6.2. Chemical synthesis of the Intermediates and Final Compounds
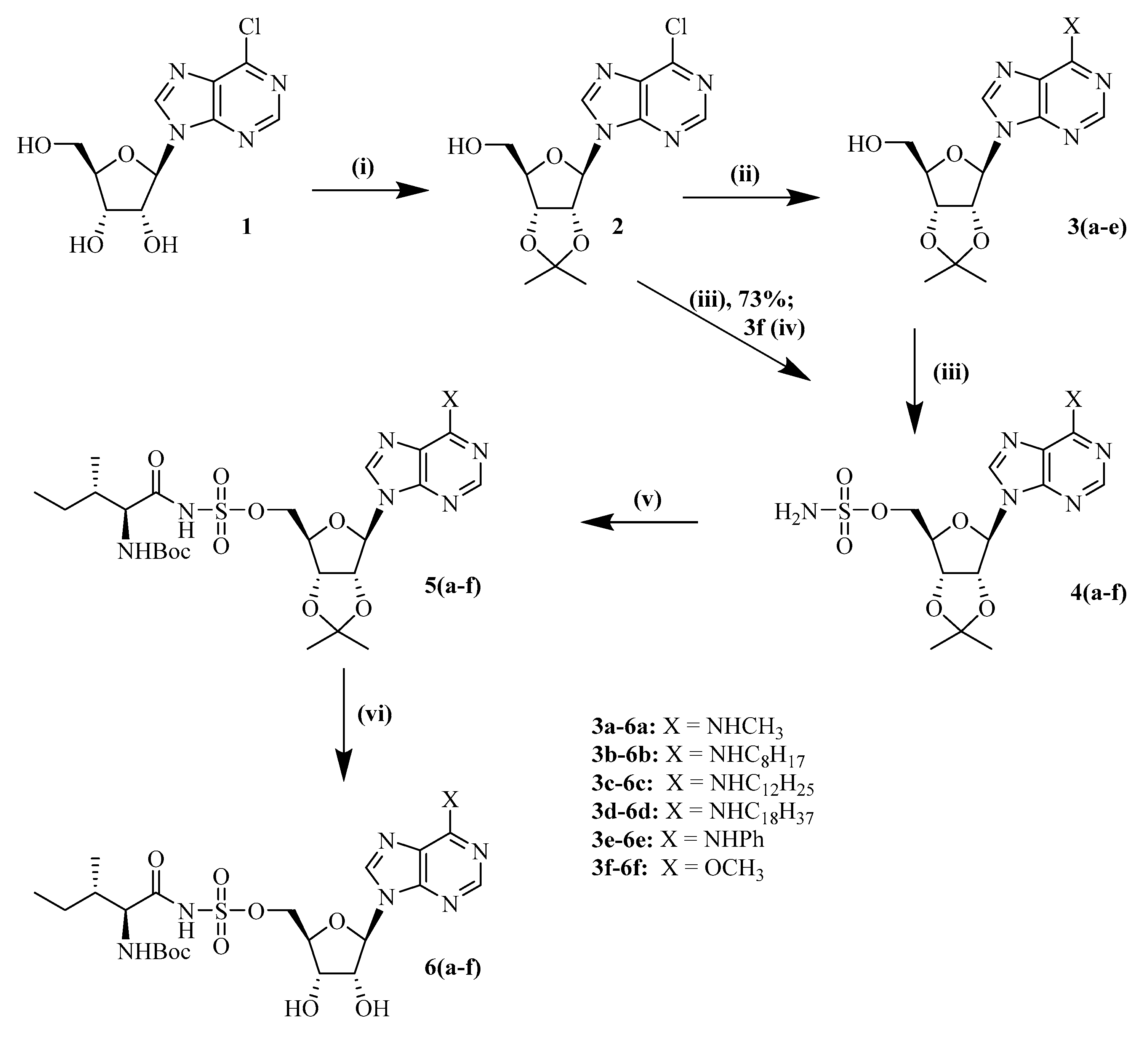
6.2.1. 2,3′-isopropylidene 6-chloropurine Riboside (2)
As reported in literature [
43], compound
1 (6-chloropurine riboside, 8 g, 0.029 mol) was stirred with a mixture of dimethoxypropane (DMP) (34.31 mL, 0.29 mol) and paratoluenesulfonic acid (PTSA) (2.66 g, 0.014 mol) in dry acetone (80 mL) at room temperature for overnight. Thin layer chromatography TLC (developed at 10% methanol in dichloromethane (DCM)) was used to monitor the reaction. Saturated sodium bicarbonate was added to quench the reaction. Afterwards, the solvent was evaporated under reduced pressure. The crude product was dissolved in DCM, and the organic layer was washed two times with saturated sodium bicarbonate and one time with brine. Column chromatography was performed with a gradient of 5% methanol in DCM to obtain the desired compound
2 at 86% yield.
1H NMR (300 MHz, CDCl
3) δ 1.37 (s, 3H, C–CH
3), 1.64 (s, 3H, C–CH
3), 3.81 (m, 1H, H-5′a), 3.97 (m, 1H, H-5′b), 4.54 (m, 1H, H-4′), 5.00 (dd,
J = 10.0, 2.6 Hz, 1H, H-3′), 5.10 (dd,
J = 6.0, 1.6 Hz, 1H, H-2′), 5.19 (dd,
J = 6.0, 4.5 Hz, 1H, H-1′), 6.00 (d,
J = 4.5 Hz, 1H, 5′OH ), 8.28 (s, 1H, H-2), 8.75 (s, 1H, H-8).
13C NMR (75 MHz, CDCl
3) δ 25.5 (C–CH
3), 27.8 (C–CH
3), 63.4 (C-5′), 81.8 (C-4′), 83.7 (C-3′), 86.8 (C-2′), 94.2 (C-1′), 114.8 (C–(CH3)
2), 145.0 (C-5), 152.0 (C-4), 152.5 (C-2). HRMS [ESI]
m/z: calcd. for C
13H
16ClN
4O
4 ([M+H]
+) 327.0854, found: 327.0835.
6.2.2. 2′,3′-isopropylidene-N6-(methyl)-adenosine (3a)
Compound 2 (200 mg, 0.613 mmol) was dissolved in 6 mL of DMSO in a microwave vial. Methylamine (40% solution in water) (1.5 equiv, 0.102 mL) and DIPEA (3 equiv, 0.32 mL, 1.84 mmol) were added, and the mixture was put in a microwave for 45 min, at 110 °C utilizing 150W power. The reaction was monitored by doing TLC in an acetone/hexane gradient (50:50 v/v). To perform the TLC, take 100 microliters of the crude reaction mixture and add 500 microliters of ethyl acetate and 500 microliters of water to an Eppendorf tube. Extract the organic layer and use this for TLC. After complete conversion, 350 mL of DCM was added, and the organic layer was washed with 100 mL potassium hydrogen sulfate (10% m/v) and 100 mL of brine. The organic layer was evaporated under vacuum, and column chromatography was performed with a gradient of 40% acetone in hexane to obtain 3a. Yield: 97% (0.19 g). 1H NMR (300 MHz, CDCl3) δ 1.31 (s, 3H, C–CH3), 1.58 (s, 3H, C–CH3), 3.10 (s, 3H, N6–CH3), 3.74 (d, J = 12.8 Hz, 1H, H-5′a), 3.91 (dd, J = 12.9, 1.6 Hz, 1H, H-5′b), 4.48 (s, 1H, H-4′), 5.06 (dd, J = 5.7, 1.3 Hz, 1H, H-3′), 5.17 (t, 1H, H-2′), 5.81 (d, J = 4.8 Hz, 1H, H-1′), 6.53 (s, 1H, N6-H), 6.75 (bs, 1H, 5′OH), 7.74 (s, 1H, H-2), 8.28 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 25.5 (C-CH3), 27.9 (C–CH3), 29.6 (N6-CH3), 63.6 (C-5′), 82.0 (C-4′), 83.4 (C-3′), 86.5 (C-2′), 94.4 (C-1′), 114.2 (C-5), 121.6 (C–(CH3)2), 139.6 (C-8), 153.1 (C-2), 156.1 (C-6). HRMS [ESI] m/z: calcd. for C14H20N5O4 ([M+H]+) 322.1510, found: 322.1512.
6.2.3. 2′,3′-isopropylidene-N6-(octyl)-adenosine (3b)
Compound 2 (200 mg, 0.613 mmol) was added to a microwave vial containing octylamine (0.152 mL, 0.92 mmol) and DIPEA (0.32 mL, 1.84 mmol) in DMSO (6 mL). The reaction condition, TLC and purification were similar as 3a. Yield: 90% (0.23 g). 1H NMR (300 MHz, CDCl3) δ 1.21–1.64 (m, 23H, C–(CH3)2, H3C–(CH2)7), 3.56 (bs, 2H, N6–CH2), 3.74 (dd, J = 12.7, 1.6 Hz, 1H, H-5′a), 4.90–3.95 (m, 1H, H-5′b), 5.06 (dd, J = 5.9, 1.2 Hz, 1H, H-4′), 5.17 (t, 1H, H-3′), 5.81 (d, J = 4.8 Hz, 1H, H-2′), 6.11 (t, 1H, H-1′), 6.78 (d, J = 11.0 Hz, 1H, 5′OH), 7.73 (s, 1H, H-2), 8.28 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 14.1- 40.9 (C–(CH3)2, H3C–(CH2)7), 63.6 (C-5′), 82.0 (C-4′), 83.4 (C-3′), 86.5 (C-2′), 94.5 (C-1′), 114.2 (C-5), 121.4 (C–(CH3)2), 139.6 (C-8), 153.1 (C-2), 155.6 (C-6). HRMS [ESI] m/z: calcd. for C21H34N5O4 ([M+H]+) 420.2605, found: 420.2597.
6.2.4. 2′,3′-isopropylidene-N6-(dodeceyl)-adenosine (3c)
Compound 2 (200 mg, 0.613 mmol) was added to a microwave vial containing dodecylamine (170.43 mg, 0.92 mmol) and DIPEA (0.32 mL, 1.84 mmol) in DMSO (6 mL). The reaction condition, TLC and purification were similar as 3a. Yield: 71% (0.21 g). 1H NMR (300 MHz, CDCl3) δ 1.24–1.70 (m, 31H, C–(CH3)2, H3C–(CH2)11), 3.2 (m, 1H, N6-H), 3.7 (d, J = 47.9 Hz, 3H, H-5′, H-4′, H-3′), 5.2 (d, J = 3.6 Hz, 2H, H-2′, H-1′), 5.78–5.83 (m, 1H, 5′OH), 7.90 (d, 1H, H-2), 8.39 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 14.2–46.1 (m, C–(CH3)2, H3C–(CH2)11), 63.6 (C-5′), 81.9 (C-4′), 83.9 (C-3′), 86.7 (C-2′), 95.3 (C-1′), 114.3 (C-5), 121.4 (C–(CH3)2), 139.3 (C-8), 153.6 (C-2). HRMS [ESI] m/z: calcd. for C25H42N5O4 ([M+H]+) 476.3231, found: 476.3227.
6.2.5. 2′,3′-isopropylidene-N6-(octadecyl)-adenosine (3d)
Compound 2 (200 mg, 0.613 mmol) was added to a microwave vial containing octadecylamine (248 mg, 0.92 mmol) and DIPEA (0.32 mL, 1.84 mmol) in DMSO (6 mL). The reaction condition, TLC and purification were similar as 3a. Yield: 57% (0.20 g). HRMS [ESI] m/z: calcd. for C31H54N5O4 ([M+H]+) 560.4170, found: 560.4174.
6.2.6. 2′,3′-isopropylidene-N6-(phenyl)-adenosine (3e)
Compound 2 (200 mg, 0.613 mmol) was added to a microwave vial containing aniline (0.083 mL, 0.92 mmol) and DIPEA (0.32 mL, 1.84 mmol) in DMSO (6 mL). The reaction condition, TLC and purification were similar as 3a. Yield: 62% (0.15 g). 1H NMR (300 MHz, CDCl3) δ 1.3–1.6 (m, 6H, C–(CH3)2), 3.7–3.9 (m, 1H, N6-H), 3.9–4.0 (m, 2H, H-5′a, H-5′b), 4.5 (m, 1H, C-4′), 4.9–5.1 (m, 1H, C-3′), 5.2 (m, 1H, C-2′), 5.9 (d, J = 4.8 Hz, 1H, C-1′), 6.0 (d, J = 4.2 Hz, 1H, 5′OH), 6.4–7.6 (m, 5H, N6–C6H5), 8.4 (s, 1H, H-2), 8.7 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 25.5 (C–(CH3)2), 27.9 (C–(CH3)2), 63.6 (C-5′), 81.8 (C-4′), 82.0 (C-3′), 83.5 (C-2′), 94.5 (C-1′), 114.3 (C-5), 114.7 (p-C-aniline), 121.0 (m-C-aniline), 122.0 (m-C-aniline), 124.3 (C-(CH3)2), 129.3 (o-C-aniline), 133.2 (o-C-aniline), 138.5 (N6-C-aniline), 140.5 (C-8), 145.0 (C-6), 148.3 (C-4), 152.3 (C-2). HRMS [ESI] m/z: calcd. for C19H22N5O4 ([M+H]+) 384.1661, found: 384.1664.
6.2.7. 2′,3′-isopropylidene-5′-O-sulfamoyl-6-chloropurine riboside20 (3f)
Formic acid (1.73 mL, 46 mmol) was added dropwise to CSI (4.0 mL, 46 mmol) in an ice bath for 10 min. After several minutes, the formation of a white solid was observed. Afterwards, acetonitrile (20 mL) was added and the solution was stirred for four hours at room temperature. Following stirring, the solution was added to compound 2 (5 g, 15 mmol) in DMA (20 mL) and was reacted overnight at room temperature. Column chromatography was performed with a gradient of 15–25% acetone in hexane to obtain desired compound at 73% (4.53 g) yield.1H NMR (300 MHz, CDCl3) δ 1.36 (s, 3H, C–CH3), 1.60 (s, 3H, C–CH3), 4.29–4.44 (m, 2H, C’5–H2), 4.60 (m, 1H, H-4′), 5.06 (dd, J = 6.2, 2.7 Hz, 1H, H-3′), 5.34 (dd, J = 6.2, 2.6 Hz, 1H, H-2′), 6.25 (d, J = 2.6 Hz, 1H, H-1′), 6.31 (d, J = 5.1 Hz, 2H, SO2-NH2), 8.39 (s, 1H, H-2), 8.73 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 25.4 (C–CH3), 27.2 (C-CH3), 69.4 (C-5′), 81.3 (C-4′), 84.4 (C-3′), 84.7 (C-2′), 91.5 (C-1′), 115.1 (C–(CH3)2), 132.0 (C-5), 144.4 (C-8), 151.2 (C-6), 152.4 (C-2). HRMS [ESI] m/z: calcd. for C13H17ClN5O6S ([M+H]+) 406.0582, found: 406.0574.
6.2.8. 2′,3′-isopropylidene-5′-O-sulfamoyl-N6-methyl-adenosine (4a)
Formic acid (0.62 mL) was added dropwise to CSI (1.42 mL) in an RBF kept in an ice bath for 10 min. After a few minutes, the formation of a white solid was observed. Afterwards, acetonitrile (20 mL) was added, and the solution was stirred for four hours at room temperature. The obtained solution of sulfamoyl chloride is used in the next reaction as such. Sulfamoyl chloride solution (2.65 mL, 1.95 mmol) was added to a solution of compound 3a (208 mg, 0.65 mmol) in DMA (10 mL) and was stirred overnight. TLC (50:50 acetone/hexane) was used to monitor the reaction. After overnight stirring, the solvent was evaporated under reduced pressure. Column chromatography was performed with a gradient of 40–50% acetone in hexane to obtain 4a. Yield: 25% (0.065 g). 1H NMR (300 MHz, CDCl3) δ 1.3 (s, 3H, C–CH3), 1.6 (s, 3H, C–CH3), 3.0 (d, J = 7.0 Hz, 3H, NH-CH3), 4.4 (s, 2H, H-5′a, H-5′b), 4.5 (s, 1H, H-4′), 5.0 (d, J = 6.8 Hz, 1H, H-3′), 5.3 (d, J = 6.2 Hz, 1H, H-2′), 6.1 (d, J = 2.5 Hz, 1H, H-1′), 6.4 (s, 1H, N6-H), 7.9 (s, 1H, H-2), 8.3 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 25.5 (C–CH3), 27.3 (C–CH3), 29.6 (N6–CH3), 69.6 (C-5′), 81.4 (C-4′), 84.4 (C-3′), 84.6 (C-2′), 90.9 (C-1′), 115.0 (C-5), 120.0 (C–(CH3)2), 139.1 (C-8), 153.6 (C-2), 155.5 (C-6). HRMS [ESI] m/z: calcd. for C14H21N6O6S ([M+H]+) 401.1238, found: 401.1236.
6.2.9. 2′,3′-isopropylidene-5′-O-sulfamoyl-N6-octyl-adenosine (4b)
Sulfamoyl chloride solution (2.8 mL, 2.04 mmol) was added to compound 3b (284 mg, 0.68 mmol, 0.33 equivalent) in DMA (10 mL). Similar reaction conditions and purification methods as described for the synthesis of compound 4a were used to obtain 4b. Yield: 97% (0.33 g). 1H NMR (300 MHz, CDCl3) δ 0.7–3.1 (m, 26H, C–(CH3)2, H3C–(CH2)7, NH–CH3), 3.5 (m, 2H, N6-H, H-5′a), 4.2–4.6 (m, 3H, SO2-NH2, H-5′b), 5.1 (dd, J = 6.3, 3.0 Hz, 1H, H-4′), 5.3 (dd, J = 6.5, 2.5 Hz, 1H, H-3′), 5.9–6.2 (m, 2H, H-2′, H-1′), 7.9 (d, J = 2.4 Hz, 1H, H-2), 8.3 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 14.1–40.9 (C–(CH3)2, H3C–(CH2)7), 69.2 (C-5′), 81.2 (C-4′), 84.2 (C-3′), 84.4 (C-2′), 90.8 (C-1′), 114.8 (C-5), 119.9 (C-(CH3)2), 138.8 (C-8), 153.6 (C-2), 155.0 (C-6). HRMS [ESI] m/z: calcd. for C21H35N6O6S ([M+H]+) 499.2333, found: 499.2329.
6.2.10. 2′,3′-isopropylidene-5′-O-sulfamoyl-N6-dodecyl-adenosine (4c)
Sulfamoyl chloride solution (2.4 mL, 1.76 mmol) was added to compound 3c (210 mg, 0.44 mmol) in DMA (10 mL). Similar reaction conditions and purification methods as described for the synthesis of compound 4a were used to obtain 4c. Yield: 69% (0.17 g). 1H NMR (300 MHz, CDCl3) δ 0.7–3.6 (m, 33H, C–(CH3)2, H3C–(CH2)11, N6-H), 4.3–4.6 (m, 2H, SO2–NH2), 5.1 (m, 3H, H-5′a, H-5′b, H-4′), 5.4 (m, 2H, H-3′, H-2′), 5.9–6.1 (m, 1H, H-1′), 7.9 (s, 1H, H-2), 8.3 (s, 1H, H-8). HRMS [ESI] m/z: calcd. for C25H43N6O6S ([M+H]+) 555.2959, found: 555.2958.
6.2.11. 2′,3′-isopropylidene-5′-O-sulfamoyl-N6-octadecyl-adenosine (4d)
Sulfamoyl chloride solution (1.95 mL, 1.44 mmol) was added to compound 3d (200 mg, 0.36 mmol) in DMA (10 mL). Similar reaction conditions and purification methods as described for the synthesis of compound 4a were used to obtain 4d. Yield: 56% (0.13 g). 1H NMR (300 MHz, CDCl3) δ 0.9–1.3 (m, 44H, C–(CH3)2, H3C–(CH2)17, N6-H), 4.3–4.6 (m, 3H, SO2–NH2, H-5′a), 5.1 (dd, J = 6.4, 3.0 Hz, 1H, H-5′b), 5.3 (dd, J = 6.3, 2.6 Hz, 1H, H-4′), 5.8–6.2 (m, 3H, H-3′, H-2′, H-1′), 7.9 (s, 1H, H-2), 8.3 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 14.4–32.2 (C–(CH3)2, H3C–(CH2)17), 69.7 (C-5′), 81.3 (C-4′), 84.2 (C-3′), 84.6 (C-2′), 91.1 (C-1′), 115.1 (C-(CH3)2), 139.2 (C-8), 153.8 (C-2). HRMS [ESI] m/z: calcd. for C25H43N6O6S ([M+H]+) 555.2959, found: 555.2958.
6.2.12. 2′,3′-isopropylidene-5′-O-sulfamoyl-N6-phenyl-adenosine (4e)
Sulfamoyl chloride solution (2 mL, 1.47 mmol) was added to compound 3e (188 mg, 0.68 mmol) in DMA (10 mL). Reaction conditions and purification methods were as described for 4a to obtain 4e. Yield: 76% (0.24 g).1H NMR (300 MHz, CDCl3) δ 1.4–1.6 (m, 6H, C-(CH3)2), 2.1–3.0 (m, 3H, N6-H, SO2NH2), 4.4 (m, 2H, H-5′a, H-5′b), 4.5–4.7 (m, 1H, H-4′), 5.1 (m, 1H, H-3′), 5.3–5.4 (m, 1H, H-2′), 6.2 (m, 1H, H-1′), 6.5 (s, 1H, p-CH-aniline), 7.4 (m, 2H, 2x m-CH-aniline), 7.7–7.8 (m, 1H, o-H-aniline), 8.1 (d, J = 3.2 Hz, 1H), 8.4 (d, J = 18.6 Hz, 1H, H-2), 8.7 (d, J = 2.1 Hz, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 25.5 (C–(CH3)2), 27.4 (C–(CH3)2), 69.6 (C-5′), 81.5 (C-4′), 84.6 (C-3′), 84.8 (C-2′), 91.2 (C-1′), 115.0 (C-5), 115.2 (p-C-aniline), 120.9 (m-C-aniline), 124.0 (m-C-aniline), 129.2 (o-C-aniline), 138.8 (N6-C-aniline), 139.9 (o-C-aniline), 144.5 (C-8), 149.2 (C-6), 151.3 (C-4), 152.5 (C-2). HRMS [ESI] m/z: calcd. for C19H22N6O6S ([M+H]+) 463.1394, found: 463.1385.
6.2.13. 2′, 3′-isopropylidene-5′-O-sulfamoyl-6-O-methyl-purine riboside (4f)
A 20% solution of sodium methoxide in methanol (0.27 mL, 0.98 mmol) was added to a cooled solution of 3f in dry methanol. The reaction was performed at 0 °C. After two hours, a few drops of glacial acetic acid were added to quench the reaction. The reaction mixture was evaporated under reduced pressure. Column chromatography was performed with a gradient of 40% acetone in hexane to yield 4f. Yield: 39% (0.15 g). 1H NMR (300 MHz, CDCl3) δ 1.3 (s, 3H, C–CH3), 1.4 (s, 3H, C–CH3), 4.1 (s, 3H, O–CH3), 4.3–4.5 (m, 2H, H-5′a, H-5′b), 4.6 (s, 1H, H-4′), 5.1 (dd, J = 6.4, 2.8 Hz, 1H, H-3′), 5.4 (dd, J = 6.2, 2.4 Hz, 1H, H-2′), 6.2 (d, J = 2.4 Hz, 1H, H-1′), 8.1 (s, 1H, H-2), 8.5 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 25.5 (C-CH3), 27.4 (C–CH3), 54.6 (O-CH3), 69.8 (C-5′), 81.5 (C-4′), 84.5 (C-3′), 84.8 (C-2′), 91.6 (C-1′), 115.2 (C-5), 122.1 (C– (CH3)2), 141.5 (C-8), 151.4 (C-2), 152.8 (C-6).
6.2.14. 2′,3′-isopropylidene-5′-O-(N-(Nα-Boc-L-isoleucyl))-sulfamoyl-N6-methyl-adenosine (5a)
Compound 4a (100 mg, 0.27 mmol) was added to a solution of Boc-Ile-Osu (133 mg, 0.405 mmol) and DBU (0.06 mL, 0.405 mmol) in DMF (12 mL). The solution was stirred overnight at room temperature. TLC was developed with a methanol/ethyl acetate mixture (10:90) to monitor the reaction. After stirring overnight, the solvent was evaporated under reduced pressure. Column chromatography was performed with a mixture of 5% methanol in ethyl acetate to obtain compound 5a. Yield: 85% (0.14 g). 1H NMR (300 MHz, CDCl3) δ 0.8–3.4 (m, 30H, Boc-Ile protons, C-(CH3)2, NH-CH3), 3.9 (s, 1H, N6-H), 4.1 (m, 1H, H-5′a), 4.2 (m, 3H, H-5′b, H-4′, H-3′), 4.5 (s, 1H, H-2′), 6.2 (d, J = 2.5 Hz, 1H, H-1′), 8.3 (d, J = 12.1 Hz, 2H, H-2, H-8). 13C NMR (75 MHz, CDCl3) δ 11.6–62.2 (C-(CH3)2), Boc-Ile carbons), 69.1 (C-5′), 81.5 (C-4′), 85.1 (C-3′), 90.7 (C-1′), 114.3 (C-5), 119.5 (C–(CH3)2), 139.6 (C-8), 148.6 (C-4), 153.7 (C-6), 155.3 (-C(O)-tBu). HRMS [ESI] m/z: calcd. for C25H39N7O9S ([M-H]-) 612.2457, found: 612.2462.
6.2.15. 2′,3′-isopropylidene-5′-O-(N-(Nα-Boc-L-isoleucyl))-sulfamoyl-N6-octyl-adenosine (5b)
Compound 4b (350 mg, 0.763 mmol) was added to a solution of Boc-Ile-Osu (376 mg, 1.145 mmol) and DBU (0.171 mL, 1.145 mmol) in DMF (12 mL). Reaction conditions and purification were similar as described for the synthesis of 5a, except that TLC and column chromatography were performed with a mixture of 30% acetone in hexane, to obtain 5b. Yield: 87% (0.47 g). 1H NMR (300 MHz, CDCl3) δ 0.8 - 3.6 (m, 44H, Boc-Ile protons, C–(CH3)2, NH(CH2)7CH3), 3.7 (d, J = 11.8 Hz, 1H, N6–H), 4.3 (m, 2H, H-5′a, H-5′b), 4.5 (d, J = 5.8 Hz, 1H, H-4′), 4.5 (s, 1H, H-3′), 4.9 (s, 1H, H-2′), 6.2 (s, 1H, H-1′), 8.2 (d, J = 15.9 Hz, 2H, SO2NH, Nα-H), 8.4 (s, 1H, H-2), 8.6 (s, 1H, H-8). HRMS [ESI] m/z: calcd. for C32H53N7O9S ([M-H]-) 710.3524, found: 710.3397.
6.2.16. 2′,3′-isopropylidene-5′-O-(N-(Nα-Boc-L-isoleucyl))-sulfamoyl-N6-dodecyl-adenosine (5c)
Compound 4c (200 mg, 0.36 mmol) was added to a solution of Boc-Ile-OSu (177 mg, 0.54 mmol) and DBU (0.08 mL, 0.54 mmol) in DMF (12 mL). Reaction conditions and purification were similar as described for the synthesis of 5a, except that TLC and column chromatography were carried out with a mixture of 35% acetone in hexane, to obtain 5c. Yield: 51% (0.14 g). 1H NMR (300 MHz, CDCl3) δ 1.4 –4.0 (m, 52H, Boc-Ile protons, C–(CH3)2, NH(CH2)11CH3), 4.2 (s, 1H, N6–H), 4.6 (s, 1H, H-5′a), 4.9 (s, 1H, H-5′b), 5.2 (s, 1H, H-4′), 5.7 (s, 1H, H-3′), 5.8 (s, 1H, H-2′), 8.9 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 11.8–44.7 (C-(CH3)2), Boc-Ile carbons), 68.7 (C-5′), 80.5 (C-4′), 85.1 (C-3′), 85.9 (C-2′), 92.5 (C-1′), 114.3 (C-5), 153.7 (C-6), 155.0 (-C(O)-tBu). HRMS [ESI] m/z: calcd. for C36H61N7O9S ([M-H]-) 766.41784, observed 766.4161.
6.2.17. 2′,3′-isopropylidene-5′-O-(N-(Nα-Boc-L-isoleucyl))-sulfamoyl-N6-octadecyl-adenosine (5d)
Compound 4d (150 mg, 0.234 mmol) was added to a solution of Boc-Ile-Osu (115 mg, 0.35 mmol) and DBU (0.05 mL, 0.35 mmol) in DMF (12 mL). Reaction conditions and were similar as described for 5a, except that TLC and column chromatography were performed with a mixture of 35% acetone in hexane, to obtain 5d. Yield: 43% (0.12 g). 1H NMR (300 MHz, CDCl3) δ 0.8 - 3.6 (m, 52H, Boc-H, C-(CH3)2, NH(CH2)17CH3), 4.0 (s, 1H, N6-H), 4.3 (s, 2H, H-5′a, H-5′b), 4.6 (s, 1H, H-4′), 5.0–5.1 (m, 1H, H-3′), 5.2 (s, 1H, H-2′), 5.4 (s, 1H, H-1′), 6.3 (s, 1H, Nα-H), 6.6 (s, 1H, SO2NH), 8.4 (s, 1H, H-2). HRMS [ESI] m/z: calcd. for C42H73N7O9S ([M-H]-) 850.5117, found: 850.5115.
6.2.18. 2′,3′-isopropylidene-5′-O-(N-(Nα-Boc-L-isoleucyl))-sulfamoyl-N6-phenyl-adenosine (5e)
Compound 4e (250 mg, 0.60 mmol) was added to a solution of Boc-Ile-Osu (295 mg, 0.90 mmol) and DBU (0.134 mL, 0.90 mmol) in DMF (12 mL). Reaction conditions and purification were similar as described for 5a, except that TLC and column chromatography were carried out with a mixture of 35% acetone in hexane to obtain 5e. Yield: 49% (0.20 g). 1H NMR (300 MHz, CDCl3) δ 1.4–2.1 (m, 22H, Boc-Ile-protons, C–(CH3)2), 4.5 (s, 1H, H-Cα), 4.6 (s, 1H, H-5′a), 4.9 (s, 1H, H-5′b), 5.2 (s, 1H, H-4′), 5.5 (d, J = 14.3 Hz, 1H, H-3′), 5.7 (s, 1H, H-2′), 6.9 (d, J = 7.7 Hz, 1H, H-1′), 7.7 (m, 1H, p-CH-aniline), 7.8 –8.0 (m, 2H, m-CH-aniline), 8.3 (d, J = 8.0 Hz, 2H, o-CH-aniline), 9.0 (s, 1H, H-2), 9.1 (s, 1H, H-8). 13C NMR (75 MHz, CDCl3) δ 11.8–62.2 (C–(CH3)2), Boc-Ile carbons), 69.2 (C-5′), 80.5 (C-4′), 85.3 (C-3′), 85.8 (C-2′), 91.3 (C-1′), 114.5 (C-5), 120.3 (p-C-aniline), 121.2 (m-C-aniline), 121.8 (m-C-aniline), 123.2 (C–(CH3)2), 123.8 (o-C-aniline), 126.9 (o-C-aniline), 139.0 (N6-C-aniline), 139.5 (C-8), 149.5 (C-6), 152.3 (C-4), 152.8 (C-2), 156.7 (-C(O)-tBu). HRMS [ESI] m/z: calcd. for C30H41N7O9S ([M-H]-) 674.2613, found: 674.2592.
6.2.19. Synthesis of 2′, 3′-isopropylidene-5′-O-sulfamoyl-(N-(Nα-Boc-L-isoleucyl)-6-O-methyl-purine riboside (5f)
Compound 4f (150 mg, 0.37 mmol) was added to a solution of Boc-Ile-Osu (184 mg, 0.54 mmol) and DBU (0.08 mL, 0.54 mmol) in DMF (12 mL). The reaction was stirred overnight at room temperature. The solvent was evaporated under reduced pressure. Column chromatography of the obtained residue was carried out with a gradient of 10–20% acetone in hexane to yield 5f. Yield: 92% (0.21 g).
6.2.20. Synthesis of 5′-O-(N-L-isoleucyl)-sulfamoyl-N6-methyl-adenosine (6a)
Compound 5a (120 mg, 0.20 mmol) was dissolved in a mixture of TFA, water and DCM (50:25:25 v/v/v) and stirred for 3 h at room temperature. TLC was performed in a methanol/DCM mixture (10:90 v/v) to check the reaction progression. After 3h, the mixture was evaporated at reduced pressure at 25 °C. RP-HPLC was performed with a C18 column with gradient elution using water/acetonitrile as the mobile phase to purify and obtain 6a. Yield: 37% (0.035 g). 1H NMR (300 MHz, D2O) δ 0.90–1.02 (m, 6H, Ile-δ-CH3, Ile-γ-CH3), 1.20-1.26 (m, 1H, Ile-γ-CH2 Ha), 1.56–1.61 (m, 1H, Ile-γ-CH2 Hb), 1.95-1.99 (m, 1H, Ile-β-CH), 3.11 (bs, 3H, N–CH3), 3.56 (d, J = 4.1 Hz, 1H, Ile-α-CH), 4.29–4.39 (m, 4H, H-5′, H-5″, H-4′, H-3′), 4.62 (t, 1H, J = 5.0 Hz, H-2′), 6.06 (s, 1H, J = 5.3 Hz, H-1′), 8.24 (s, 1H, H-2), 8.45 (s, 1H, H-8). 13C NMR (75 MHz, D2O) δ 12.1 (Ile- δ -CH3), 15.5 (Ile-γ-CH3), 25.7 (Ile- δ-CH2), 38.2 (Ile-β-CH), 61.5 (Ile-α-CH), 69.0 (C-5′), 72.0 (C-4′), 76.2 (C-3′), 84.2 (C-2′), 89.4 (C-1′), 120.6 (C-5), 140.6 (C-8), 153.9 (C-2), 156.7 (C-6), 174.9 (-C(C=O, Ile). HRMS [ESI] m/z: calcd. for C17H27N7O7S ([M-H]-) 472.1620, found: 472.1633.
6.2.21. Synthesis of 5′-O-(N-L-isoleucyl)-sulfamoyl-N6-octyl-adenosine (6b)
Compound 5b (300 mg, 0.42 mmol) was treated like compound 5a regarding reaction condition and purification to obtain 6b. Yield: 21% (0.051 g). 1H NMR (300 MHz, MeOD) δ 0.90–0.95 (m, 6H, CH3-Octyl chain and Ile-δ-CH3), 1.03 (d, J = 7.0 Hz, 3H, Ile-γ-CH3), 1.32-1.46 (m, 11H, Octyl chain and Ile-γ-CH2 Ha), 1.58–1.62 (m, 1H, Ile-γ-CH2 Hb), 1.70–172 (m, 2H CH2-Octyl chain), 1.97–2.01 (m, 1H, Ile-β-CH), 3.56 (d, J = 3.7 Hz, 1H, Ile-α-CH), 3.58–3.62 (m, 2H, NH-CH2 octyl chain), 4.31–4.42 (m, 4H, H-5′a, H-5′b, H-4′, H-3′), 4.64 (t, J = 5.1 Hz, 1H, H-2′), 6.10 (d, J = 5.4 Hz, 1H, H-1′), 8.25 (s, 1H, H-2), 8.48 (s, 1H, H-8). 13C NMR (75 MHz, D2O) δ 12.1 (Ile- δ -CH3), 14.4 (C8 alkyl chain), 15.5 (Ile-γ-CH3), 23.7(C8 alkyl chain), 25.6 (Ile- δ-CH2), 28.0–33.0 (C8 alkyl chain), 38.2 (Ile-β-CH), 41.7 (C8 alkyl chain), 61.5 (Ile-α-CH), 69.1 (C-5′), 72.0 (C-4′), 76.2 (C-3′), 84.3 (C-2′), 89.3 (C-1′), 120.4 (C-5), 140.4 (C-8), 149.7 (C-6), 154.0 (C-4), 156.1 (C-2), 175.0 (C=O, Ile). HRMS [ESI] m/z: calcd. for C24H41N7O7S ([M+H]+) 572.2861, found 572.2864.
6.2.22. 5′-O-(N-L-isoleucyl)-sulfamoyl-N6-dodecyl-adenosine (6c)
Compound 5c (90 mg, 0.12 mmol) was treated in the same way as compound 5a regarding reaction condition and purification to obtain 6c. Yield: 11% (0.008 g). 1H NMR (300 MHz, MeOD) δ 0.87–0.90 (m, 6H, CH3-dodecyl chain and Ile-δ-CH3), 1.04 (d, J = 7.0 Hz, 3H,), 1.28–1.30 (m, 31H, dodecyl chain, Ile-γ-CH3), 1.68–1.73 (m, 4H, Ile-β-CH, dodecyl chain), 3.37–3.39 (m, 2H, NH-CH2 dodecyl chain), 3.71–3.77 (m, 3H, Ile-α-CH), 4.51–4.66 (m, 5H, H-5′a, H-5′b, H-3′, H-4′), 4.89–4.90 (s, 1H, H-2′), 6.55 (s, 1H, H-1′), 8.31 (d, J = 4.8 Hz, 1H, H-2), 8.56 (s, 1H, H-8). HRMS [ESI] m/z: calcd. for C28H49N7O7S ([M-H]-): 626.3341, found: 626.3338.
6.2.23. 5′-O-(N-L-isoleucyl)-sulfamoyl-N6-octadecyl-adenosine (6d)
Compound 5d (80 mg, 0.09 mmol) was treated in the same way as compound 5a to obtain 6d. Yield: 19% (0.012 g). 1H NMR (300 MHz, MeOD) δ 0.89-0.98 (m, 8H, CH3-octadecyl chain and Ile-δ-CH3), 1.04 (d, J = 7.0 Hz, 3H, Ile-γ-CH3), 1.30 (s, 31H, octadecyl chain), 1.55–1.61 (m, 1H, Ile-γ-CH2 Hb), 1.69-171 (m, 2H CH2-Octadecyl chain), 1.97-2.01 (m, 1H, Ile-β-CH), 3.56-362 (m, 3H, Ile-α-CH and NH-CH2 octadecyl chain), 4.31–4.41 (m, 5H, H-5′a, H-5′b, H-3′, H-2′, H-4′), 6.09 (d, J = 5.4 Hz, H-1′), 8.25 (d, J = 4.8 Hz, 1H, H-2), 8.48 (s, 1H, H-8). HRMS [ESI] m/z: calcd. for C34H61N7O7S ([M+H]+) 712.4426, found: 712.4470.
6.2.24. Synthesis of 5′-O-(N-L-isoleucyl)-sulfamoyl-N6-phenyl-adenosine (6e)
Compound 5e (150 mg, 0.22 mmol) was treated as for 5a to obtain 6e. Yield: 18% (0.021 g). 1H NMR (300 MHz, MeOD) δ 0.90–0.95 (m, 3H, Ile-δ-CH3,), 1.03 (d, J = 6.9 Hz, 3H, Ile-γ-CH3), 1.20–1.33 (m, 1H, Ile-γ-CH2 Ha), 1.56–1.60 (m, 1H, Ile-γ-CH2 Hb), 1.97–2.01 (m, 1H, Ile-β-CH), 3.61 (d, J = 3.9 Hz, 1H, Ile-α-CH), 4.37–4.45 (m, 5H, H-5′, H-5″, H-4′, H-3′, H-2′), 6.14 (d, J = 5.1 Hz, 1H, H-1′), 7.14 (t, J = 7.1 Hz, 1H, p-CH-aniline), 7.39 (t, J = 7.5 Hz, 2H, m-CH-aniline), 7.76 (d, J = 8.1 Hz, 2H, o-CH-aniline), 8.38 (s, 1H, H-2), 8.57 (s, 1H, H-8). 13C NMR (75 MHz, D2O) δ 10.5 (Ile- δ -CH3), 13.9 (Ile-γ-CH3), 23.9 (Ile- δ-CH2), 36.5 (Ile-β-CH), 59.8 (Ile-α-CH), 67.5 (C-5′), 70.2 (C-4′), 74.4 (C-3′), 82.5 (C-2′), 87.7 (C-1′), 119.4 (C-5), 120.7 (o-CH-aniline), 123.4 (p-CH-aniline), 128.3 (m-CH-aniline), 138.3 (NH-CH-aniline), 139.8 (C-8), 142.6 (C-6), 149.0 (C-4), 152.0 (C-2), 173.7 (C=O, Ile). HRMS [ESI] m/z: calcd. for C22H29N7O7S ([M-H]-): 534.1776, found 534.1777.
6.2.25. Synthesis of 5′-O-(N-L-isoleucyl)-sulfamoyl-6-O-methyl-purine riboside (6f)
Compound 5f (100 mg, 0.16 mmol) was added to a fresh solution of TFA/water/DCM (50:25:25 v/v), and the reaction was stirred for 3 h at room temperature. TLC, with an acetone/hexane gradient, was used to monitor the reaction. The reaction mixture was evaporated under reduced pressure at 25 °C. The product was purified with RP-HPLC using a C18 column and gradient elution, with water/acetonitrile as the mobile phase, to obtain 6f. Yield: 55% (0.042 g). 1H NMR (300 MHz, MeOD) δ 0.91–1.03 (m, 6H, Ile-δ-CH3, Ile-γ-CH3), 1.28–1.29 (m, 1H, Ile-γ-CH2 Ha), 1.54–1.56 (m, 1H, Ile-γ-CH2 Hb), 1.95–1.97 (m, 1H, Ile-β-CH), 3.58 (d, J = 4.0 Hz, 1H, Ile-α-CH), 4.18 (s, 3H, O-CH3), 4.32–4.43 (m, 4H, H-5′a, H-5′b, H-4′, H-3′), 4.66–4.69 (m, 1H, H-2′), 6.17 (d, J = 5.2 Hz, 1H, H-1′), 8.53 (s, 1H, H-2), 8.68 (s, 1H, H-8). 13C NMR (75 MHz, D2O) δ 12.1 (Ile- δ -CH3), 15.4 (Ile-γ-CH3), 25.7 (Ile- δ-CH2), 38.2 (Ile-β-CH), 54.8 (O-Me), 61.4 (Ile-α-CH), 69.0 (C-5′), 72.0 (C-4′), 76.2 (C-3′), 84.4 (C-2′), 89.8 (C-1′), 143.5 (C-2), 153.5 (C-2). HRMS [ESI] m/z: calcd. for C17H26N6O8S ([M-H]-) 473.1460, found: 473.1471.
The synthesis of compound 2′,3′,5′-tri-O-TBDMS-adenosine (8), 2′,3′-di-O-TBDMS adenosine (9), and 2′,3′-di-O-TBDMS-5′-O-sulfamoyl adenosine (10) was performed by following reported procedure20 and obtained at 88, 84 and 97% yields respectively.
6.2.26. 5′-O-[N-(N-Boc)leucyl]sulfamoyl adenosine (11)
Compound 10 (300 mg, 0.52 mmol), Boc-Leu-OSu (1.2 equivalent, 205.85 mg, 0.63 mmol), DBU (1 equivalent, 0.08 mL, 0.52 mmol) were added together using DMF (10 mL) as solvent. The reaction, which after a short period of time turned pink, was let to react at room temperature and overnight. A small sample was work-upped with EtOAc and 10% KHSO4 for TLC analysis which was later developed with 1% MeOH in EtOAc and sprayed with ammonium molybdate (Rf = 0.73). The solvents were evaporated after completion of the reaction and the obtained wine-red dense liquid was partitioned between water and EtOAc. A small amount of 10% KHSO4 was added in the first wash to assure that the pH from the aqueous layer had pH 5-6. The organic layers were collected, dried over MgSO4, filtered and, lastly, the solvent was evaporated. The residue was purified using silica gel column chromatography with elution at 1.5% MeOH:EtOAc. The UV-active fractions were collected and further dried. Yield: 77%. 1H NMR (300 MHz, MeOD) δ -0.42 (3H, s, CH3-Si), -0.18 (3H, m, CH3-Si), 0.11–0.12 (6H, m, CH3-Si), 0.66 (9H, s, tBu CH3), 0.84–0.86 (6H, m, Leu-δ’-CH3, Leu-δ″), 0.92 (9H, s, tBu CH3), 1.36 (10H, s, Boc-tBu CH3 Leu-γ-CH), 1.47–1.63 (2H, m, Leu-ß-CH2), 3.77-3.78 (1H, m, Leu-α-CH), 4.02–4.14 (3H, m, H5″, H5’, H3’), 4.32 (1H, d, J = 7.4 Hz, H2’), 4.89–4.93 (1H, m, H4’), 5.95 (1H, d, J = 7.4 Hz, H1’), 7.26 (2H, bs, NH2), 8.13 (1H, s, H8), 8.47 (1H, s, H2). 13C NMR (300 MHz, MeOD), δ -5.6 (CH3-Si), -5.7 (CH3-Si), -4.6 (CH3-Si), 17.5 (tBu C(CH3)3), 17.9 (tBu C(CH3)3), 22.1 (Leu-δ’-CH3), 23.4 (Leu-δ″-CH3), 24.5 (Leu-γ-CH), 25.6 (tBu C(CH3)3), 25.9 (tBu C(CH3)3), 28.4 (Boc-tBu CH3), 43.0 (Leu-ß-CH2), 54.9 (Leu-α-CH), 66.9 (C-5′), 73.4 (C-3′), 74.7 (C-2′), 77.5 (C-4′), 84.1 (Boc-tertiary C), 86.1 (C-1′), 119.0 (C-5), 139.7 (C-8)150.0 (C-4), 152.8 (C-2), 155.1 (Boc- carbonyl C), 156.1(C-6), 177.0 (CONH). HRMS [ESI] m/z: calcd. for C33H60N7O9S1Si2 ([M-H]-) 786.3717, found: 786.3720.
6.2.27. 2′,3′-di-O-TBDMS-5′-O-(N-leucyl)sulfamoyl adenosine (12)
Selective Boc deprotection was performed by adding a 10 mL solution of TFA:DCM:water (2:1:1) to the Boc and TBDMS protected compound 9 (273 mg). The reaction was first started at 0 °C and then held at room temperature for 2 h. For TLC (20% MeOH in EtOAc plus a few drops of TEA was used as mobile phase; Rf = 0.3), the sample was first evaporated in high-vacuum, then the dried compound was dissolved and evaporated two times after dissolving with EtOH to assure that there was no TFA left, as it could interfere with TLC analysis. Upon completion of the reaction, TFA and DCM were evaporated, and the resulting residue was purified by silica gel column chromatography. 1H NMR (300 MHz, MeOD) δ -0.36 (3H, s, CH3-Si), 0.06 (3H, m, CH3-Si), 0.11–0.14 (6H, m, CH3-Si), 0.69 (9H, s, tBu CH3), 0.88–0.93 (15H, m, tBu CH3, Leu-δ’-CH3, Leu-δ″), 1.47–1.53 (1H, m, Leu-γ-CH), 1.69–1.77 (2H, m, Leu-ß-CH2), 3.48–3.53 (1H, m, Leu-α-CH), 4.24–4.39 (4H, m, H5″, H5’, H3’, H2’), 4.76–4.80 (1H, m, H4’), 6.06 (1H, d, J = 7.1 Hz, H1’), 8.14 (1H, s, H8), 8.53 (1H, s, H2). 13C NMR (300 MHz, MeOD), δ -5.2 (CH3–Si), -4.3 (CH3–Si), -4.2 (CH3–Si), 18.7 (tBu C(CH3)3), 18.9 (tBu C(CH3)3), 22.3 (Leu-δ’-CH3), 23.4 (Leu-δ″-CH3), 25.8 (Leu-γ-CH), 26.2 (tBu C(CH3)3), 26.4 (tBu C(CH3)3), 43.3 (Leu-ß-CH2), 55.9 (Leu-α-CH), 69.1 (C-5′), 74.6 (C-3′), 77.6 (C-2′), 85.9 (C-4′), 88.5 (C-1′), 120.1 (C-5), 141.6 (C-8), 151.0 (C-4), 153.9 (C-2), 157.3(C-6), 178.6 (CONH). HRMS [ESI] m/z: calcd. for C28H52N7O7S1Si2 ([M-H]-) 686.3193, found 686.3195.
6.2.28. 2′,3′-di-O-TBDMS-5′-O-[Nα-(p-nitrobenzyloxycarbonyl)leucyl]sulfamoyl adenosine (13)
For coupling of the leucyladenosine derivative and the promoiety, the dried compound 12 (116 mg, 0.17 mmol) was mixed with 4-nitrobenzyl chloroformate (1.2 equivalent, 43.62 mg, 0.204 mmol) and DIPEA (3 equivalent, 0.09 mL, 0.51 mmol) and dissolved in DFM. The reaction was left at room temperature overnight. A predeveloped TLC was eluted at 5% MeOH in DCM in the presence of a few drops of TEA (Rf = 0.42). The solvent was evaporated in order to obtain a residue and silica gel column chromatography was used to purify the compound. Yield: 62%. 1H NMR (300 MHz, MeOD) δ -0.21–0.20 (3H, s, CH3–Si), 0.11–0.14 (3H, m, CH3–Si), 0.29–0.31 (6H, m, CH3–Si), 0.86–0.92 (9H, m, Leu-δ’-CH3, Leu-δ″-CH3, Leu-γ-CH, Leu-ß-CH2), 1.01–1.11 (18H, m, 2·tBu CH3), 4.43–4.57 (6H, m, H5″, H5’, H4’, H3’, H2’, Leu-α-CH), 5.34 (2H, s, Bn CH2), 6.21–6.26 (1H, m, H1’), 7.69-7.71 (2H, d, o-2CH), 8.31-8.33 (3H, d, H8, m-2H), 8.70 (1H, m, H2). 13C NMR (300 MHz, MeOD), δ -5.2 (CH3–Si), -4.3 (CH3–Si), -4.2 (CH3–Si), 22.1 (Leu-δ’-CH3), 23.4 (Leu-δ″-CH3), 23.8 (Leu-γ-CH), 26.2 (tBu C(CH3)3), 26.4 (tBu C(CH3)3), 42.5 (Leu-ß-CH2), 55.2 (Leu-α-CH), 61.1 (C-5′), 66.1 (Bn), 74.6 (C-3′), 77.4 (C-2′), 85.9 (C-4′), 88.3 (C-1′), 120.1 (C-5), 124.5 (o-CH), 130.0 (m-CH),141.5 (C), 145.5 (p-CH), 148.8 (C-4), 151.0 (C-2), 153.9 (C-6) 157.3 (COO-Bn), 177.95 (CONH). HRMS [ESI] m/z: calcd. for C36H58N8O11S1Si2 ([M-H]-) 865.3411, found: 865.3431.
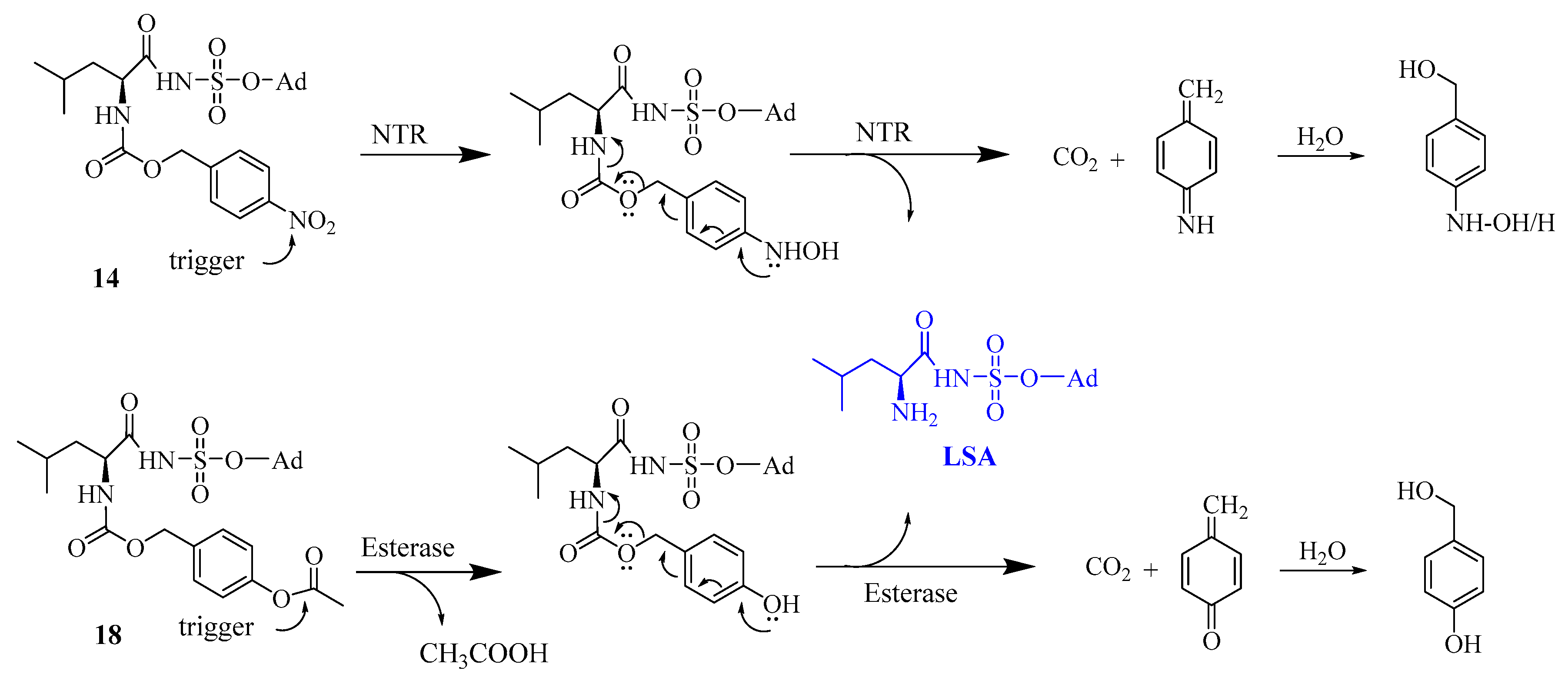
6.2.29. 5′-O-[Nα-(p-nitrobenzyloxycarbonyl)leucyl]sulfamoyl adenosine (14)
First, compound 13 (160 mg, 0.19 mmol) was dissolved in THF (5 mL) and Et3N·3HF (2 mL) was added to start the reaction at room temperature for overnight. The reaction progress was checked with TLC which was predeveloped in 10% MeOH: DCM in the presence of TEA (Rf = 0.48). The reaction mixture was purified using silica gel chromatography. Fractions corresponding to compound 14 were collected, evaporated and further purified using reverse phase HPLC. 1H NMR (300 MHz, MeOD) δ 0.89–0.95 (6H, m, Leu-δ’-CH3, Leu-δ″-CH3) 1.70–1.78 (1H, m, Leu-γ-CH), 1.90–1.92 (2H, m, Leu-ß-CH2), 4.19–4.38 (5H, m, H5″, H5’, H4’, H3’, Leu-α-CH), 4.63–4.66 (1H, m, H2’), 5.18–5.22 (2H, m, Bn-CH2), 6.06–6.08 (1H, d, H1’, J = 5.73), 7.55–7.57 (2H, d, o-2CH, J = 8.70), 8.17–8.20 (3H, m, H8, m-2CH), 8.49 (1H, s, H2). 13C NMR (300 MHz, MeOD), 20.3 (Leu-δ’-CH3), 21.6 (Leu-δ″-CH3), 24.3 (Leu-γ-CH), 40.7 (Leu-ß-CH2), 59.4 (Leu-α-CH), 64.5 (C-5′), 67.7 (Bn), 70.5 (C-3′), 74.3 (C-2′), 82.7 (C-4′), 87.4 (C-1′), 118.4 (C-5), 122.8 (m-CH), 127.4 (o-CH), 139.4 (ipso-CH), 144.3 (C-8), 147.1 (C-4), 149.2 (p-CH), 152.2 (C-2), 155.5 (C-6), 171.2 (CO–NαH), 178.02 (CONH). HRMS [ESI] m/z: calcd. for C24H30N8O11S1 ([M-H]-) 637.1682 found: 637.1689.
6.2.30. p-Acetoxybenzylchloroformate (16)
Take triphosgene (134mg, 0.45 mmol) in a round bottom flask and dry using high vacuum; then at 0 °C add DIPEA (0.16 mL, 0.9 mmol, 2 equiv.) and 5 mL THF. In another round bottom flask weigh 75 mg of p-acetoxybenzylalcohol and add 5 mL THF, then add this solution to triphosgene at 0 °C. Let the reaction continue at 0 °C for four hours. Use the reaction mixture without purification for the next reaction. The formation of the compound was confirmed by NMR. 1H NMR (300 MHz, CDCl3) δ 2.29 (s, 3H, acetyl CH3), 5.09(s, 2H, benzyl CH2), 7.35–7.38 (m, 2H, meta to acetyl), 7.04–7.09 (m, 2H, ortho to acetyl); 13C NMR (75 MHz, CDCl3) δ 21.1 (acetyl CH3), 66.5 (benzyl CH2), 121.7 (ortho C to acetyl), 128.7 (meta C to acetyl), 133.9 (para C to acetyl) 150.6 (Acetoxy-C), 156.3 (carbonyl C chloroformate), 171.2 (acetyl carbonyl C).
6.2.31. 2′,3′-di-O-TBDMS-5′-O-[Nα-(p-acetoxybenzyloxycarbonyl)leucyl]sulfamoyl adenosine (17)
The synthesis was performed like compound 13, instead of p-nitrobenzyl chloroformate we used 16 as a reactant. The compound was obtained as a white solid in 62% yield. HRMS [ESI] m/z: calcd. for C38H60N7O11S1Si2 ([M-H]-) 878.3615, found: 878.3627.
6.2.32. 5′-O-[Nα-(p-acetyloxybenzyloxycarbonyl)leucyl]sulfamoyl adenosine (18)
The synthesis was performed like compound 14, and the compound was obtained as a white solid in 28% yield. 1H NMR (300 MHz, MeOD) δ 0.92-1.29 (9H, m, Leu-δ’-CH3, Leu-δ″-CH3, Leu-γ-CH, Leu-ß-CH2), 2.25 (3H, s, CH3-Acetyl), 4.07–4.57 (6H, m, H5″, H5’, H4’, H3’, H2’, Leu-α-CH), 5.02–5.05 (2H, m, Bn CH2), 6.07-6.08 (1H, d, H1’, J = 6.67), 7.02-7.03 (2H, d, o-2CH, J = 8.10), 7.35–7.36 (2H, d, m-2CH, J = 9.06), 8.19 (1H, s, H8), 8.51 (1H, s, H2). 13C NMR (300 MHz, MeOD), 20.9 (CH3-Ac), 22.00 (Leu-δ’-CH3), 23.7 (Leu-δ″-CH3), 26.1 (Leu-γ-CH), 43.1 (Leu-ß-CH2), 57.2 (Leu-α-CH), 66.7 (C-5′), 69.2 (Bn), 72.3 (C-3′), 76.1 (C-2′), 84.5 (C-4′), 89.0 (C-1′), 120.1 (C-5), 122.7 (m-CH), 129.8 (o-CH), 136.1 (ipso-CH), 141.2 (C-8), 150.9 (C-4), 151.8 (p-CH), 153.8 (C-2), 157.2 (C-6), 158.5 (NH-COO) 171.2 (CO-Ac), 181.4 (CONH). HRMS [ESI] m/z: calcd. for C26H34N7O11S1 ([M+H]+) 652.2031, found: 652.2043.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}