FtsA interaction in Escherichia coli. Proc. Natl. Acad. Sci. USA 2018, 115, E6855–E6862. [Google Scholar] [CrossRef]
- Du, S.; Pichoff, S.; Kruse, K.; Lutkenhaus, J. FtsZ filaments have the opposite kinetic polarity of microtubules. Proc. Natl. Acad. Sci. USA 2018, 115, 10768–10773. [Google Scholar] [CrossRef]
- Wang, M.; Fang, C.; Ma, B.; Luo, X.; Hou, Z. Regulation of cytokinesis: FtsZ and its accessory proteins. Curr. Genet. 2020, 66, 43–49. [Google Scholar] [CrossRef]
- Arumugam, S.; Petrašek, Z.; Schwille, P. MinCDE exploits the dynamic nature of FtsZ filaments for its spatial regulation. Proc. Natl. Acad. Sci. USA 2014, 111, E1192–E1200. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Zeng, W. Structures of the nucleoid occlusion protein SlmA bound to DNA and the C-terminal domain of the cytoskeletal protein FtsZ. Proc. Natl. Acad. Sci. USA 2016, 113, 4988–4993. [Google Scholar] [CrossRef]
- Vedyaykin, A.; Rumyantseva, N.; Khodorkovskii, M.; Vishnyakov, I. SulA is able to block cell division in Escherichia coli by a mechanism different from sequestration. Biochem. Biophys. Res. Commun. 2020, 525, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.P.; Osawa, M. FtsZ Constriction Force—Curved Protofilaments Bending Membranes. Subcell Biochem. 2017, 84, 139–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lyu, Z.; Miguel, A.; McQuillen, R.; Huang, K.C.; Xiao, J. GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science 2017, 355, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Bisson-Filho, A.W.; Hsu, Y.P.; Squyres, G.R.; Kuru, E.; Wu, F.; Jukes, C.; Sun, Y.; Dekker, C.; Holden, S.; VanNieuwenhze, M.S.; et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 2017, 355, 739–743. [Google Scholar] [CrossRef]
- Dewar, S.J.; Dorazi, R. Control of division gene expression in Escherichia coli. FEMS Microbiol. Lett. 2000, 187, 1–7. [Google Scholar] [CrossRef]
- Tamames, J.; González-Moreno, M.; Mingorance, J.; Valencia, A.; Vicente, M. Bringing gene order into bacterial shape. Trends Genet. 2001, 17, 124–126. [Google Scholar] [CrossRef]
- de la Fuente, A.; Palacios, P.; Vicente, M. Transcription of the Escherichia coli dcw cluster: Evidence for distal upstream transcripts being involved in the expression of the downstream ftsZ gene. Biochimie 2001, 83, 109–115. [Google Scholar] [CrossRef]
- Snyder, L.A.; Shafer, W.M.; Saunders, N.J. Divergence and transcriptional analysis of the division cell wall (dcw) gene cluster in Neisseria spp. Mol. Microbiol. 2003, 47, 431–442. [Google Scholar] [CrossRef]
- Real, G.; Henriques, A.O. Localization of the Bacillus subtilis murB gene within the dcw cluster is important for growth and sporulation. J. Bacteriol. 2006, 188, 1721–1732. [Google Scholar] [CrossRef]
- Massidda, O.; Nováková, L.; Vollmer, W. From models to pathogens: How much have we learned about Streptococcus pneumoniae cell division? Environ. Microbiol. 2013, 15, 3133–3157. [Google Scholar] [CrossRef]
- Santini, T.; Turchi, L.; Ceccarelli, G.; Di Franco, C.; Beccari, E. Transcriptional analysis of ftsZ within the dcw cluster in Bacillus mycoides. BMC Microbiol. 2013, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.; Thoelken, C.; Volk, M.; Remes, B.; Lechner, M.; Klug, G. The Conserved Dcw Gene Cluster of R. sphaeroides is Preceded by an Uncommonly Extended 5′ Leader Featuring the sRNA UpsM. PLoS ONE 2016, 11, e0165694. [Google Scholar] [CrossRef] [PubMed]
- Bobay, B.G.; Andreeva, A.; Mueller, G.A.; Cavanagh, J.; Murzin, A.G. Revised structure of the AbrB N-terminal domain unifies a diverse superfamily of putative DNA-binding proteins. FEBS Lett. 2005, 579, 5669–5674. [Google Scholar] [CrossRef] [PubMed]
- Coles, M.; Djuranovic, S.; Söding, J.; Frickey, T.; Koretke, K.; Truffault, V.; Martin, J.; Lupas, A.N. AbrB-like transcription factors assume a swapped hairpin fold that is evolutionarily related to double-psi beta barrels. Structure 2005, 13, 919–928. [Google Scholar] [CrossRef]
- Eraso, J.M.; Markillie, L.M.; Mitchell, H.D.; Taylor, R.C.; Orr, G.; Margolin, W. The highly conserved MraZ protein is a transcriptional regulator in Escherichia coli. J. Bacteriol. 2014, 196, 2053–2066. [Google Scholar] [CrossRef]
- Fisunov, G.Y.; Evsyutina, D.V.; Semashko, T.A.; Arzamasov, A.A.; Manuvera, V.A.; Letarov, A.V.; Govorun, V.M. Binding site of MraZ transcription factor in Mollicutes. Biochimie 2016, 125, 59–65. [Google Scholar] [CrossRef]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Depoorter, E.; De Canck, E.; Peeters, C.; Wieme, A.D.; Cnockaert, M.; Zlosnik, J.E.A.; LiPuma, J.J.; Coenye, T.; Vandamme, P. Burkholderia cepacia Complex Taxon K: Where to Split? Front. Microbiol. 2020, 11, 1594. [Google Scholar] [CrossRef]
- Jones, A.M.; Dodd, M.E.; Govan, J.R.; Barcus, V.; Doherty, C.J.; Morris, J.; Webb, A.K. Burkholderia cenocepacia and Burkholderia multivorans: Influence on survival in cystic fibrosis. Thorax 2004, 59, 948–951. [Google Scholar] [CrossRef]
- Drevinek, P.; Mahenthiralingam, E. Burkholderia cenocepacia in cystic fibrosis: Epidemiology and molecular mechanisms of virulence. Clin. Microbiol. Infect. 2010, 16, 821–830. [Google Scholar] [CrossRef]
- Holden, M.T.; Seth-Smith, H.M.; Crossman, L.C.; Sebaihia, M.; Bentley, S.D.; Cerdeño-Tárraga, A.M.; Thomson, N.R.; Bason, N.; Quail, M.A.; Sharp, S.; et al. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J. Bacteriol. 2009, 191, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.A.; Ramos, C.G.; Leitão, J.H. Burkholderia cepacia Complex: Emerging Multihost Pathogens Equipped with a Wide Range of Virulence Factors and Determinants. Int. J. Microbiol. 2011, 2011, 607575. [Google Scholar] [CrossRef] [PubMed]
- Horsley, A.; Webb, K.; Bright-Thomas, R.; Govan, J.; Jones, A. Can early Burkholderia cepacia complex infection in cystic fibrosis be eradicated with antibiotic therapy? Front. Cell Infect. Microbiol. 2011, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Scoffone, V.C.; Chiarelli, L.R.; Trespidi, G.; Mentasti, M.; Riccardi, G.; Buroni, S. Burkholderia cenocepacia Infections in Cystic Fibrosis Patients: Drug Resistance and Therapeutic Approaches. Front. Microbiol. 2017, 8, 1592. [Google Scholar] [CrossRef]
- Scoffone, V.C.; Spadaro, F.; Udine, C.; Makarov, V.; Fondi, M.; Fani, R.; De Rossi, E.; Riccardi, G.; Buroni, S. Mechanism of resistance to an antitubercular 2-thiopyridine derivative that is also active against Burkholderia cenocepacia. Antimicrob. Agents Chemother. 2014, 58, 2415–2417. [Google Scholar] [CrossRef][Green Version]
- Scoffone, V.C.; Ryabova, O.; Makarov, V.; Iadarola, P.; Fumagalli, M.; Fondi, M.; Fani, R.; De Rossi, E.; Riccardi, G.; Buroni, S. Efflux-mediated resistance to a benzothiadiazol derivative effective against Burkholderia cenocepacia. Front. Microbiol. 2015, 6, 815. [Google Scholar] [CrossRef][Green Version]
- Hogan, A.M.; Scoffone, V.C.; Makarov, V.; Gislason, A.S.; Tesfu, H.; Stietz, M.S.; Brassinga, A.K.C.; Domaratzki, M.; Li, X.; Azzalin, A.; et al. Competitive Fitness of Essential Gene Knockdowns Reveals a Broad-Spectrum Antibacterial Inhibitor of the Cell Division Protein FtsZ. Antimicrob. Agents Chemother. 2018, 62, e01231-18. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Scoffone, V.C.; Trespidi, G.; Barbieri, G.; Riabova, O.; Monakhova, N.; Porta, A.; Manina, G.; Riccardi, G.; Makarov, V.; et al. Chemical, Metabolic, and Cellular Characterization of a FtsZ Inhibitor Effective Against Burkholderia cenocepacia. Front. Microbiol. 2020, 11, 562. [Google Scholar] [CrossRef]
- Costabile, G.; Provenzano, R.; Azzalin, A.; Scoffone, V.C.; Chiarelli, L.R.; Rondelli, V.; Grillo, I.; Zinn, T.; Lepioshkin, A.; Savina, S.; et al. PEGylated mucus-penetrating nanocrystals for lung delivery of a new FtsZ inhibitor against Burkholderia cenocepacia infection. Nanomedicine 2020, 23, 102113. [Google Scholar] [CrossRef]
- Buroni, S.; Makarov, V.; Scoffone, V.C.; Trespidi, G.; Riccardi, G.; Chiarelli, L.R. The cell division protein FtsZ as a cellular target to hit cystic fibrosis pathogens. Eur. J. Med. Chem. 2020, 190, 112132. [Google Scholar] [CrossRef]
- Winsor, G.L.; Khaira, B.; Van Rossum, T.; Lo, R.; Whiteside, M.D.; Brinkman, F.S. The Burkholderia Genome Database: Facilitating flexible queries and comparative analyses. Bioinformatics 2008, 24, 2803–2804. [Google Scholar] [CrossRef] [PubMed]
- Kingsford, C.L.; Ayanbule, K.; Salzberg, S.L. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007, 8, R22. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Yasuda, S.; Horiuchi, K.; Park, J.T. A promoter for the first nine genes of the Escherichia coli mra cluster of cell division and cell envelope biosynthesis genes, including ftsI and ftsW. J. Bacteriol. 1997, 179, 5802–5811. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Flärdh, K.; Garrido, T.; Vicente, M. Contribution of individual promoters in the ddlB-ftsZ region to the transcription of the essential cell-division gene ftsZ in Escherichia coli. Mol. Microbiol. 1997, 24, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Ohashi, T.; Corbin, L.; Erickson, H.P. High-resolution crystal structures of Escherichia coli FtsZ bound to GDP and GTP. Acta Crystallogr. F Struct. Biol. Commun. 2020, 76, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Cordell, S.C.; Robinson, E.J.; Lowe, J. Crystal structure of the SOS cell division inhibitor SulA and in complex with FtsZ. Proc. Natl. Acad. Sci. USA 2003, 100, 7889–7894. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Mukherjee, A.; Lutkenhaus, J. Recruitment of ZipA to the division site by interaction with FtsZ. Mol. Microbiol. 1999, 31, 1853–1861. [Google Scholar] [CrossRef]
- Tsang, M.J.; Bernhardt, T.G. A role for the FtsQLB complex in cytokinetic ring activation revealed by an ftsL allele that accelerates division. Mol. Microbiol. 2015, 95, 925–944. [Google Scholar] [CrossRef]
- Krupka, M.; Sobrinos-Sanguino, M.; Jiménez, M.; Rivas, G.; Margolin, W. Escherichia coli ZipA Organizes FtsZ Polymers into Dynamic Ring-Like Protofilament Structures. mBio 2018, 9, e01008-18. [Google Scholar] [CrossRef]
- Busiek, K.K.; Eraso, J.M.; Wang, Y.; Margolin, W. The early divisome protein FtsA interacts directly through its 1c subdomain with the cytoplasmic domain of the late divisome protein FtsN. J. Bacteriol. 2012, 194, 1989–2000. [Google Scholar] [CrossRef]
- Dajkovic, A.; Mukherjee, A.; Lutkenhaus, J. Investigation of regulation of FtsZ assembly by SulA and development of a model for FtsZ polymerization. J. Bacteriol. 2008, 190, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Szwedziak, P.; Wang, Q.; Freund, S.M.; Löwe, J. FtsA forms actin-like protofilaments. EMBO J. 2012, 31, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Olland, A.; Falcoz, P.E.; Kessler, R.; Massard, G. Should cystic fibrosis patients infected with Burkholderia cepacia complex be listed for lung transplantation? Interact. Cardiovasc. Thorac. Surg. 2011, 13, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Mori, H.; Itoh, T.; Gojobori, T. Genome plasticity as a paradigm of eubacteria evolution. J. Mol. Evol. 1997, 44, S57–S64. [Google Scholar] [CrossRef]
- Palacios, P.; Vicente, M.; Sánchez, M. Dependency of Escherichia coli cell-division size, and independency of nucleoid segregation on the mode and level of ftsZ expression. Mol. Microbiol. 1996, 20, 1093–1098. [Google Scholar] [CrossRef]
- Siefert, J.L.; Fox, G.E. Phylogenetic mapping of bacterial morphology. Microbiology 1998, 144, 2803–2808. [Google Scholar] [CrossRef]
- Mingorance, J.; Tamames, J.; Vicente, M. Genomic channeling in bacterial cell division. J. Mol. Recognit. 2004, 17, 481–487. [Google Scholar] [CrossRef]
- Vicente, M.; Gomez, M.J.; Ayala, J.A. Regulation of transcription of cell division genes in the Escherichia coli dcw cluster. Cell. Mol. Life Sci. 1998, 54, 317–324. [Google Scholar] [CrossRef]
- Mengin-Lecreulx, D.; Ayala, J.; Bouhss, A.; van Heijenoort, J.; Parquet, C.; Hara, H. Contribution of the Pmra promoter to expression of genes in the Escherichia coli mra cluster of cell envelope biosynthesis and cell division genes. J. Bacteriol. 1998, 180, 4406–4412. [Google Scholar] [CrossRef]
- Beall, B.; Lutkenhaus, J. Sequence analysis, transcriptional organization, and insertional mutagenesis of the envA gene of Escherichia coli. J. Bacteriol. 1987, 169, 5408–5415. [Google Scholar] [CrossRef]
- Maeda, T.; Tanaka, Y.; Takemoto, N.; Hamamoto, N.; Inui, M. RNase III mediated cleavage of the coding region of mraZ mRNA is required for efficient cell division in Corynebacterium glutamicum. Mol. Microbiol. 2016, 99, 1149–1166. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Milam, S.L.; Erickson, H.P. SulA inhibits assembly of FtsZ by a simple sequestration mechanism. Biochemistry 2012, 51, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Karimova, G.; Dautin, N.; Ladant, D. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 2005, 187, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Loose, M.; Mitchison, T.J. The bacterial cell division proteins FtsA and FtsZ self-organize into dynamic cytoskeletal patterns. Nat. Cell Biol. 2014, 16, 38–46. [Google Scholar] [CrossRef]
- Ohashi, T.; Hale, C.A.; de Boer, P.A.; Erickson, H.P. Structural evidence that the P/Q domain of ZipA is an unstructured, flexible tether between the membrane and the C-terminal FtsZ-binding domain. J. Bacteriol. 2002, 184, 4313–4315. [Google Scholar] [CrossRef][Green Version]
- Schmidt, K.L.; Peterson, N.D.; Kustusch, R.J.; Wissel, M.C.; Graham, B.; Phillips, G.J.; Weiss, D.S. A predicted ABC transporter, FtsEX, is needed for cell division in Escherichia coli. J. Bacteriol. 2004, 186, 785–793. [Google Scholar] [CrossRef]
- Du, S.; Pichoff, S.; Lutkenhaus, J. FtsEX acts on FtsA to regulate divisome assembly and activity. Proc. Natl. Acad. Sci. USA 2016, 113, E5052–E5061. [Google Scholar] [CrossRef]
- Corbin, B.D.; Wang, Y.; Beuria, T.K.; Margolin, W. Interaction between cell division proteins FtsE and FtsZ. J. Bacteriol. 2007, 189, 3026–3035. [Google Scholar] [CrossRef]
- Grenga, L.; Guglielmi, G.; Melino, S.; Ghelardini, P.; Paolozzi, L. FtsQ interaction mutants: A way to identify new antibacterial targets. N. Biotechnol. 2010, 27, 870–881. [Google Scholar] [CrossRef]
- Alexeeva, S.; Gadella, T.W.; Verheul, J.; Verhoeven, G.S.; den Blaauwen, T. Direct interactions of early and late assembling division proteins in Escherichia coli cells resolved by FRET. Mol. Microbiol. 2010, 77, 384–398. [Google Scholar] [CrossRef]
- Zou, Y.; Li, Y.; Dillon, J.R. The distinctive cell division interactome of Neisseria gonorrhoeae. BMC Microbiol. 2017, 17, 232. [Google Scholar] [CrossRef] [PubMed]
- Jenul, C.; Sieber, S.; Daeppen, C.; Mathew, A.; Lardi, M.; Pessi, G.; Hoepfner, D.; Neuburger, M.; Linden, A.; Gademann, K.; et al. Biosynthesis of fragin is controlled by a novel quorum sensing signal. Nat. Commun. 2018, 9, 1297. [Google Scholar] [CrossRef] [PubMed]
- Stachel, S.; An, G.; Flores, C.; Nester, E. A Tn3 lacZ transposon for the random generation of beta-galactosidase gene fusions: Application to the analysis of gene expression in Agrobacterium. EMBO J. 1985, 4, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Król, E.; Scheffers, D.J. FtsZ polymerization assays: Simple protocols and considerations. J. Vis. Exp. 2013, 16, e50844. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trespidi, G.; Scoffone, V.C.; Barbieri, G.; Riccardi, G.; De Rossi, E.; Buroni, S. Molecular Characterization of the Burkholderia cenocepacia dcw Operon and FtsZ Interactors as New Targets for Novel Antimicrobial Design. Antibiotics 2020, 9, 841. https://doi.org/10.3390/antibiotics9120841
Trespidi G, Scoffone VC, Barbieri G, Riccardi G, De Rossi E, Buroni S. Molecular Characterization of the Burkholderia cenocepacia dcw Operon and FtsZ Interactors as New Targets for Novel Antimicrobial Design. Antibiotics. 2020; 9(12):841. https://doi.org/10.3390/antibiotics9120841
Chicago/Turabian StyleTrespidi, Gabriele, Viola Camilla Scoffone, Giulia Barbieri, Giovanna Riccardi, Edda De Rossi, and Silvia Buroni. 2020. "Molecular Characterization of the Burkholderia cenocepacia dcw Operon and FtsZ Interactors as New Targets for Novel Antimicrobial Design" Antibiotics 9, no. 12: 841. https://doi.org/10.3390/antibiotics9120841
APA StyleTrespidi, G., Scoffone, V. C., Barbieri, G., Riccardi, G., De Rossi, E., & Buroni, S. (2020). Molecular Characterization of the Burkholderia cenocepacia dcw Operon and FtsZ Interactors as New Targets for Novel Antimicrobial Design. Antibiotics, 9(12), 841. https://doi.org/10.3390/antibiotics9120841