DnaG Primase—A Target for the Development of Novel Antibacterial Agents


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
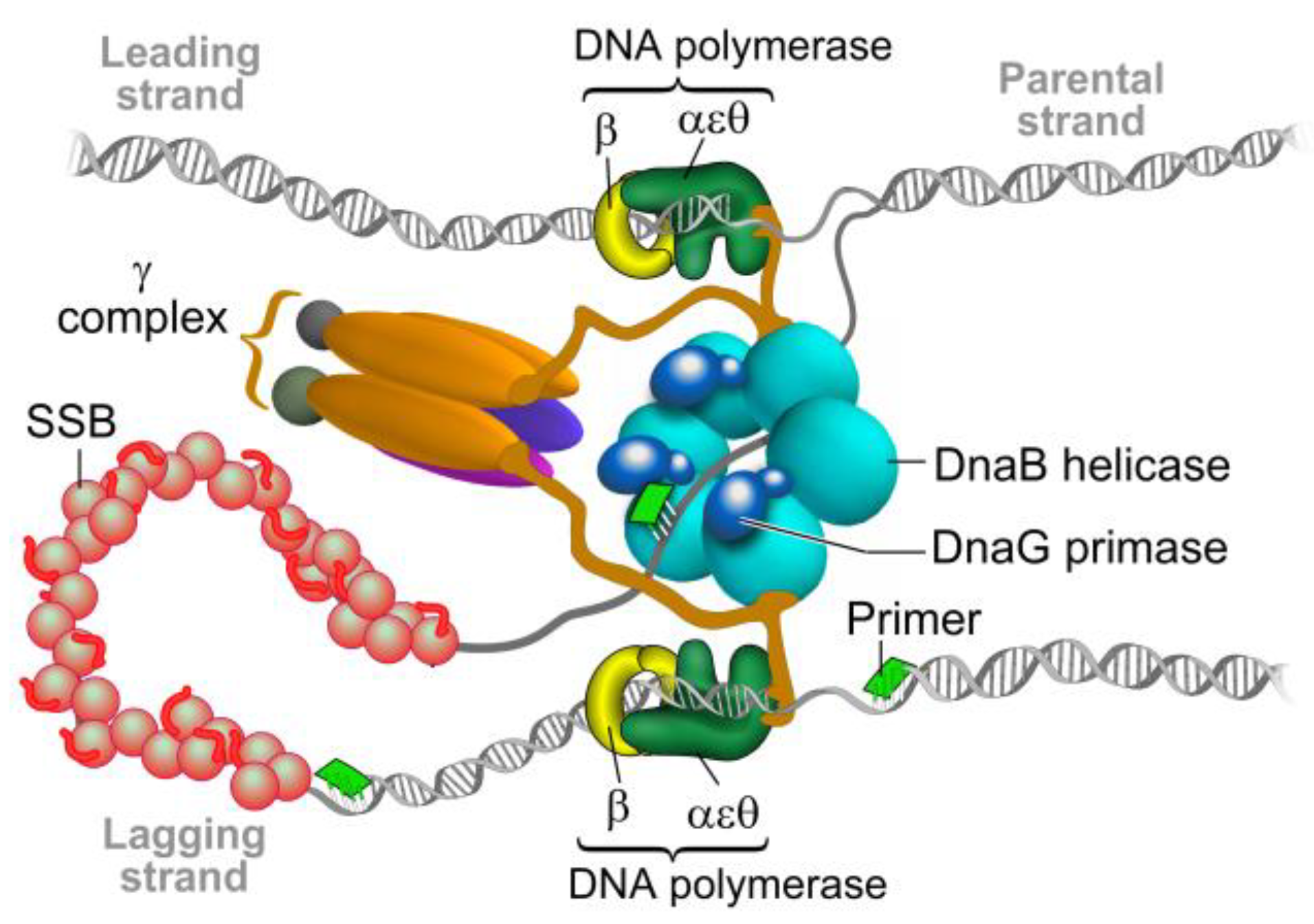
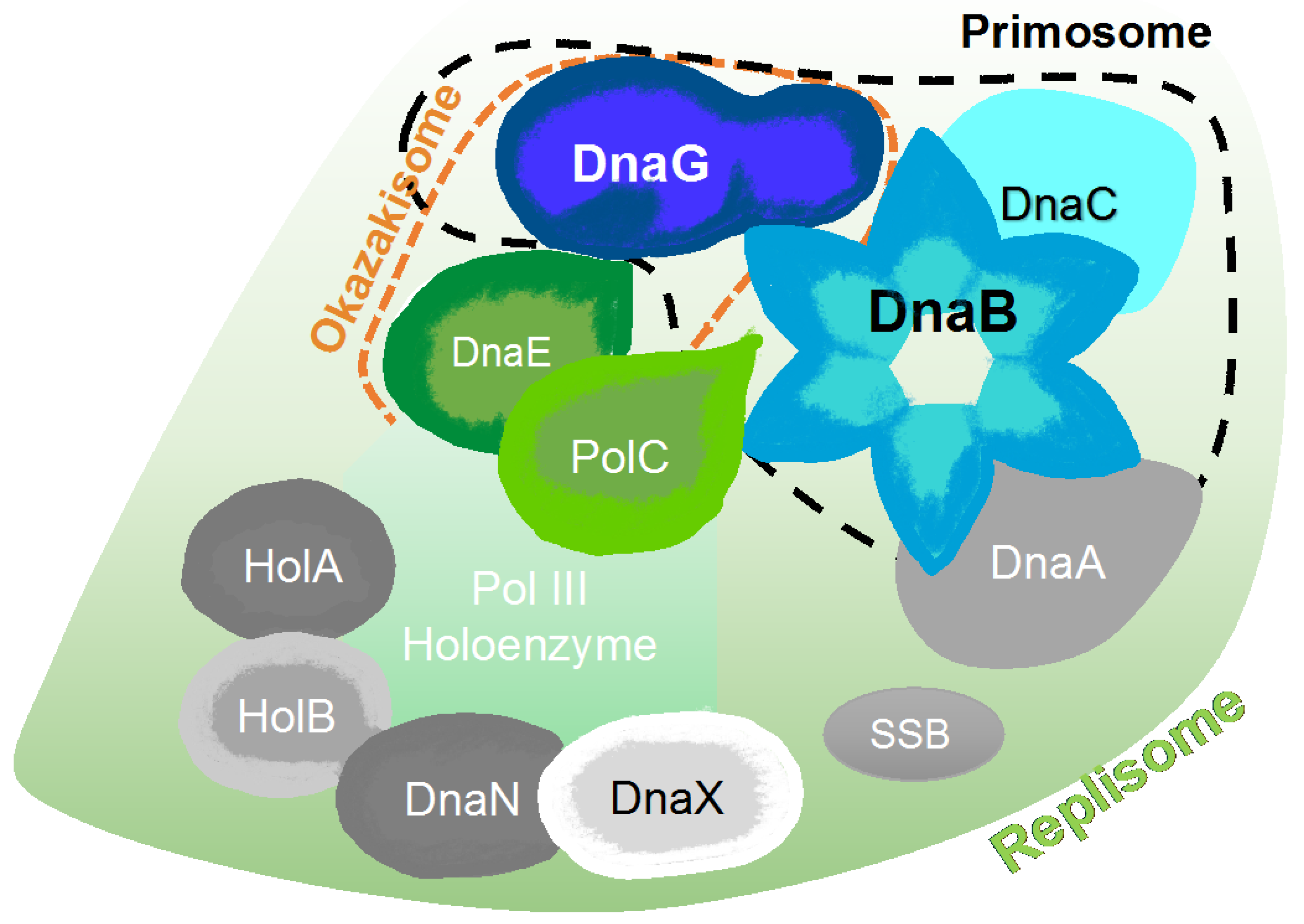
2. The Bacterial Replisome as a Multiple-Drug Target
Examples for Unique Potential DNA Replication Targets
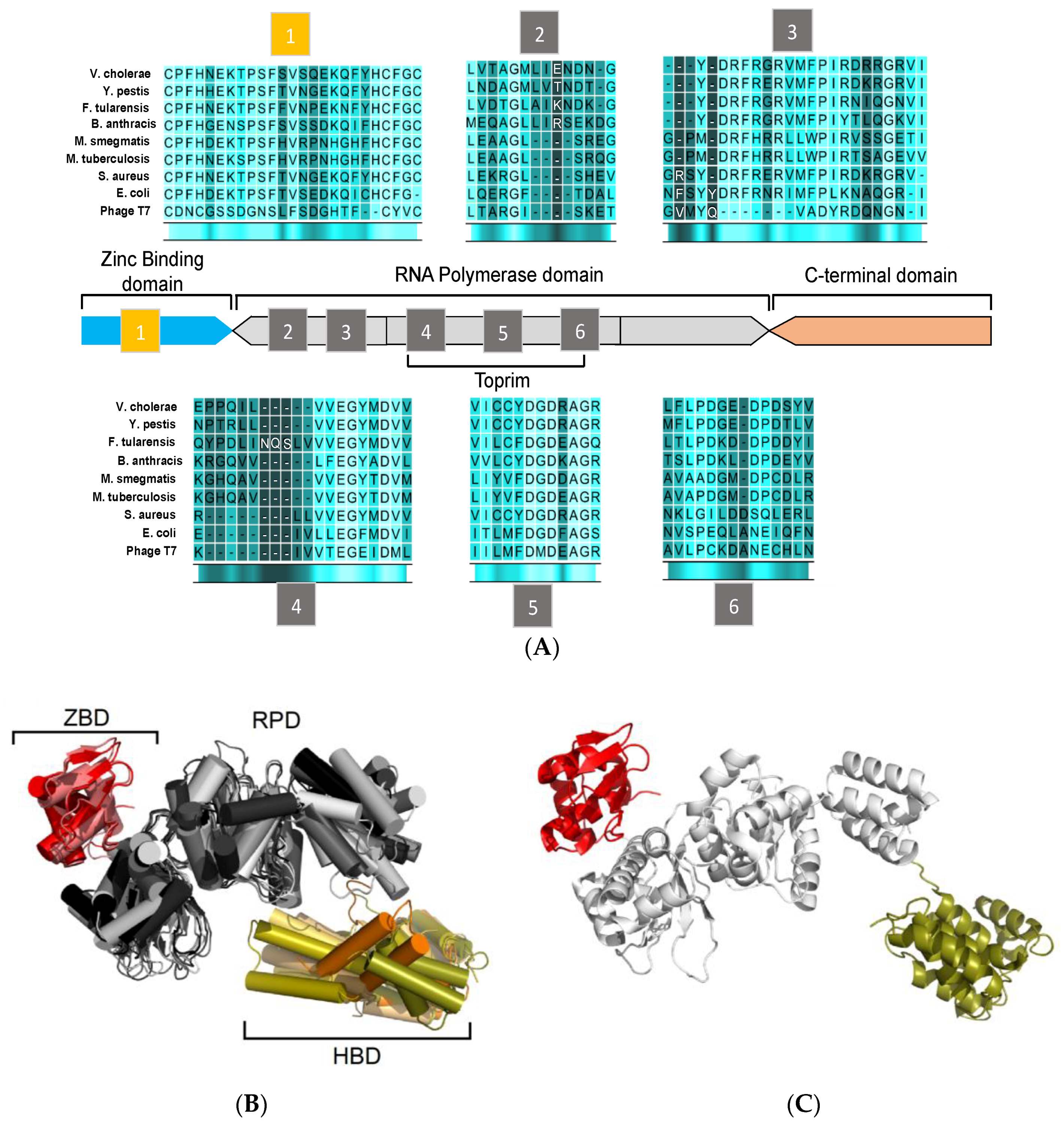
3. Structural Features of DnaG Primase: Opportunities for Drug Targeting
4. Challenges in Targeting DnaG Primase
5. Potential Types of Inhibition of DNA Primase
6. Primase Interactions: An Opportunity to Disrupt Essential Activities at the DNA Replication Fork
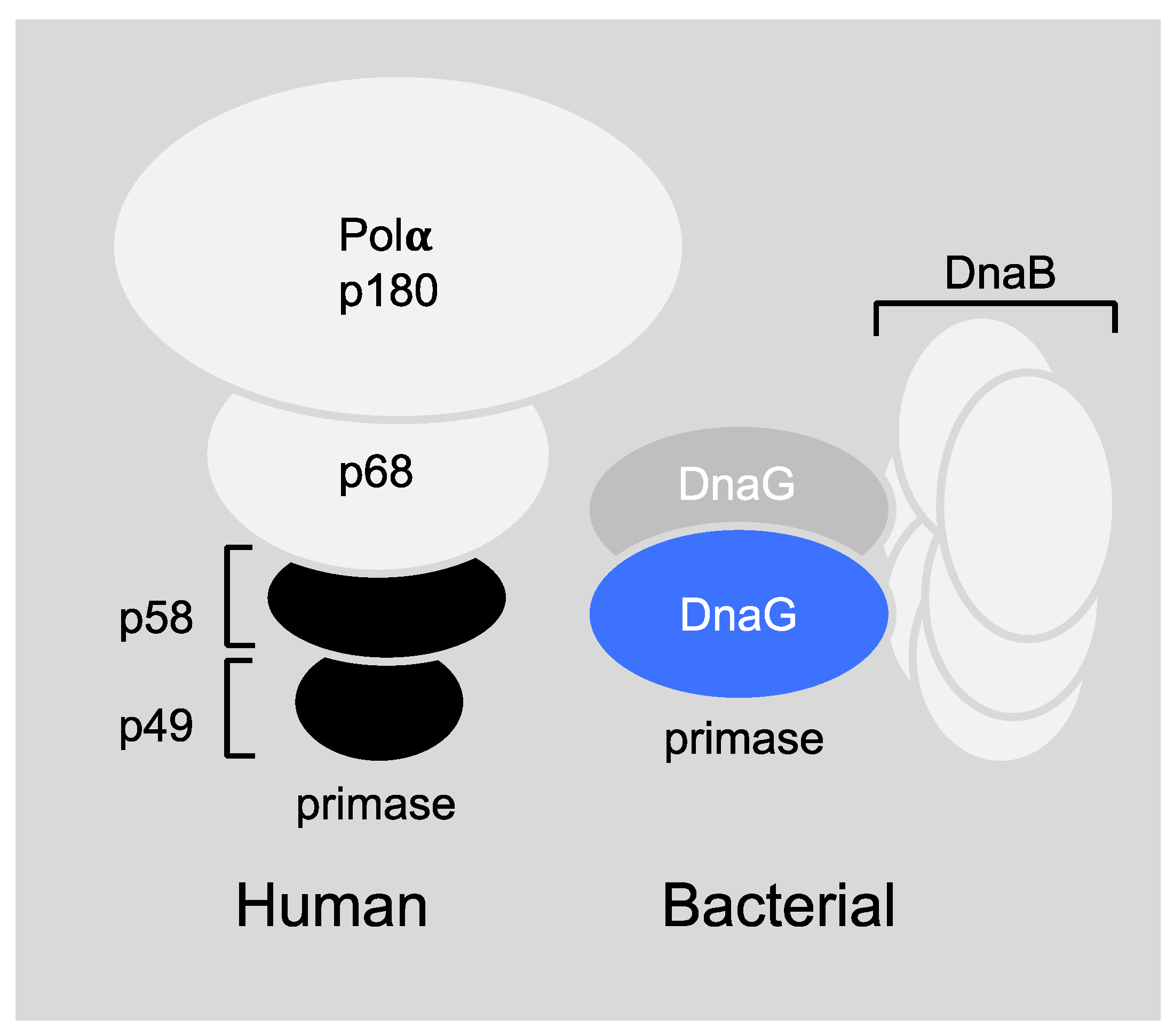
6.1. Primase-Helicase Interactions
6.2. Primase-SSB Interactions
6.3. Primase-Polymerase Interactions
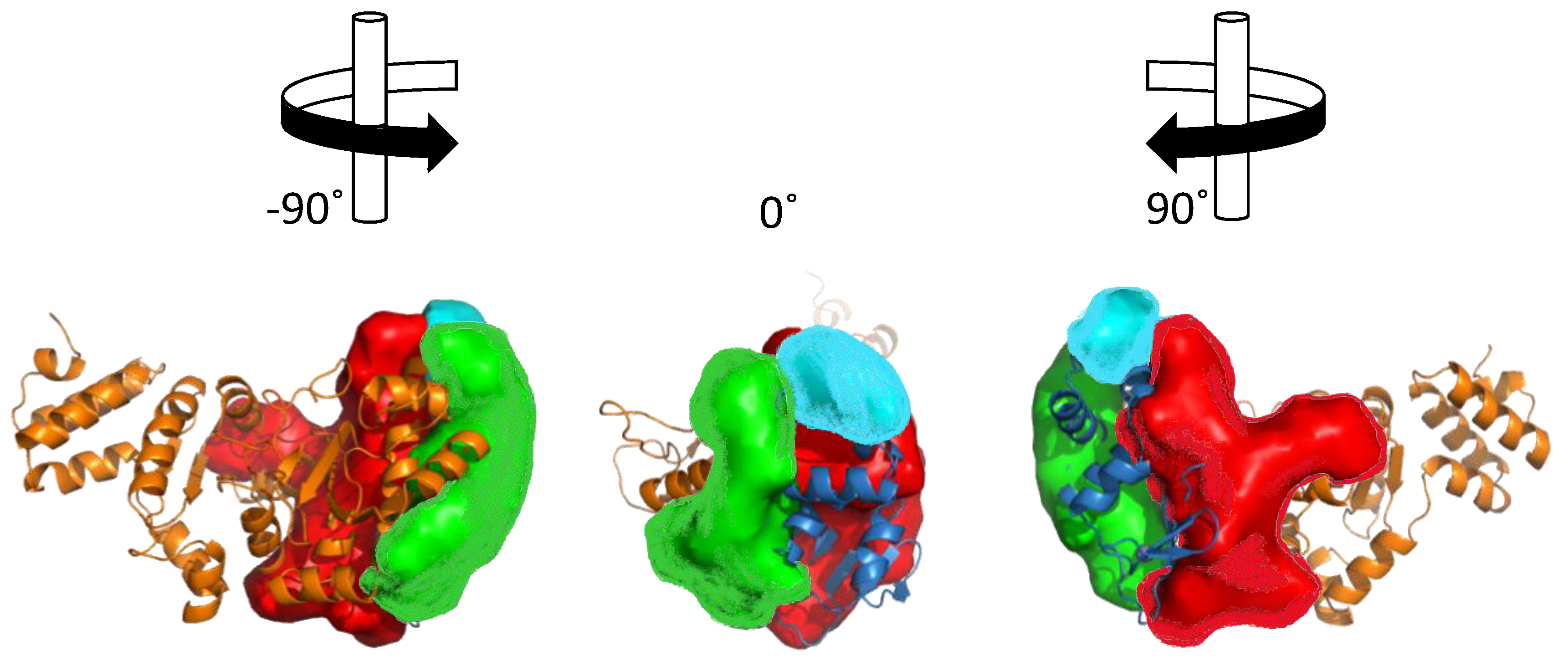
6.4. DNA-Primase Interactions at the Okazaki Fragment Start Sites: A Novel Drug Target
7. The Development of Novel Therapeutic Approaches
8. Approaches in Screening for DNA Primase Inhibitors
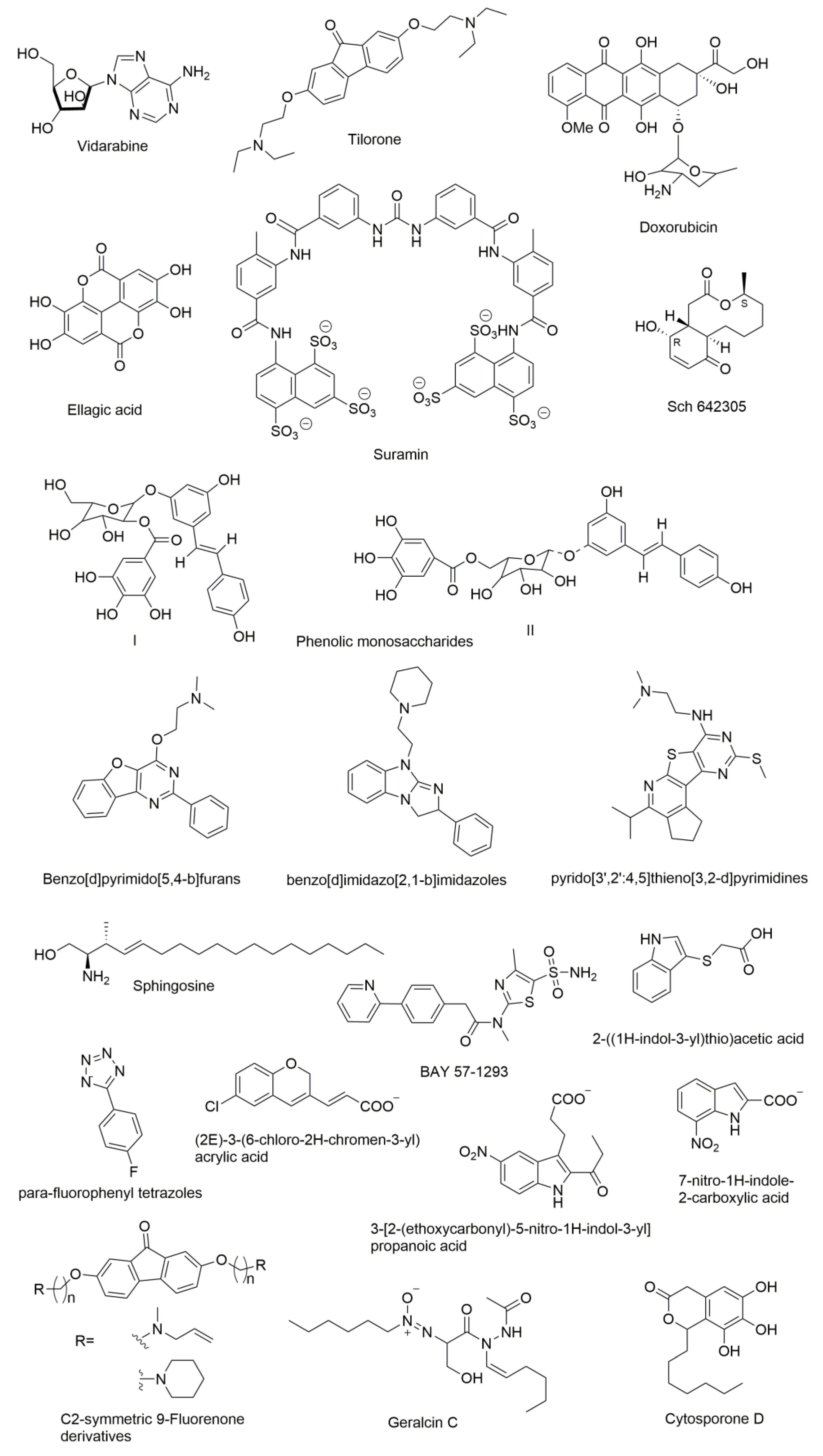
9. Molecules that were Found to Inhibit DNA Primase
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reiche, M.A.; Warner, D.F.; Mizrahi, V. Targeting DNA replication and repair for the development of novel therapeutics against tuberculosis. Front. Mol. Biosci. 2017, 4, 75. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Marians, K.J. Understanding how the replisome works. Nat. Struct. Mol. Biol. 2008, 15, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.Y.; O’Donnell, M. Snapshot: The replisome. Cell 2010, 141, 1088. [Google Scholar] [CrossRef] [PubMed]
- Frick, D.N.; Richardson, C.C. DNA primases. Annu. Rev. Biochem. 2001, 70, 39–80. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, A.; Baker, T.A. DNA Replication, 2nd ed.; University Science Books: Sausalito, CA, USA, 2005. [Google Scholar]
- Kelman, Z.; O’Donnell, M. DNA polymerase III holoenzyme: Structure and function of a chromosomal replicating machine. Annu. Rev. Biochem. 1995, 64, 171–200. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Bell, S.P. Polymerases and the replisome: Machines within machines. Cell 1998, 92, 295–305. [Google Scholar] [CrossRef]
- Van Oijen, A.M.; Loparo, J.J. Single-molecule studies of the replisome. Annu. Rev. Biophys. 2010, 39, 429–448. [Google Scholar] [CrossRef] [PubMed]
- Van Eijk, E.; Wittekoek, B.; Kuijper, E.J.; Smits, W.K. DNA replication proteins as potential targets for antimicrobials in drug-resistant bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Ditse, Z.; Lamers, M.H.; Warner, D.F. DNA replication in mycobacterium tuberculosis. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Mitscher, L.A. Bacterial topoisomerase inhibitors: Quinolone and pyridone antibacterial agents. Chem. Rev. 2005, 105, 559–592. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C. Mechanisms of action of antimicrobials: Focus on fluoroquinolones. Clin. Infect. Dis. 2001, 32, S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C. Mode of action of fluoroquinolones. Drugs 1999, 58 (Suppl. 2), 6–10. [Google Scholar] [CrossRef]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 2008, 52, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Hiasa, H.; Kerns, R.; Malik, M.; Mustaev, A.; Zhao, X. Quinolones: Action and resistance updated. Curr. Top. Med. Chem. 2009, 9, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Osheroff, N. Type ii topoisomerases as targets for quinolone antibacterials: Turning dr. Jekyll into mr. Hyde. Curr. Pharm. Des. 2001, 7, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Kling, A.; Lukat, P.; Almeida, D.V.; Bauer, A.; Fontaine, E.; Sordello, S.; Zaburannyi, N.; Herrmann, J.; Wenzel, S.C.; Konig, C.; et al. Antibiotics. Targeting dnan for tuberculosis therapy using novel griselimycins. Science 2015, 348, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Banos-Mateos, S.; van Roon, A.M.; Lang, U.F.; Maslen, S.L.; Skehel, J.M.; Lamers, M.H. High-fidelity DNA replication in mycobacterium tuberculosis relies on a trinuclear zinc center. Nat. Commun. 2017, 8, 855. [Google Scholar] [CrossRef] [PubMed]
- Champney, S.W. New Antibiotic Targets; Springer: New York, NY, USA, 2008; Volume 142, pp. 11–23. [Google Scholar]
- Wohlkonig, A.; Chan, P.F.; Fosberry, A.P.; Homes, P.; Huang, J.; Kranz, M.; Leydon, V.R.; Miles, T.J.; Pearson, N.D.; Perera, R.L.; et al. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 2010, 17, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, H.E.; Maitland, M.L.; Dolan, M.E.; Cox, N.J.; Ratain, M.J. Cancer pharmacogenomics: Strategies and challenges. Nat. Rev. Genet. 2013, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Arezi, B.; Kuchta, R.D. Eukaryotic DNA primase. Trends Biochem. Sci. 2000, 25, 572–576. [Google Scholar] [CrossRef]
- Lee, S.J.; Zhu, B.; Akabayov, B.; Richardson, C.C. Zinc-binding domain of the bacteriophage t7 DNA primase modulates binding to the DNA template. J. Biol. Chem. 2012. [Google Scholar] [CrossRef] [PubMed]
- Akabayov, B.; Lee, S.J.; Akabayov, S.R.; Rekhi, S.; Zhu, B.; Richardson, C.C. DNA recognition by the DNA primase of bacteriophage t7: A structure-function study of the zinc-binding domain. Biochemistry 2009, 48, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Wigley, D.B. Structure of the zinc-binding domain of bacillus stearothermophilus DNA primase. Structure 2000, 8, 231–239. [Google Scholar] [CrossRef]
- Corn, J.E.; Pease, P.J.; Hura, G.L.; Berger, J.M. Crosstalk between primase subunits can act to regulate primer synthesis in trans. Mol. Cell 2005, 20, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Leipe, D.D.; Koonin, E.V. Toprim—A conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, old family nucleases and RecR proteins. Nucleic Acids Res. 1998, 26, 4205–4213. [Google Scholar] [CrossRef] [PubMed]
- Keck, J.L.; Roche, D.D.; Lynch, A.S.; Berger, J.M. Structure of the RNA polymerase domain of E. coli primase. Science 2000, 287, 2482–2486. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A.; Steitz, J.A. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6498–6502. [Google Scholar] [CrossRef] [PubMed]
- Stamford, N.P.; Lilley, P.E.; Dixon, N.E. Enriched sources of Escherichia coli replication proteins. The DnaG primase is a zinc metalloprotein. Biochim. Biophys. Acta 1992, 1132, 17–25. [Google Scholar] [CrossRef]
- Rymer, R.U.; Solorio, F.A.; Tehranchi, A.K.; Chu, C.; Corn, J.E.; Keck, J.L.; Wang, J.D.; Berger, J.M. Binding mechanism of metal·NTP substrates and stringent-response alarmones to bacterial DnaG-type primases. Structure 2012, 20, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Godson, G.N.; Schoenich, J.; Sun, W.; Mustaev, A.A. Identification of the magnesium ion binding site in the catalytic center of Escherichia coli primase by iron cleavage. Biochemistry 2000, 39, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Biswas, T.; Tsodikov, O.V. Structures of the catalytic domain of bacterial primase DnaG in complexes with DNA provide insight into key priming events. Biochemistry 2018, 57, 2084–2093. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Luo, H.; Liu, Z.; Yang, M.; Pang, X.; Sun, F.; Wang, G. Structural insight into the specific DNA template binding to DnaG primase in bacteria. Sci. Rep. 2017, 7, 659. [Google Scholar] [CrossRef] [PubMed]
- Kuchta, R.D.; Stengel, G. Mechanism and evolution of DNA primases. Biochim. Biophys. Acta 2010, 1804, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Tougu, K.; Peng, H.; Marians, K.J. Identification of a domain of Escherichia coli primase required for functional interaction with the DnaB helicase at the replication fork. J. Biol. Chem. 1994, 269, 4675–4682. [Google Scholar] [PubMed]
- Oakley, A.J.; Loscha, K.V.; Schaeffer, P.M.; Liepinsh, E.; Pintacuda, G.; Wilce, M.C.; Otting, G.; Dixon, N.E. Crystal and solution structures of the helicase-binding domain of Escherichia coli primase. J. Biol. Chem. 2005, 280, 11495–11504. [Google Scholar] [CrossRef] [PubMed]
- Syson, K.; Thirlway, J.; Hounslow, A.M.; Soultanas, P.; Waltho, J.P. Solution structure of the helicase-interaction domain of the primase DnaG: A model for helicase activation. Structure 2005, 13, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Abdulrehman, S.A.; Verma, V.; Mazumder, M.; Dhar, S.K.; Gourinath, S. Crystal structure and mode of helicase binding of the c-terminal domain of primase from Helicobacter pylori. J. Bacteriol. 2013, 195, 2826–2838. [Google Scholar] [CrossRef] [PubMed]
- Su, X.C.; Schaeffer, P.M.; Loscha, K.V.; Gan, P.H.; Dixon, N.E.; Otting, G. Monomeric solution structure of the helicase-binding domain of Escherichia coli DnaG primase. FEBS J. 2006, 273, 4997–5009. [Google Scholar] [CrossRef] [PubMed]
- Shortridge, M.D.; Griep, M.A.; Powers, R. (1)H, (1)(3)C, and (1)(5)N NMR assignments for the helicase interaction domain of Staphylococcus aureus DnaG primase. Biomol. NMR Assign. 2012, 6, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.; Eliason, W.K.; Steitz, T.A. The crystal structure of the Thermus aquaticus DnaB helicase monomer. Nucleic Acids Res. 2007, 35, 4728–4736. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xi, J.; Zhuang, Z.; Benkovic, S.J. The oligomeric t4 primase is the functional form during replication. J. Biol. Chem. 2005, 280, 25416–25423. [Google Scholar] [CrossRef] [PubMed]
- Kuron, A.; Korycka-Machala, M.; Brzostek, A.; Nowosielski, M.; Doherty, A.; Dziadek, B.; Dziadek, J. Evaluation of DNA primase DnaG as a potential target for antibiotics. Antimicrob. Agents Chemother. 2014, 58, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Marians, K.J. Prokaryotic DNA replication. Annu. Rev. Biochem. 1992, 61, 673–719. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Hite, R.K.; Hamdan, S.M.; Xie, X.S.; Richardson, C.C.; van Oijen, A.M. DNA primase acts as a molecular brake in DNA replication. Nature 2006, 439, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Biswas, T.; Resto-Roldan, E.; Sawyer, S.K.; Artsimovitch, I.; Tsodikov, O.V. A novel non-radioactive primase-pyrophosphatase activity assay and its application to the discovery of inhibitors of mycobacterium tuberculosis primase DnaG. Nucleic Acids Res. 2013, 41, e56. [Google Scholar] [CrossRef] [PubMed]
- Mitsuya, H.; Yarchoan, R.; Broder, S. Molecular targets for aids therapy. Science 1990, 249, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Mitsuya, H.; Weinhold, K.J.; Furman, P.A.; St Clair, M.H.; Lehrman, S.N.; Gallo, R.C.; Bolognesi, D.; Barry, D.W.; Broder, S. 3′-azido-3′-deoxythymidine (bw a509u): An antiviral agent that inhibits the infectivity and cytopathic effect of human t-lymphotropic virus type iii/lymphadenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. USA 1985, 82, 7096–7100. [Google Scholar] [CrossRef] [PubMed]
- Schauer, G.D.; Huber, K.D.; Leuba, S.H.; Sluis-Cremer, N. Mechanism of allosteric inhibition of HIV-1 reverse transcriptase revealed by single-molecule and ensemble fluorescence. Nucleic Acids Res. 2014, 42, 11687–11696. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Leung, C.H.; Chan, D.S.; Ma, V.P.; Ma, D.L. DNA-binding small molecules as inhibitors of transcription factors. Med. Res. Rev. 2013, 33, 823–846. [Google Scholar] [CrossRef] [PubMed]
- Noirot-Gros, M.F.; Dervyn, E.; Wu, L.J.; Mervelet, P.; Errington, J.; Ehrlich, S.D.; Noirot, P. An expanded view of bacterial DNA replication. Proc. Natl. Acad. Sci. USA 2002, 99, 8342–8347. [Google Scholar] [CrossRef] [PubMed]
- Corn, J.E.; Berger, J.M. Regulation of bacterial priming and daughter strand synthesis through helicase-primase interactions. Nucleic Acids Res. 2006, 34, 4082–4088. [Google Scholar] [CrossRef] [PubMed]
- Bird, L.E.; Pan, H.; Soultanas, P.; Wigley, D.B. Mapping protein-protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry 2000, 39, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Tougu, K.; Marians, K.J. The interaction between helicase and primase sets the replication fork clock. J. Biol. Chem. 1996, 271, 21398–21405. [Google Scholar] [CrossRef] [PubMed]
- Mitkova, A.V.; Khopde, S.M.; Biswas, S.B. Mechanism and stoichiometry of interaction of DnaG primase with DnaB helicase of Escherichia coli in RNA primer synthesis. J. Biol. Chem. 2003, 278, 52253–52261. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.B.; Ratnakar, P.V.; Mohanty, B.K.; Bastia, D. Direct physical interaction between DnaG primase and DnaB helicase of Escherichia coli is necessary for optimal synthesis of primer RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 12902–12907. [Google Scholar] [CrossRef] [PubMed]
- Thirlway, J.; Soultanas, P. In the Bacillus stearothermophilus DnaB-DnaG complex, the activities of the two proteins are modulated by distinct but overlapping networks of residues. J. Bacteriol. 2006, 188, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Marians, K.J. Identification of a region of Escherichia coli DnaB required for functional interaction with DnaG at the replication fork. J. Biol. Chem. 2000, 275, 26187–26195. [Google Scholar] [CrossRef] [PubMed]
- Biswas, T.; Tsodikov, O.V. Hexameric ring structure of the n-terminal domain of mycobacterium tuberculosis DnaB helicase. FEBS J. 2008, 275, 3064–3071. [Google Scholar] [CrossRef] [PubMed]
- Dickey, T.H.; Altschuler, S.E.; Wuttke, D.S. Single-stranded DNA-binding proteins: Multiple domains for multiple functions. Structure 2013, 21, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Ha, T. Need for speed: Mechanical regulation of a replicative helicase. Cell 2007, 129, 1249–1250. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Tuteja, R. Unraveling DNA helicases. Motif, structure, mechanism and function. Eur. J. Biochem. FEBS 2004, 271, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Causer, R.J.; Dixon, N.E. Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Curr. Drug Targets 2012, 13, 352–372. [Google Scholar] [CrossRef] [PubMed]
- Shereda, R.D.; Kozlov, A.G.; Lohman, T.M.; Cox, M.M.; Keck, J.L. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 289–318. [Google Scholar] [CrossRef] [PubMed]
- Naue, N.; Beerbaum, M.; Bogutzki, A.; Schmieder, P.; Curth, U. The helicase-binding domain of Escherichia coli DnaG primase interacts with the highly conserved c-terminal region of single-stranded DNA-binding protein. Nucleic Acids Res. 2013, 41, 4507–4517. [Google Scholar] [CrossRef] [PubMed]
- Chilingaryan, Z.; Headey, S.J.; Lo, A.T.Y.; Xu, Z.Q.; Otting, G.; Dixon, N.E.; Scanlon, M.J.; Oakley, A.J. Fragment-based discovery of inhibitors of the bacterial DnaG-SSB interaction. Antibiotics 2018, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.C.; Marians, K.J. Replication fork reactivation downstream of a blocked nascent leading strand. Nature 2006, 439, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Sheaff, R.J.; Kuchta, R.D.; Ilsley, D. Calf thymus DNA polymerase alpha-primase: “Communication” and primer-template movement between the two active sites. Biochemistry 1994, 33, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.A.; Zechner, E.L.; Marians, K.J. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. I. Multiple effectors act to modulate okazaki fragment size. J. Biol. Chem. 1992, 267, 4030–4044. [Google Scholar] [PubMed]
- Nakai, H.; Richardson, C.C. The effect of the t7 and Escherichia coli DNA-binding proteins at the replication fork of bacteriophage t7. J. Biol. Chem. 1988, 263, 9831–9839. [Google Scholar] [PubMed]
- Zechner, E.L.; Wu, C.A.; Marians, K.J. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. III. A polymerase-primase interaction governs primer size. J. Biol. Chem. 1992, 267, 4054–4063. [Google Scholar] [PubMed]
- Ogawa, T.; Arai, K.; Okazaki, T. Site selection and structure of DNA-linked rna primers synthesized by the primosome in phage phi x174 DNA replication in vitro. J. Biol. Chem. 1983, 258, 13353–13358. [Google Scholar] [PubMed]
- Yuzhakov, A.; Kelman, Z.; O’Donnell, M. Trading places on DNA—A three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell 1999, 96, 153–163. [Google Scholar] [CrossRef]
- Sanders, G.M.; Dallmann, H.G.; McHenry, C.S. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol. Cell 2010, 37, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Titok, M.; Suski, C.; Dalmais, B.; Ehrlich, S.D.; Janniere, L. The replicative polymerases PoLC and DnaE are required for theta replication of the Bacillus subtilis plasmid pBS72. Microbiology 2006, 152, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Dervyn, E.; Suski, C.; Daniel, R.; Bruand, C.; Chapuis, J.; Errington, J.; Janniere, L.; Ehrlich, S.D. Two essential DNA polymerases at the bacterial replication fork. Science 2001, 294, 1716–1719. [Google Scholar] [CrossRef] [PubMed]
- Rannou, O.; Le Chatelier, E.; Larson, M.A.; Nouri, H.; Dalmais, B.; Laughton, C.; Janniere, L.; Soultanas, P. Functional interplay of DnaE polymerase, DnaG primase and dnac helicase within a ternary complex, and primase to polymerase hand-off during lagging strand DNA replication in Bacillus subtilis. Nucleic Acids Res. 2013, 41, 5303–5320. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef] [PubMed]
- Tabor, S.; Richardson, C.C. Template recognition sequence for RNA primer synthesis by gene 4 protein of bacteriophage t7. Proc. Natl. Acad. Sci. USA 1981, 78, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Kusakabe, T.; Richardson, C.C. Template recognition and ribonucleotide specificity of the DNA primase of bacteriophage t7. J. Biol. Chem. 1997, 272, 5943–5951. [Google Scholar] [CrossRef] [PubMed]
- Mendelman, L.V.; Richardson, C.C. Requirements for primer synthesis by bacteriophage t7 63-kda gene 4 protein. Roles of template sequence and t7 56-kda gene 4 protein. J. Biol. Chem. 1991, 266, 23240–23250. [Google Scholar] [PubMed]
- Mendelman, L.V.; Beauchamp, B.B.; Richardson, C.C. Requirement for a zinc motif for template recognition by the bacteriophage t7 primase. EMBO J. 1994, 13, 3909–3916. [Google Scholar] [PubMed]
- Lee, S.J.; Zhu, B.; Hamdan, S.M.; Richardson, C.C. Mechanism of sequence-specific template binding by the DNA primase of bacteriophage t7. Nucleic Acids Res. 2010, 38, 4372–4383. [Google Scholar] [CrossRef] [PubMed]
- Afek, A.; Ilic, S.; Horton, J.; Lukatsky, D.B.; Gordan, R.; Akabayov, B. DNA sequence context controls the binding and processivity of the t7 DNA primase. iScience 2018, 2, 141–147. [Google Scholar] [CrossRef]
- Abraham, E.P.; Chain, E. An enzyme from bacteria able to destroy penicillin. 1940. Rev. Infect. Dis. 1988, 10, 677–678. [Google Scholar] [PubMed]
- Hsueh, P.R.; Teng, L.J.; Chen, C.Y.; Chen, W.H.; Yu, C.J.; Ho, S.W.; Luh, K.T. Pandrug-resistant Acinetobacter baumannii causing nosocomial infections in a university hospital, Taiwan. Emerg. Infect. Dis. 2002, 8, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Gootz, T.D. The global problem of antibiotic resistance. Crit. Rev. Immunol. 2010, 30, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P. Drug discovery: Chemical beauty contest. Nature 2012, 481, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.E.; van Helden, P. Handbook of Tuberculosis: Clinics, Diagnostics, Therapy, and Epidemiology; Wiley-Blackwell: Hoboken, NJ, USA, 2008. [Google Scholar]
- Singh, M.; Tam, B.; Akabayov, B. NMR-fragment based virtual screening: A brief overview. Molecules 2018, 23, 233. [Google Scholar] [CrossRef]
- Koepsell, S.A.; Hanson, S.; Hinrichs, S.H.; Griep, M.A. Fluorometric assay for bacterial primases. Anal. Biochem. 2005, 339, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Riccardi, G. New tuberculosis drugs on the horizon. Curr. Opin. Microbiol. 2011, 14, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Macarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, B.; Flipo, M.; Blondiaux, N.; Crauste, C.; Malaquin, S.; Leroux, F.; Piveteau, C.; Villeret, V.; Brodin, P.; Villoutreix, B.O.; et al. Ligand efficiency driven design of new inhibitors of Mycobacterium tuberculosis transcriptional repressor EthR using fragment growing, merging, and linking approaches. J. Med. Chem. 2014, 57, 4876–4888. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Fragment-based lead discovery grows up. Nat. Rev. Drug Discov. 2013, 12, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.P.; Chung, C.W.; Danielson, U.H.; Egner, U.; Hennig, M.; Hubbard, R.E.; Nar, H. Biophysics in drug discovery: Impact, challenges and opportunities. Nat. Rev. Drug Discov. 2016, 15, 679–698. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Lepre, C.A. Library design for NMR-based screening. Drug Discov. Today 2001, 6, 133–140. [Google Scholar] [CrossRef]
- Peng, J.W.; Lepre, C.A.; Fejzo, J.; Abdul-Manan, N.; Moore, J.M. Nuclear magnetic resonance-based approaches for lead generation in drug discovery. Methods Enzymol. 2001, 338, 202–230. [Google Scholar] [PubMed]
- Kuchta, R.D.; Ilsley, D.; Kravig, K.D.; Schubert, S.; Harris, B. Inhibition of DNA primase and polymerase alpha by arabinofuranosylnucleoside triphosphates and related compounds. Biochemistry 1992, 31, 4720–4728. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.B.; Cheng, Y.C. Inhibition of DNA primase by nucleoside triphosphates and their arabinofuranosyl analogs. Mol. Pharmacol. 1987, 31, 146–151. [Google Scholar] [PubMed]
- Frick, D.N.; Richardson, C.C. Interaction of bacteriophage t7 gene 4 primase with its template recognition site. J. Biol. Chem. 1999, 274, 35889–35898. [Google Scholar] [CrossRef] [PubMed]
- Biswas, T.; Green, K.D.; Garneau-Tsodikova, S.; Tsodikov, O.V. Discovery of inhibitors of Bacillus anthracis primase DnaG. Biochemistry 2013, 52, 6905–6910. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.R.; Larson, M.A.; Hinrichs, S.H.; Narayanasamy, P. Development of potential broad spectrum antimicrobials using c2-symmetric 9-fluorenone alkyl amine. Bioorg. Med. Chem. Lett. 2016, 26, 1997–1999. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Mierzwa, R.; Xu, L.; He, L.; Terracciano, J.; Patel, M.; Gullo, V.; Black, T.; Zhao, W.; Chan, T.M.; et al. Isolation and structure elucidation of sch 642305, a novel bacterial DNA primase inhibitor produced by Penicillium verrucosum. J. Nat. Prod. 2003, 66, 1527–1530. [Google Scholar] [CrossRef] [PubMed]
- Hegde, V.R.; Pu, H.; Patel, M.; Black, T.; Soriano, A.; Zhao, W.; Gullo, V.P.; Chan, T.M. Two new bacterial DNA primase inhibitors from the plant Polygonum cuspidatum. Bioorg. Med. Chem. Lett. 2004, 14, 2275–2277. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.F.; Wagenaar, M.M.; Singh, M.P.; Janso, J.E.; Clardy, J. The cytosporones, new octaketide antibiotics isolated from an endophytic fungus. Org. Lett. 2000, 2, 4043–4046. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, G.; Martin, M.T.; Iorga, B.I.; Adelin, E.; Servy, C.; Cortial, S.; Ouazzani, J. Isolation and characterization of unusual hydrazides from Streptomyces sp. Impact of the cultivation support and extraction procedure. J. Nat. Prod. 2013, 76, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Adelin, E.; Martin, M.T.; Cortial, S.; Retailleau, P.; Lumyong, S.; Ouazzani, J. Bioactive polyketides isolated from agar-supported fermentation of Phomopsis sp. Cmu-lma, taking advantage of the scale-up device, platotex. Phytochemistry 2013, 93, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Louise-May, S.; Thanassi, J.A.; Podos, S.D.; Cheng, J.; Thoma, C.; Liu, C.; Wiles, J.A.; Nelson, D.M.; Phadke, A.S.; et al. Small molecule inhibitors of E. coli primase, a novel bacterial target. Bioorg. Med. Chem. Lett. 2007, 17, 2807–2810. [Google Scholar] [CrossRef] [PubMed]
- Simbulan, C.M.; Tamiya-Koizumi, K.; Suzuki, M.; Shoji, M.; Taki, T.; Yoshida, S. Sphingosine inhibits the synthesis of RNA primers by primase in vitro. Biochemistry 1994, 33, 9007–9012. [Google Scholar] [CrossRef] [PubMed]
- Tamiya-Koizumi, K.; Murate, T.; Suzuki, M.; Simbulan, C.M.; Nakagawa, M.; Takemura, M.; Furuta, K.; Izuta, S.; Yoshida, S. Inhibition of DNA primase by sphingosine and its analogues parallels with their growth suppression of cultured human leukemic cells. Biochem. Mol. Biol. Int. 1997, 41, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Kleymann, G.; Fischer, R.; Betz, U.A.; Hendrix, M.; Bender, W.; Schneider, U.; Handke, G.; Eckenberg, P.; Hewlett, G.; Pevzner, V.; et al. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat. Med. 2002, 8, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Ilic, S.; Akabayov, S.R.; Arthanari, H.; Wagner, G.; Richardson, C.C.; Akabayov, B. Identification of DNA primase inhibitors via a combined fragment-based and virtual screening. Sci. Rep. 2016, 6, 36322. [Google Scholar] [CrossRef] [PubMed]
- Kaguni, J.M. The macromolecular machines that duplicate the Escherichia coli chromosome as targets for drug discovery. Antibiotics 2018, 7, 23. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilic, S.; Cohen, S.; Singh, M.; Tam, B.; Dayan, A.; Akabayov, B. DnaG Primase—A Target for the Development of Novel Antibacterial Agents. Antibiotics 2018, 7, 72. https://doi.org/10.3390/antibiotics7030072
Ilic S, Cohen S, Singh M, Tam B, Dayan A, Akabayov B. DnaG Primase—A Target for the Development of Novel Antibacterial Agents. Antibiotics. 2018; 7(3):72. https://doi.org/10.3390/antibiotics7030072
Chicago/Turabian StyleIlic, Stefan, Shira Cohen, Meenakshi Singh, Benjamin Tam, Adi Dayan, and Barak Akabayov. 2018. "DnaG Primase—A Target for the Development of Novel Antibacterial Agents" Antibiotics 7, no. 3: 72. https://doi.org/10.3390/antibiotics7030072
APA StyleIlic, S., Cohen, S., Singh, M., Tam, B., Dayan, A., & Akabayov, B. (2018). DnaG Primase—A Target for the Development of Novel Antibacterial Agents. Antibiotics, 7(3), 72. https://doi.org/10.3390/antibiotics7030072