Anti-Tubercular Activity of Substituted 7-Methyl and 7-Formylindolizines and In Silico Study for Prospective Molecular Target Identification

 ,
,  , , ,
, , ,  ,
,  , , , ,
, , , ,  , ,
, ,  , and
, and
Abstract
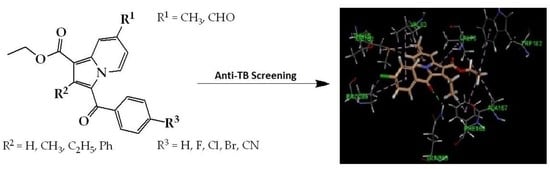
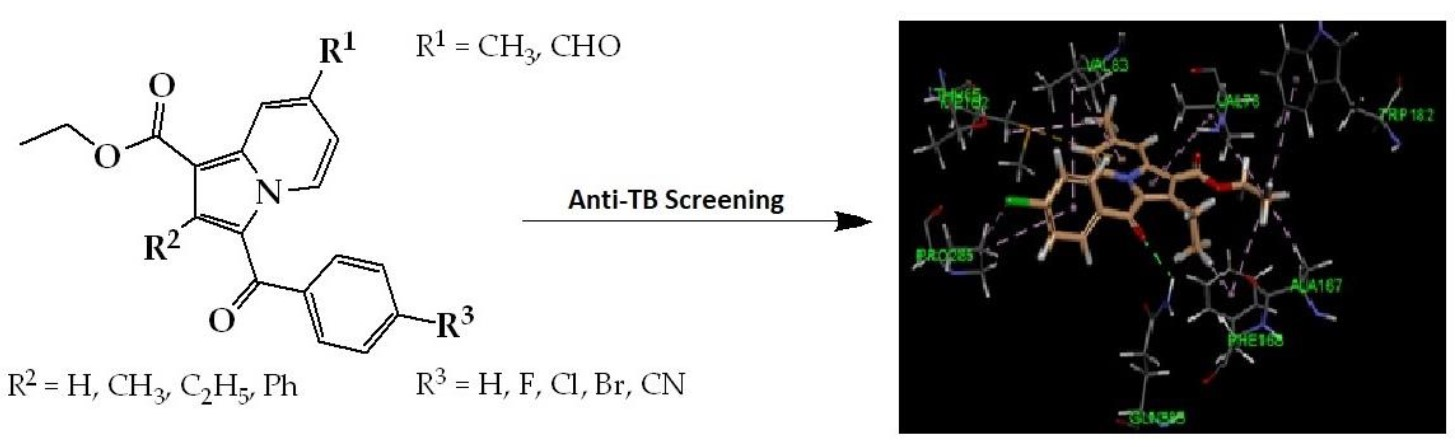
1. Introduction
2. Results and Discussion
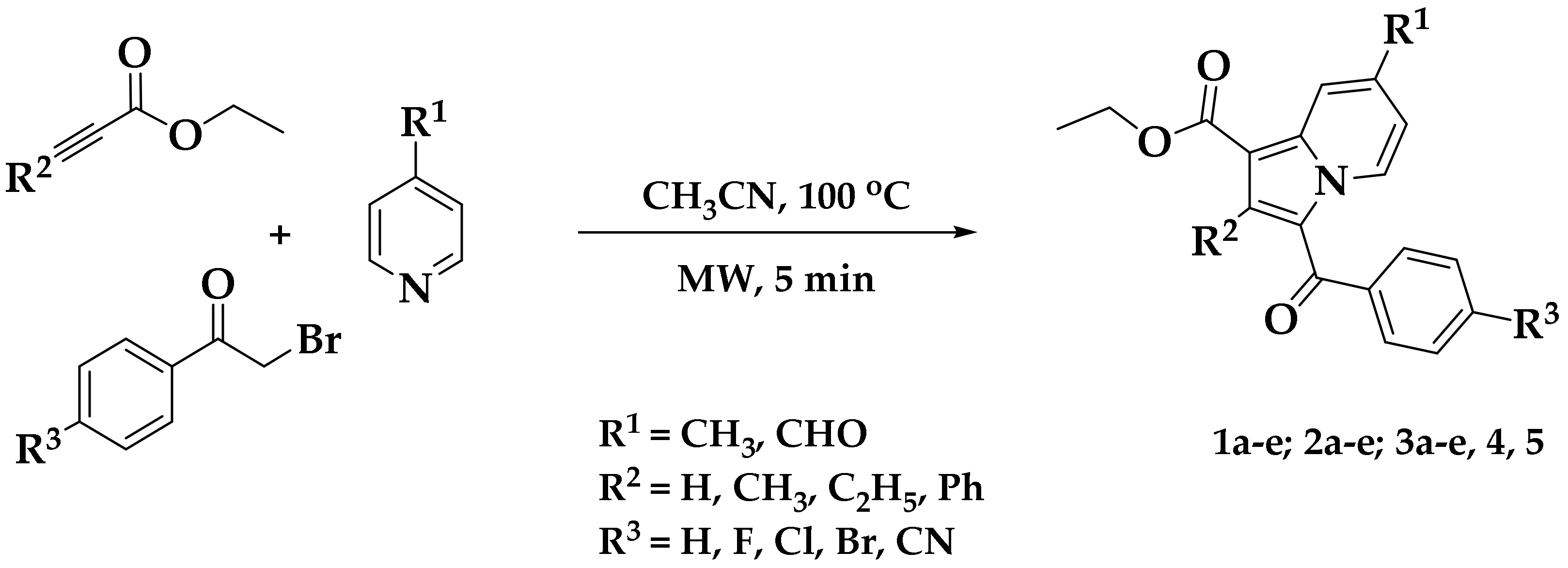
2.1. Chemistry
2.2. Anti-Tubercular Activity
2.3. Safety Studies
2.4. Molecular Modeling
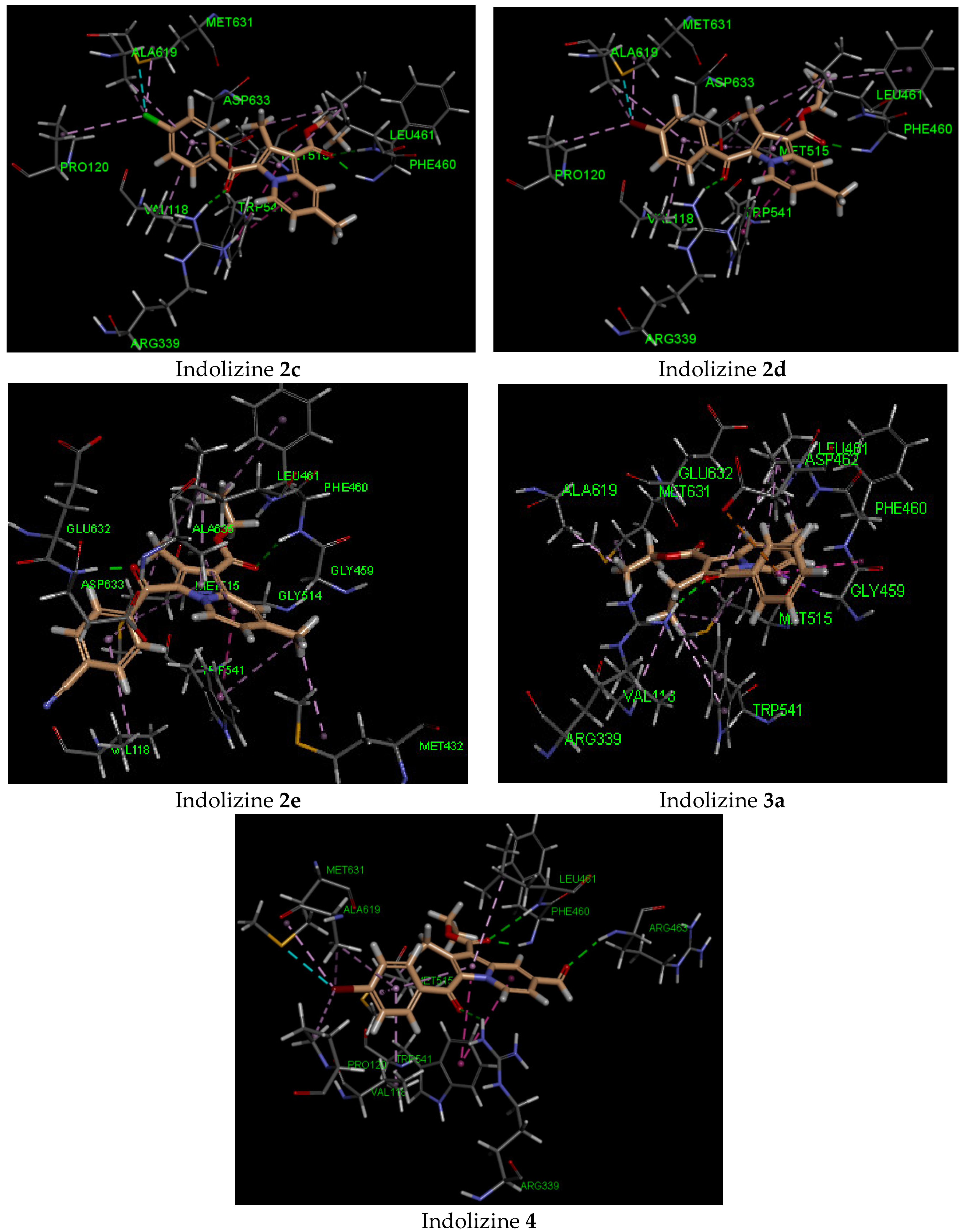
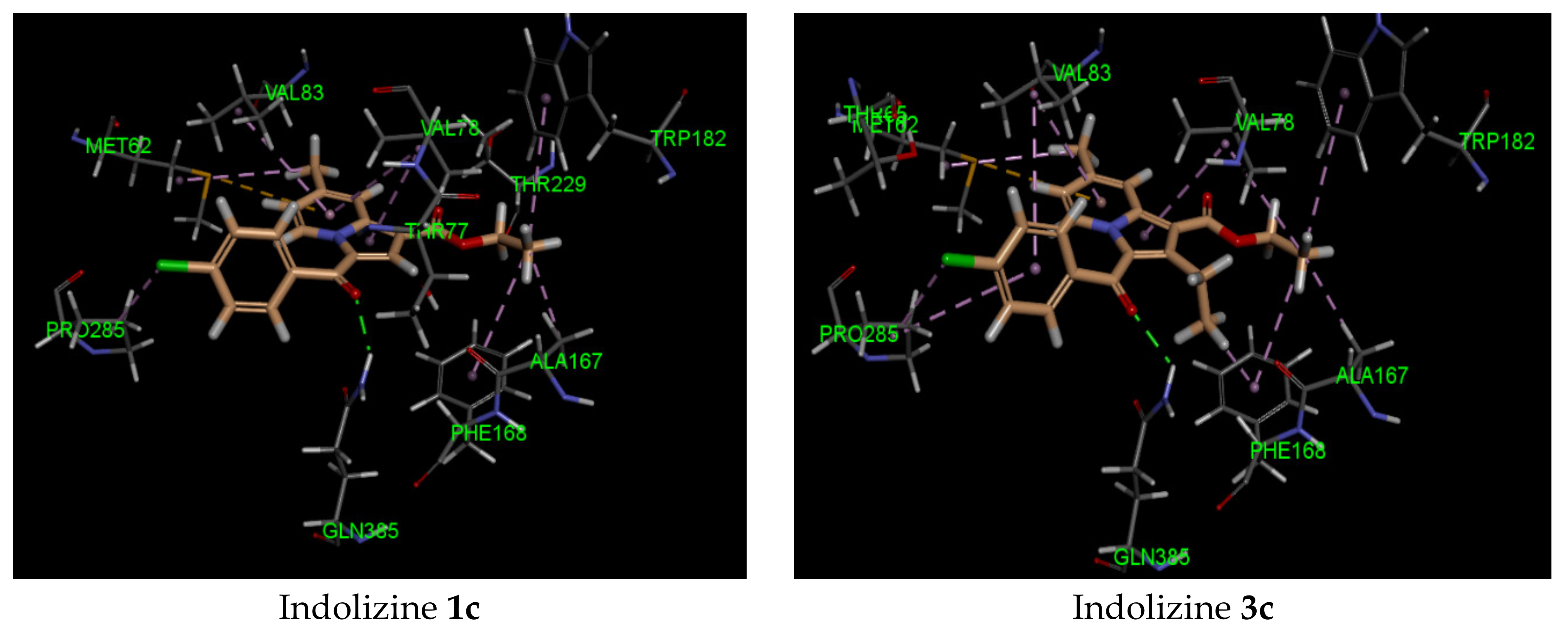
2.5. Molecular Docking Analysis with CYP121 Receptor
2.6. Molecular Docking Analysis with Malate Synthase Receptor
2.7. Molecular Docking Analysis with GyrB ATPase Receptor
3. Materials and Methods
3.1. General
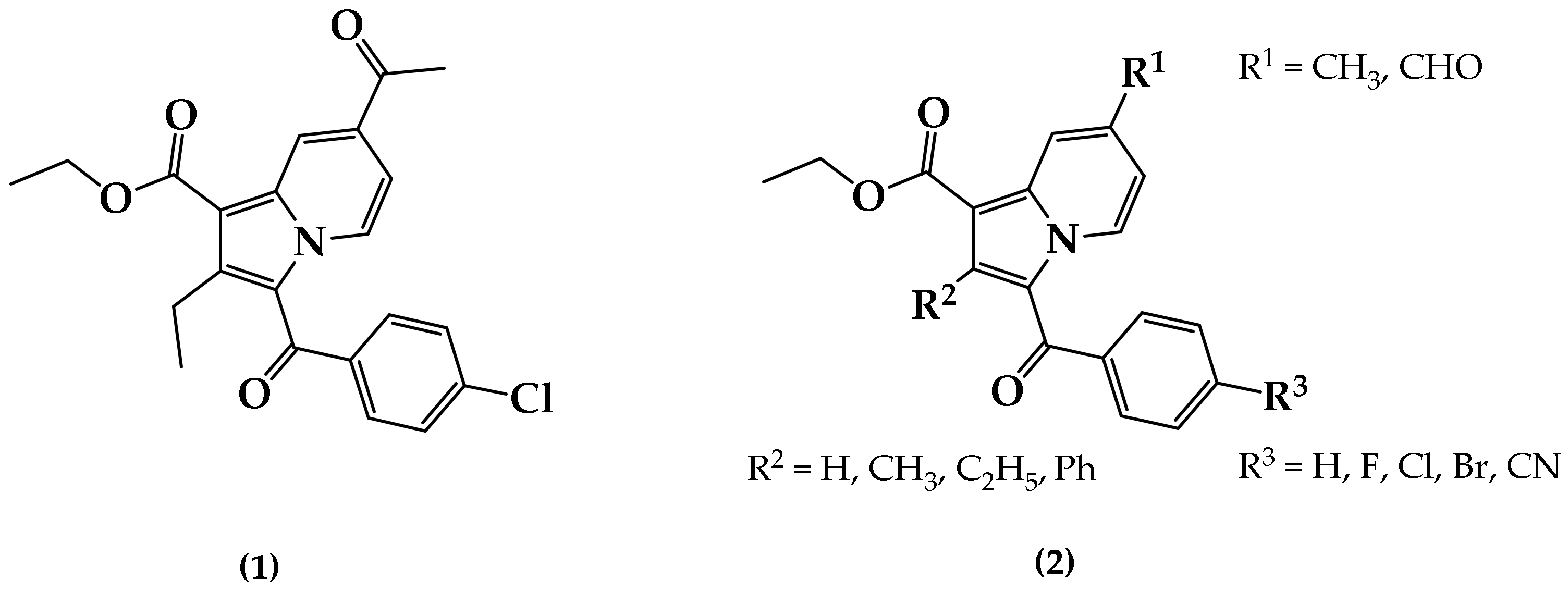
3.2. Synthetic Procedure for the Construction of Ethyl 3-(4-Bromobenzoyl)-7-formyl-2-methylindolizine-1-carboxylate (4)
Methyl 3-(4-Fluorobenzoyl)-7-methyl-2-phenylindolizine-1-carboxylate (5)
3.3. Anti-Tubercular Activity by Resazurin Microplate Assay (REMA)
3.4. Safety Studies
3.5. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Executive Summary_21Sept2018. Available online: http://www.who.int/tb/publications/global_report/Exec_summary_21Sept2018.v1.1.pdf?ua=1 2018 (accessed on 29 September 2019).
- Marcos, A.E. The global situation of MDR-TB. Tuberculosis 2003, 83, 44–51. [Google Scholar]
- Caminero, J.A.; Sotgiu, G.; Zumla, A.; Migliori, G.B. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect. Dis. 2010, 10, 621–629. [Google Scholar] [CrossRef]
- Hu, Y.; Xu, L.; He, Y.L.; Pang, Y.; Lu, N.; Liu, J.; Shen, J.; Zhu, D.M.; Feng, X.; Wang, Y.W.; et al. Prevalence and molecular characterization of second-line drugs resistance among multidrug-resistant Mycobacterium tuberculosis isolates in Southwest of China. BioMed Res. Int. 2017, 2017, 4563826. [Google Scholar] [CrossRef]
- Parida, S.K.; Axelsson-Robertson, R.; Rao, M.V.; Singh, N.; Master, I.; Lutckii, A.; Keshavjee, S.; Andersson, J.; Zumla, A.; Maeurer, M. Totally drug-resistant tuberculosis and adjunct therapies. J. Int. Med. 2015, 277, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Cox, E.; Laessig, K. FDA Approval of Bedaquiline—The benefit–risk balance for drug-resistant Tuberculosis. N. Engl. J. Med. 2014, 371, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Barry Iii, C.E. Timing is everything for compassionate use of delamanid. Nat. Med. 2015, 21, 211. [Google Scholar] [CrossRef] [PubMed]
- Vaught, J.L.; Carson, J.R.; Carmosin, R.J.; Blum, P.S.; Persico, F.J.; Hageman, W.E.; Shank, R.P.; Raffa, R.B. Antinociceptive action of McN-5195 in rodents: A structurally novel (indolizine) analgesic with a nonopioid mechanism of action. J. Pharmacol. Exp. Ther. 1990, 255, 1–10. [Google Scholar]
- Butler, M.S. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [Google Scholar] [CrossRef]
- Chandrashekharappa, S.; Padmashali, B.; Venugopala, K.N.; Kulkarni, R.S.; Venugopala, R.; Odhav, B. Synthesis and characterization of ethyl 7-acetyl-2-substituted 3-(substituted benzoyl)indolizine-1-carboxylates for in vitro anticancer activity. Asian J. Chem. 2016, 28, 1043–1048. [Google Scholar]
- Mederski, W.; Beier, N.; Burgdorf, L.T.; Gericke, R.; Klein, M.; Tsaklakidis, C. Indolizine Derivatives and the Use Thereof as Antidiabetics. U.S. Patent 8,106,067, 8 January 2012. [Google Scholar]
- Cingolani, G.M.; Claudi, F.; Massi, M.; Venturi, F. Indolizine derivatives with biological activity VI 1-(2-aminoethyl)-3-benzyl-7-methoxy-2-methylindolizine, benanserin structural analogue. Cingolani 1990, 25, 709–712. [Google Scholar] [CrossRef]
- Hagishita, S.; Yamada, M.; Shirahase, K.; Okada, T.; Murakami, Y.; Ito, Y.; Matsuura, T.; Wada, M.; Kato, T.; Ueno, M.; et al. Potent inhibitors of secretory phospholipase A2: Synthesis and inhibitory activities of indolizine and indene derivatives. J. Med. Chem. 1996, 39, 3636–3658. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekharappa, S.; Venugopala, K.N.; Tratrat, C.; Mahomoodally, F.M.; Aldhubiab, B.E.; Haroun, M.; Venugopala, R.; Mohan, M.K.; Kulkarni, R.S.; Attimarad, M.V.; et al. Efficient synthesis and characterization of novel indolizines: Exploration of in vitro COX-2 inhibitory activity and molecular modelling studies. New J. Chem. 2018, 42, 4893–4901. [Google Scholar] [CrossRef]
- Jaisankar, P.; Pal, B.; Manna, K.N.; Pradhan, P.K.; Medda, S.; Basu, M.K.; Giri, V.S. Synthesis of antileishmanial (5R)-(-)-5-carbomethoxy-3-formyl-5,6-dihydroindolo-[2,3-a]-indolizine. ARKIVOC 2003, 9, 150–157. [Google Scholar]
- Hazra, A.; Mondal, S.; Maity, A.; Naskar, S.; Saha, P.; Paira, R.; Sahu, K.B.; Paira, P.; Ghosh, S.; Sinha, C.; et al. Amberlite-IRA-402 (OH) ion exchange resin mediated synthesis of indolizines, pyrrolo [1,2-a] quinolines and isoquinolines: Antibacterial and antifungal evaluation of the products. Eur. J. Med. Chem. 2011, 46, 2132–2140. [Google Scholar] [CrossRef] [PubMed]
- Olejnikova, P.; Birosova, L.; Svorc, L. Antimicrobial and antimutagenic properties of newly synthesized derivatives of indolizine. Sci. Pharm. 2009, 77, 216. [Google Scholar] [CrossRef]
- Nasir, A.I.; Gundersen, L.-L.; Rise, F.; Antonsen, Ø.; Kristensen, T.; Langhelle, B.; Bast, A.; Custers, I.; Haenen, G.R.M.M.; Wikström, H. Inhibition of lipid peroxidation mediated by indolizines. Bioorg. Med. Chem. Lett. 1998, 8, 1829–1832. [Google Scholar] [CrossRef]
- Mishra, B.B.; Tiwari, V.K. Natural products in drug discovery: Clinical evaluations and investigations. Oppor. Chall. Scope Nat. Prod. Med. Chem. 2011, 661, 1–61. [Google Scholar]
- Chandrashekharappa, S.; Venugopala, K.N.; Gleiser, R.M.; Chetram, A.; Padmashali, B.; Kulkarni, R.S.; Venugopala, R.; Odhav, B. Greener synthesis of indolizine analogues using water as a base and solvent: Study for larvicidal activity against Anopheles Arab. Chem. Biol. Drug Des. 2016, 88, 899–904. [Google Scholar]
- Chandrashekharappa, S.; Venugopala, K.N.; Nayak, S.K.; Gleiser, R.M.; García, D.A.; Kumalo, H.M.; Kulkarni, R.S.; Mahomoodally, F.M.; Venugopala, R.; Mohan, M.K.; et al. One-pot microwave assisted synthesis and structural elucidation of novel ethyl 3-substituted-7-methylindolizine-1-carboxylates with larvicidal activity against Anopheles Arab. J. Mol. Struct. 2018, 1156, 377–384. [Google Scholar] [CrossRef]
- Smith, S.C.; Clarke, E.D.; Ridley, S.M.; Bartlett, D.; Greenhow, D.T.; Glithro, H.; Klong, A.Y.; Mitchell, G.; Mullier, G.W. Herbicidal indolizine-5,8-diones: Photosystem I redox mediators. Pest Manag. Sci. 2005, 61, 16–24. [Google Scholar] [CrossRef]
- Dannhardt, G.; Meindl, W.; Gussmann, S.; Ajili, S.; Kappe, T. Anti-mycobacterial 7-hydroxy-2,3-dihydro-1H-indolizin-5-ones. Eur. J. Med. Chem. 1987, 22, 505–510. [Google Scholar] [CrossRef]
- Danac, R.; Mangalagiu, I.I. Antimycobacterial activity of nitrogen heterocycles derivatives: Bipyridine derivatives. Part III. Eur. J. Med. Chem. 2014, 74, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekharappa, S.; Venugopala, K.N.; Venugopala, R.; Padmashali, B. Qualitative anti-tubercular activity of synthetic ethyl 7-acetyl2-substituted-3-(4-substituted benzoyl) indolizine-1-carboxylate analogues. J. Appl. Pharm. Sci. 2019, 9, 124–128. [Google Scholar]
- Olaru, A.M.; Vasilache, V.; Danac, R.; Mangalagiu, I.I. Antimycobacterial activity of nitrogen heterocycles derivatives: 7-(pyridine-4-yl)-indolizine derivatives. Part VII(8-12). J. Enzyme Inhib. Med. Chem. 2017, 32, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Al-Attraqchi, O.H.; Tratrat, C.; Nayak, S.K.; Morsy, M.A.; Aldhubiab, B.E.; Attimarad, M.; Nair, A.B.; Sreeharsha, N.; Venugopala, R.; et al. Novel series of methyl 3-(substituted benzoyl)-7-substituted-2-phenylindolizine-1-carboxylates as promising anti-inflammatory agents: Molecular modeling studies. Biomolecules 2019, 9, 661. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Albericio, F.; Coovadia, Y.M.; Kruger, H.G.; Glenn, E.M.; Pillay, M.; Govender, T. Total synthesis of a depsidomycin analogue by convergent solid phase peptide synthesis and macrolactonization strategy for anti-tubercular activity. J. Pep. Sci. 2011, 17, 683–689. [Google Scholar]
- Venugopala, K.N.; Nayak, S.K.; Pillay, M.; Prasanna, R.; Coovadia, Y.M.; Odhav, B. Synthesis and antitubercular activity of 2-(substituted phenyl/benzyl-amino)-6-(4-chlorophenyl)-5-(methoxycarbonyl)-4-methyl-3,6-dihydropyrimidin-1-ium chlorides. Chem. Biol. Drug Des. 2013, 81, 219–227. [Google Scholar]
- Venugopala, K.N.; Dharma Rao, G.B.; Bhandary, S.; Pillay, M.; Chopra, D.; Aldhubiab, B.E.; Attimarad, M.; Alwassil, O.I.; Harsha, S.; Mlisana, K. Design, synthesis, and characterization of (1-(4-aryl)-1H-1,2,3-triazol-4-yl)methyl, substituted phenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates against Mycobacterium tuberculosis. Drug Des. Dev. Ther. 2016, 10, 2681–2690. [Google Scholar] [CrossRef]
- Khedr, M.A.; Pillay, M.; Chandrashekharappa, S.; Chopra, D.; Aldhubiab, B.E.; Attimarad, M.; Alwassil, O.I.; Mlisana, K.; Odhav, B.; Venugopala, K.N. Molecular modeling studies and anti-TB activity of trisubstituted indolizine analogues; molecular docking and dynamic inputs. J. Biomol. Struct. Dyn. 2018, 36, 2163–2178. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Chandrashekharappa, S.; Pillay, M.; Bhandary, S.; Kandeel, M.; Mahomoodally, F.M.; Morsy, M.A.; Chopra, D.; Aldhubiab, B.E.; Attimarad, M.; et al. Synthesis and structural elucidation of novel benzothiazole derivatives as anti-tubercular agents: In-silico screening for possible target identification. Med. Chem. 2019, 15, 311–326. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Chandrashekharappa, S.; Pillay, M.; Abdallah, H.H.; Mahomoodally, F.M.; Bhandary, S.; Chopra, D.; Attimarad, M.; Aldhubiab, B.E.; Nair, A.B.; et al. Computational, crystallographic studies, cytotoxicity and anti-tubercular activity of substituted 7-methoxy-indolizine analogues. PLoS ONE 2019, 14, e0217270. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Khedr, M.A.; Pillay, M.; Nayak, S.K.; Chandrashekharappa, S.; Aldhubiab, B.E.; Harsha, S.; Attimard, M.; Odhav, B. Benzothiazole analogs as potential anti-TB agents: Computational input and molecular dynamics. J. Biomol. Struct. Dyn. 2019, 37, 1830–1842. [Google Scholar] [CrossRef] [PubMed]
- Taban, I.M.; Elshihawy, H.; Torun, B.; Zucchini, B.; Williamson, C.J.; Altuwairigi, D.; Ngu, A.S.T.; McLean, K.J.; Levy, C.W.; Sood, S.; et al. Novel Aryl Substituted Pyrazoles as Small Molecule Inhibitors of Cytochrome P450 CYP121A1: Synthesis and Antimycobacterial Evaluation. J. Med. Chem. 2017, 60, 10257–10267. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.A.; McLean, K.J.; Surade, S.; Yang, Y.Q.; Leys, D.; Ciulli, A.; Munro, A.W.; Abell, C. Application of fragment screening and merging to the discovery of inhibitors of the Mycobacterium tuberculosis cytochrome P450 CYP121. Angew. Chem. Int. Ed. 2012, 51, 9311–9316. [Google Scholar] [CrossRef] [PubMed]
- Hameed, S.P.; Solapure, S.; Mukherjee, K.; Nandi, V.; Waterson, D.; Shandil, R.; Balganesh, M.; Sambandamurthy, V.K.; Raichurkar, A.K.; Deshpande, A.; et al. Optimization of pyrrolamides as mycobacterial GyrB ATPase inhibitors: Structure-activity relationship and in vivo efficacy in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 61–70. [Google Scholar]
- Martin, A.; Morcillo, N.; Lemus, D.; Montoro, E.; Telles, M.A.; Simboli, N.; Pontino, M.; Porras, T.; Leon, C.; Velasco, M.; et al. Multicenter study of MTT and resazurin assays for testing susceptibility to first-line anti-tuberculosis drugs. Int. J. Tuberc. Lung Dis. 2005, 9, 901–906. [Google Scholar]
- Mossman, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Mol Formula (Mol Mass) | R1 | R2 | R3 | Yield (%) a | m.p. (°C) | cLogP b |
|---|---|---|---|---|---|---|---|
| 4 | C20H16NO4Br (413) | CHO | CH3 | Br | 86 | 105–106 | 5.2033 |
| 5 | C24H18NO3F (387) | CH3 | C6H5 | F | 88 | 167–168 | 6.3602 |
| Entry | R1 | R2 | R3 | MIC (µg/mL) | |
|---|---|---|---|---|---|
| H37Rv | MDR–MTB b | ||||
| 1a a | CH3 | H | H | >32 | - |
| 1b a | CH3 | H | F | >32 | - |
| 1c a | CH3 | H | Cl | >32 | - |
| 1d a | CH3 | H | Br | >32 | - |
| 1e a | CH3 | H | CN | >32 | - |
| 2a a | CH3 | CH3 | H | >32 | - |
| 2b a | CH3 | CH3 | F | >32 | - |
| 2c a | CH3 | CH3 | Cl | 16 ± 0.02 c,e | >32 |
| 2d a | CH3 | CH3 | Br | 16 ± 0.02 c,e | 32 ± 0.02 c,e |
| 2e a | CH3 | CH3 | CN | 16 ± 0.02 d,e | 32 ± 0.02 c,d |
| 3a a | CH3 | CH2CH3 | H | 16 ± 0.02 c,d | >32 |
| 3b a | CH3 | CH2CH3 | F | >32 | - |
| 3c a | CH3 | CH2CH3 | Cl | >32 | - |
| 3d a | CH3 | CH2CH3 | Br | >32 | - |
| 3e a | CH3 | CH2CH3 | CN | >32 | - |
| 4 | CHO | CH3 | Br | 4 ± 0.02 c,e | 32 ± 0.02 c,e |
| 5 | CH3 | C6H5 | F | 32 ± 0.02 c,d | >32 |
| Entry | CYP 121A1 (PDB 5OP9) | Malate Synthase (PDB 5CBB) | DNA GyrB ATPase (PDB 4B6C) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CDocker E. (Kcal/mol) | Residue Interactions | CDocker E. (Kcal/mol) | Residue Interactions | CDocker E. (Kcal/mol) | Residue Interactions | ||||
| H-Bond (Dist. Å, atom) | Pi-Bond | H-Bond (Dist. Å, atom) | Pi-Bond | H-Bond (Dist. Å, atom) | Pi-Bond | ||||
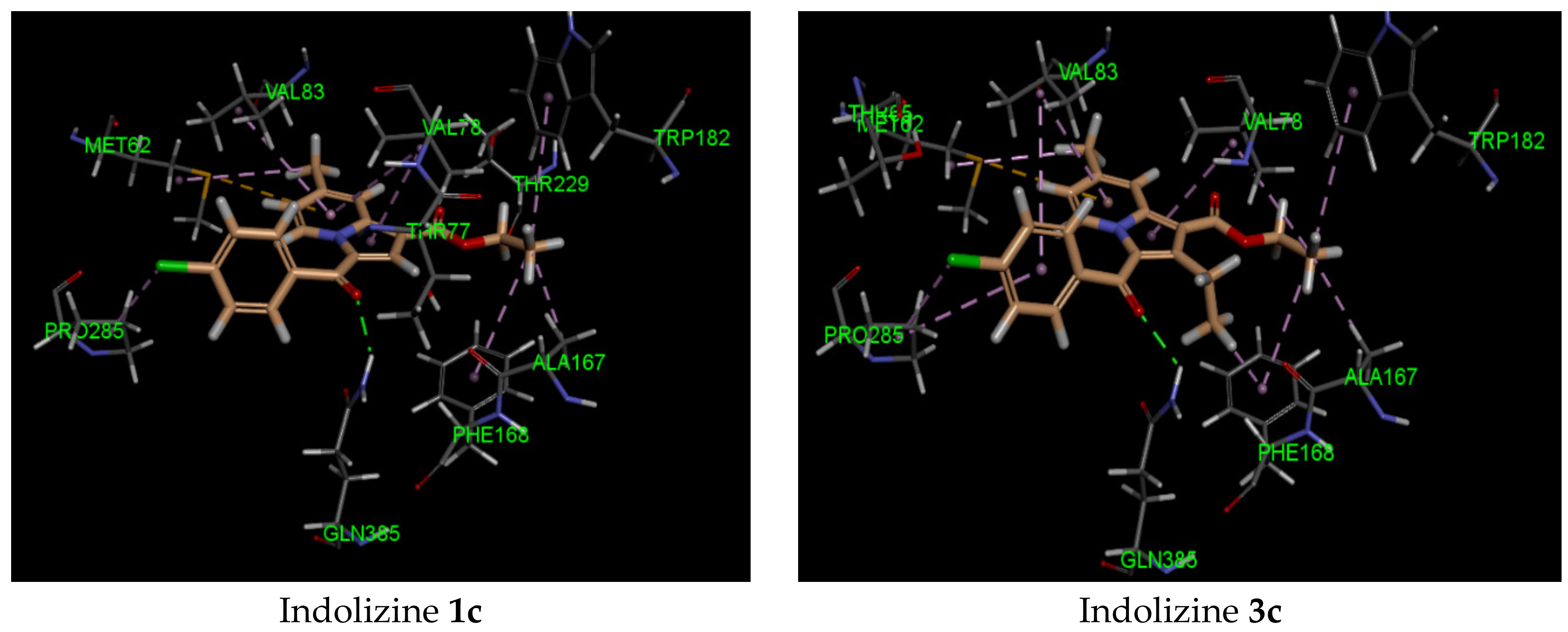
| 2c | −42 | Gln 385 (2.07, CO benzoyl) | Phe168 | −44 | Arg 339 (2.72, CO benzoyl) Phe 460 (1.90, CO ester) Leu 461 (2.57, CO ester) | Try 541 | −34 | Gln 102 (2.27, CO benzoyl) | Glu 56 |
| 2d | −42 | Gln 385 (1.90, CO benzoyl) | Phe168 | −45 | Arg 339 (2.71, CO benzoyl) Phe 460 (1.99, CO ester) | Try 541 | −33 | Gln 102 (2.32, CO benzoyl) | Glu 56 |
| 2e | −41 | Gln 385 (1.98, CO benzoyl) | Phe168 | −41 | Phe 460 (2.48, CO ester) Asp 633 (1.83, CO benzoyl) | Try 541 | −37 | Gln 102 (2.16, CO ester) | Arg 82 |
| 3a | −38 | Gln 385 (2.27, CO benzoyl) | Phe168 | −31 | Arg 339 (2.36, CO benzoyl) | Asp 462 | −34 | Gln 102 (2.21, CO benzoyl) | Arg 82 |
| 3b | −41 | His 343 (2.28, F) | Phe168 | −36 | Arg 339 (2.62, CO benzoyl) Phe 460 (2.31, CO ester) Leu 461 (2.68, CO ester) Asp 462 (2.93, CO ester) | Try 541 | −33 | Gln 102 (2.45, CO benzoyl) | Arg 82 |
| 3c | −41 | Gln 385 (2.21, CO benzoyl) | Met 62 | −40 | Arg 339 (2.74, CO benzoyl) Phe 460 (1.94, CO ester) Leu 461 (2.62, CO ester) | Try 541 | −34 | Gln 102 (2.45, CO benzoyl) | Glu 56 |
| 3d | −41 | Gln 385 (2.24, CO benzoyl) | Met 62 | −37 | Arg 339 (2.56, CO benzoyl) Phe 460 (2.25, CO ester)) Leu 461 (2.64, CO ester) Asp 462 (2.94, CO ester) | Try 541 | −33 | Gln 102 (2.45, CO benzoyl) | Arg 82 |
| 3e | −39 | Gln 385 (2.94, CN) | - | −36 | Arg 339 (2.55, CO benzoyl) Phe 460 (2.14, CO ester) Leu 461 (2.44, CO ester) Asp 462 (2.92, CO ester) | Try 541 | −33 | Gln 102 (2.26, CO benzoyl) | Arg 82 |
| 4 | −44 | Gln 385 Asn 85 (2.55, CO formyl) | Phe168 | −50 | Arg 339 (2.72, CO benzoyl) Phe 460 (1.91, CO ester) Leu 461 (2.57, CO ester) Arg 463 (2.34, CO formyl) | Try 541 | −45 | Gly 83 (2.13, CO formyl)Gln 102 (2.70, CO benzoyl) Arg 141 (3.04, CO ester) | Glu 56 |
| 5 | −40 | Gln 385 (2.02, CO benzoyl) | Phe168 | −18 | Arg 339 (2.32, CO benzoyl) Arg 463 (2.66, F) Asp 633 (2.37, O ester) | Asp 462 | −27 | - | Arg 82 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venugopala, K.N.; Tratrat, C.; Pillay, M.; Mahomoodally, F.M.; Bhandary, S.; Chopra, D.; Morsy, M.A.; Haroun, M.; Aldhubiab, B.E.; Attimarad, M.; et al. Anti-Tubercular Activity of Substituted 7-Methyl and 7-Formylindolizines and In Silico Study for Prospective Molecular Target Identification. Antibiotics 2019, 8, 247. https://doi.org/10.3390/antibiotics8040247
Venugopala KN, Tratrat C, Pillay M, Mahomoodally FM, Bhandary S, Chopra D, Morsy MA, Haroun M, Aldhubiab BE, Attimarad M, et al. Anti-Tubercular Activity of Substituted 7-Methyl and 7-Formylindolizines and In Silico Study for Prospective Molecular Target Identification. Antibiotics. 2019; 8(4):247. https://doi.org/10.3390/antibiotics8040247
Chicago/Turabian StyleVenugopala, Katharigatta N., Christophe Tratrat, Melendhran Pillay, Fawzi M. Mahomoodally, Subhrajyoti Bhandary, Deepak Chopra, Mohamed A. Morsy, Michelyne Haroun, Bandar E. Aldhubiab, Mahesh Attimarad, and et al. 2019. "Anti-Tubercular Activity of Substituted 7-Methyl and 7-Formylindolizines and In Silico Study for Prospective Molecular Target Identification" Antibiotics 8, no. 4: 247. https://doi.org/10.3390/antibiotics8040247
APA StyleVenugopala, K. N., Tratrat, C., Pillay, M., Mahomoodally, F. M., Bhandary, S., Chopra, D., Morsy, M. A., Haroun, M., Aldhubiab, B. E., Attimarad, M., Nair, A. B., Sreeharsha, N., Venugopala, R., Chandrashekharappa, S., Alwassil, O. I., & Odhav, B. (2019). Anti-Tubercular Activity of Substituted 7-Methyl and 7-Formylindolizines and In Silico Study for Prospective Molecular Target Identification. Antibiotics, 8(4), 247. https://doi.org/10.3390/antibiotics8040247