Screening of E. coli β-clamp Inhibitors Revealed that Few Inhibit Helicobacter pylori More Effectively: Structural and Functional Characterization

Abstract
:1. Introduction
2. Results and Discussions
2.1. Screening of E. coli β-clamp Drugs/Inhibitors
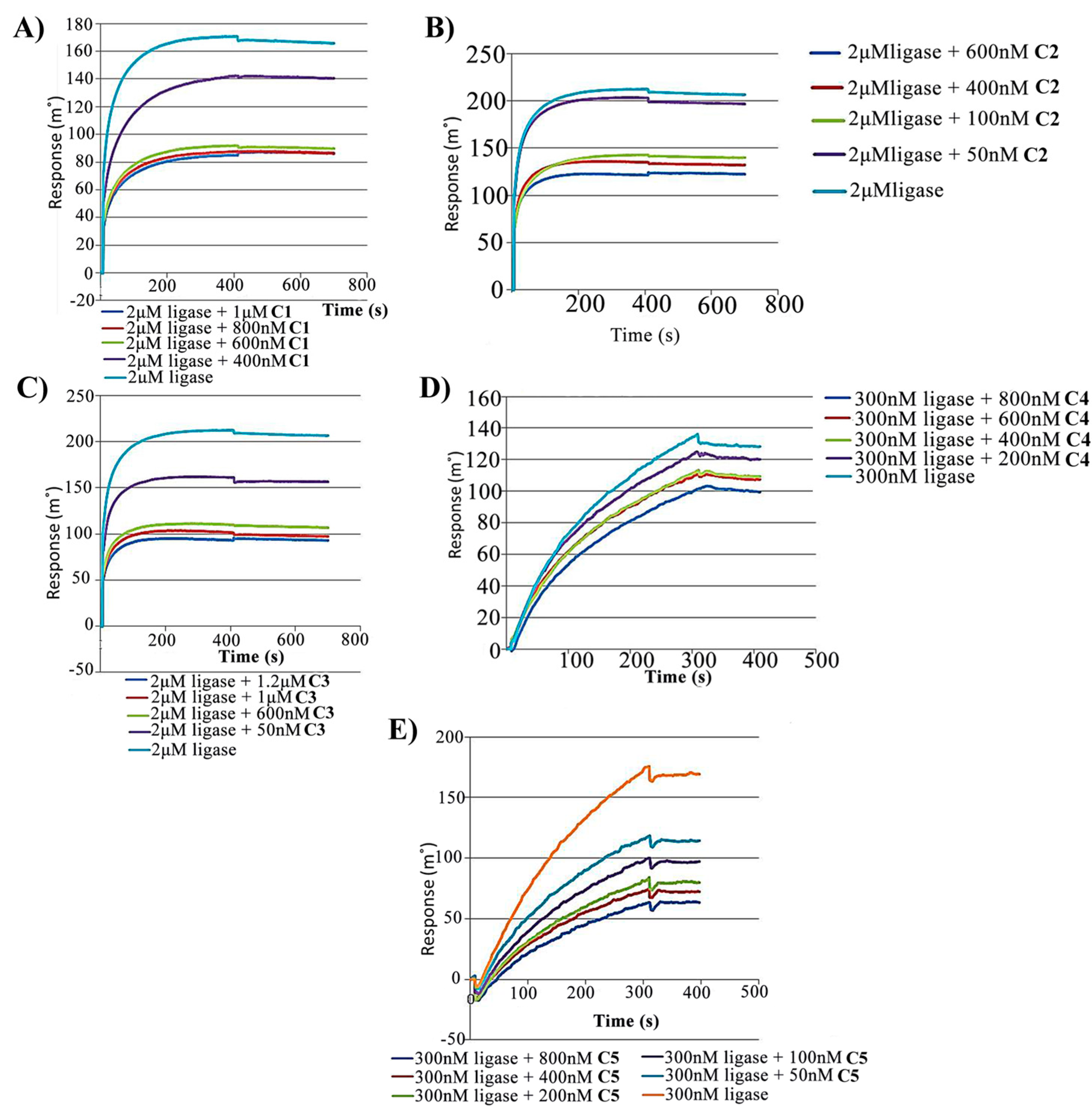
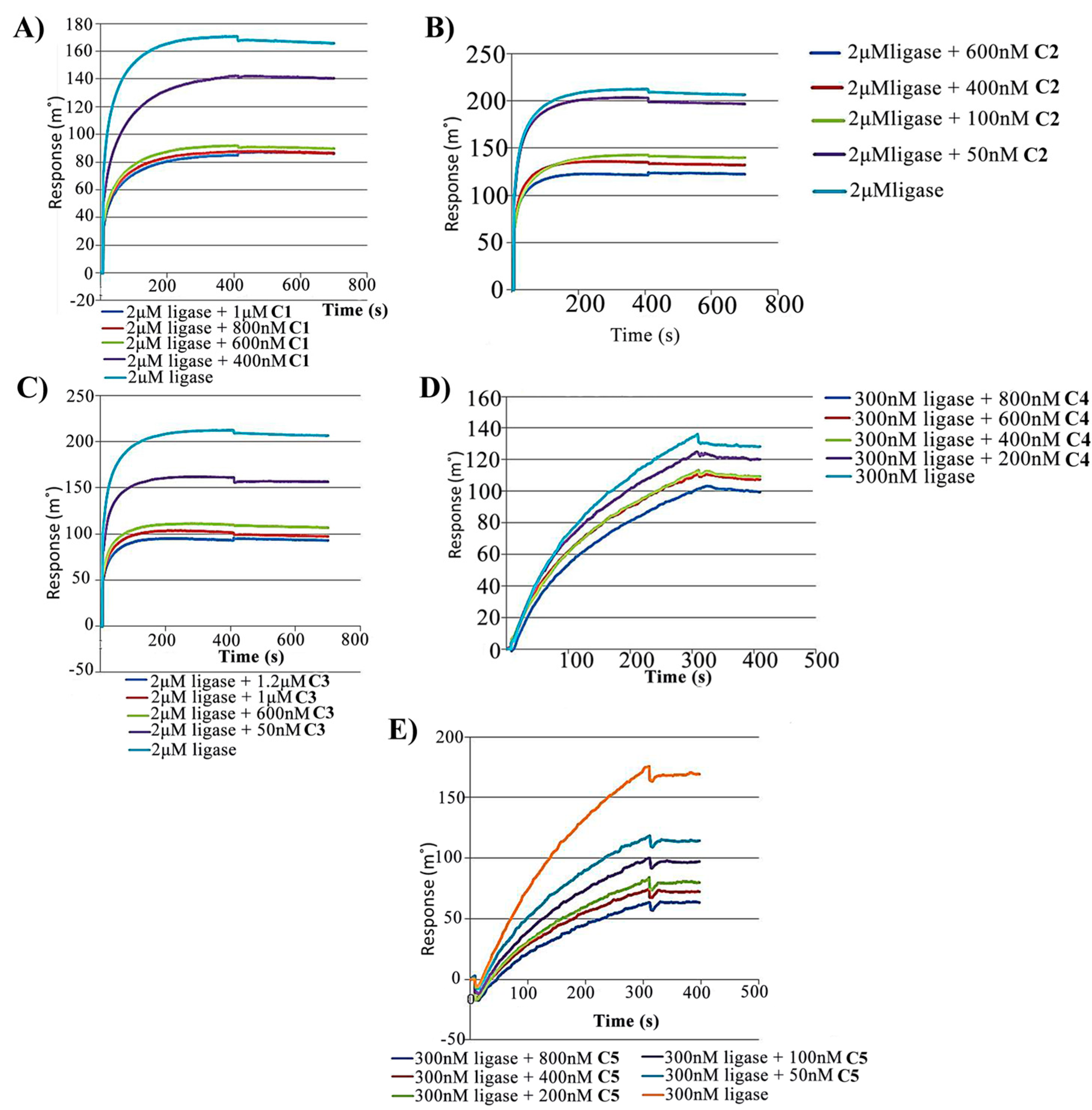
2.2. Competitive Inhibition Using Surface Competition Assay
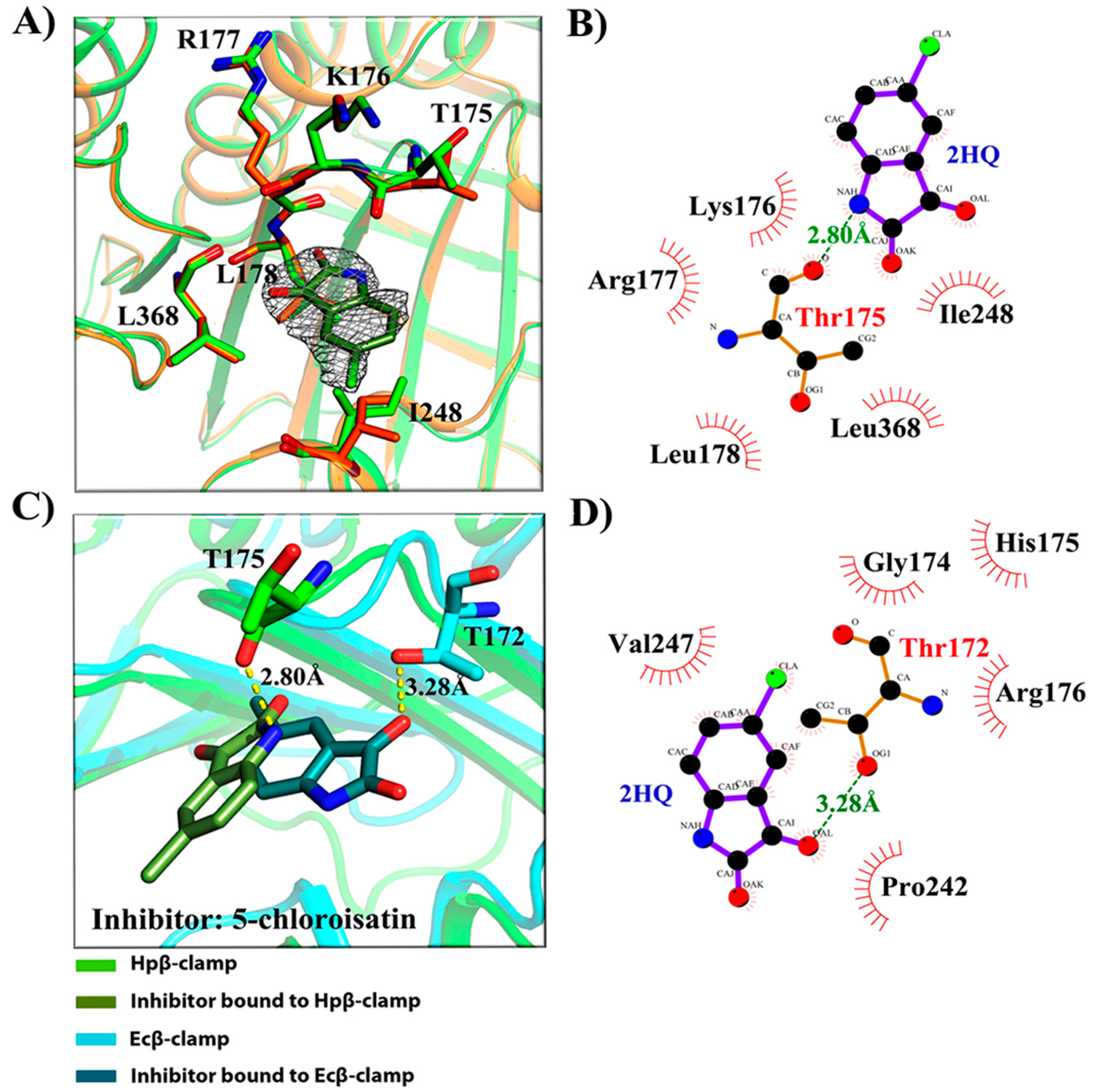
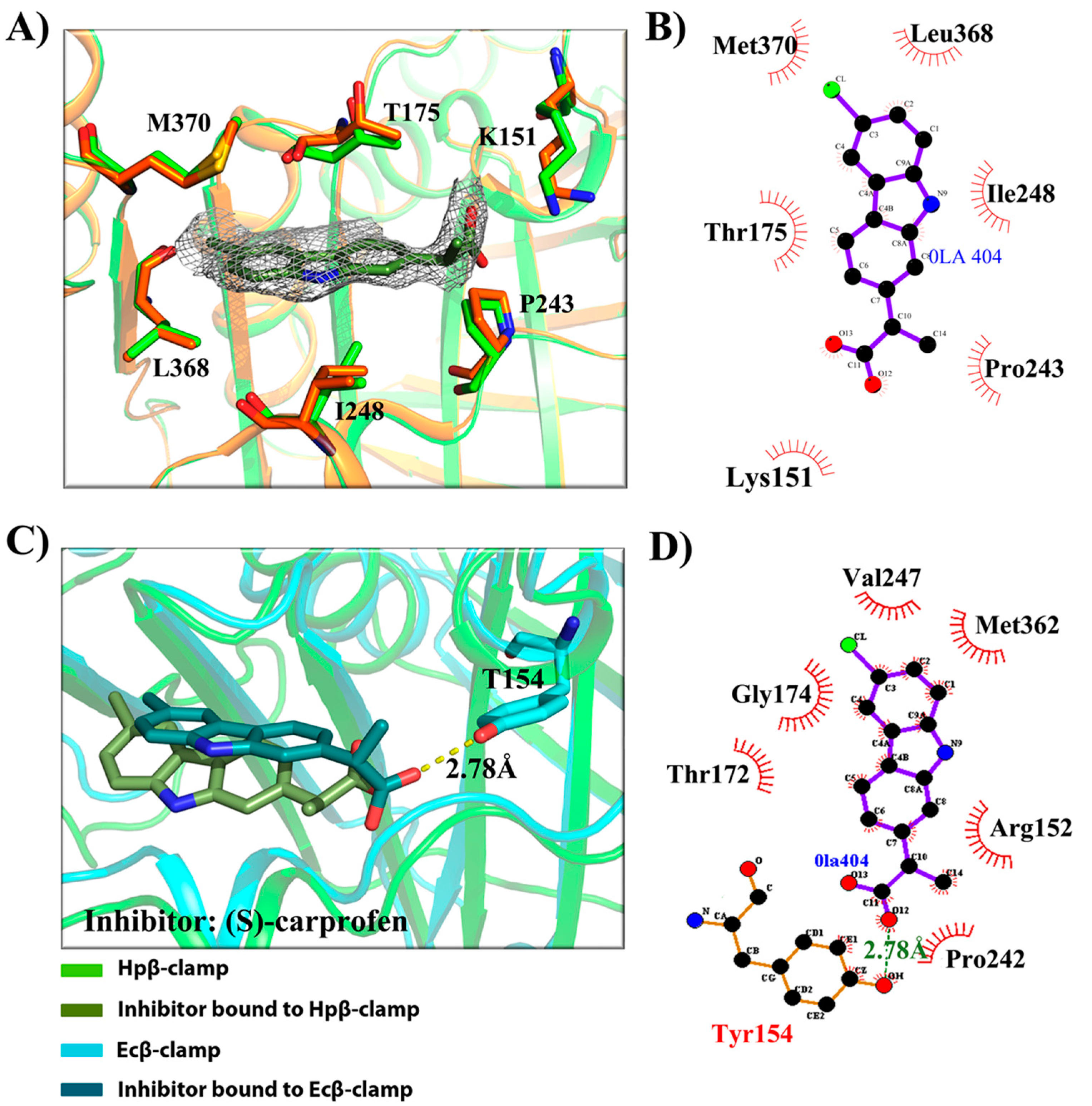
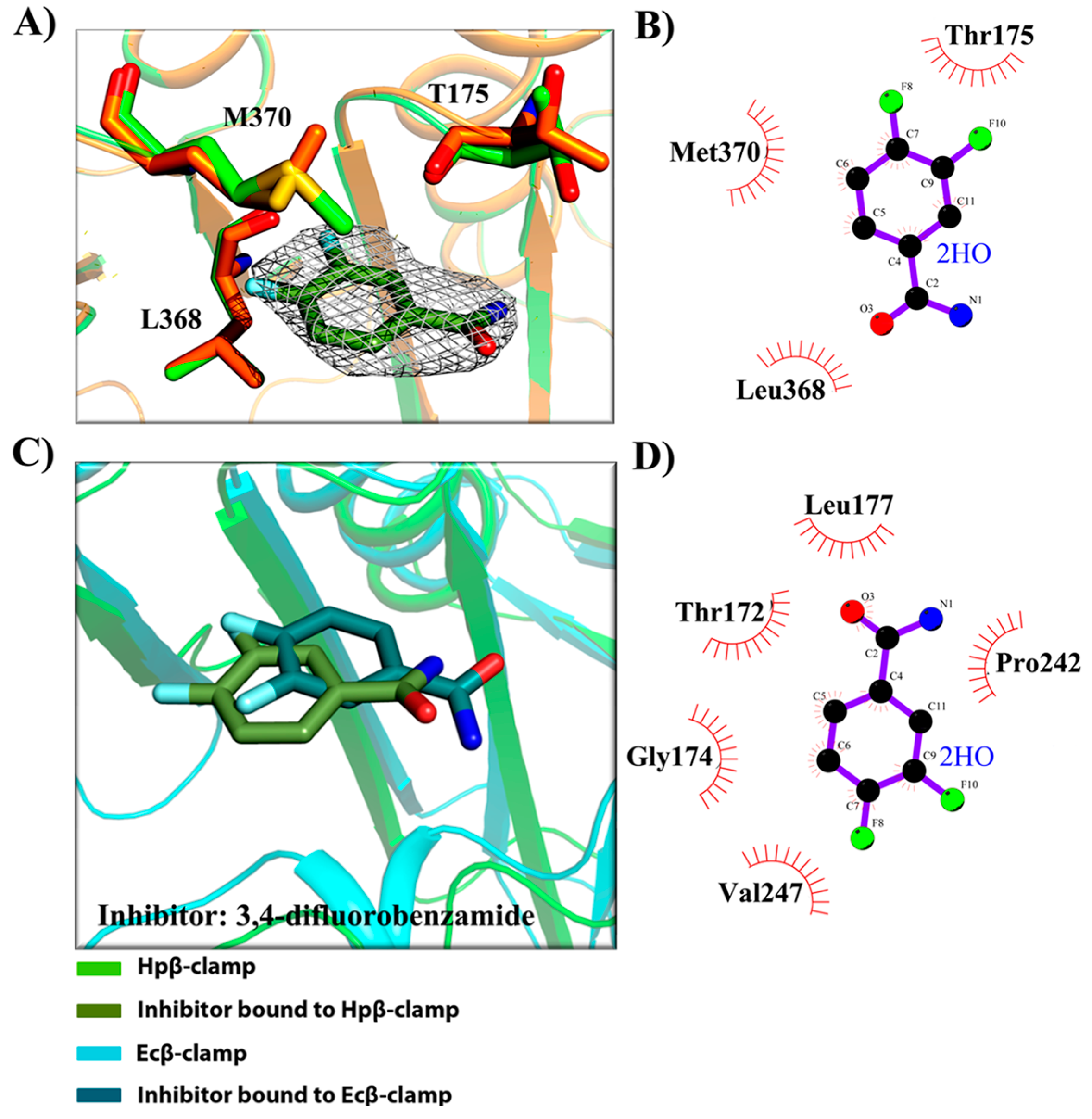
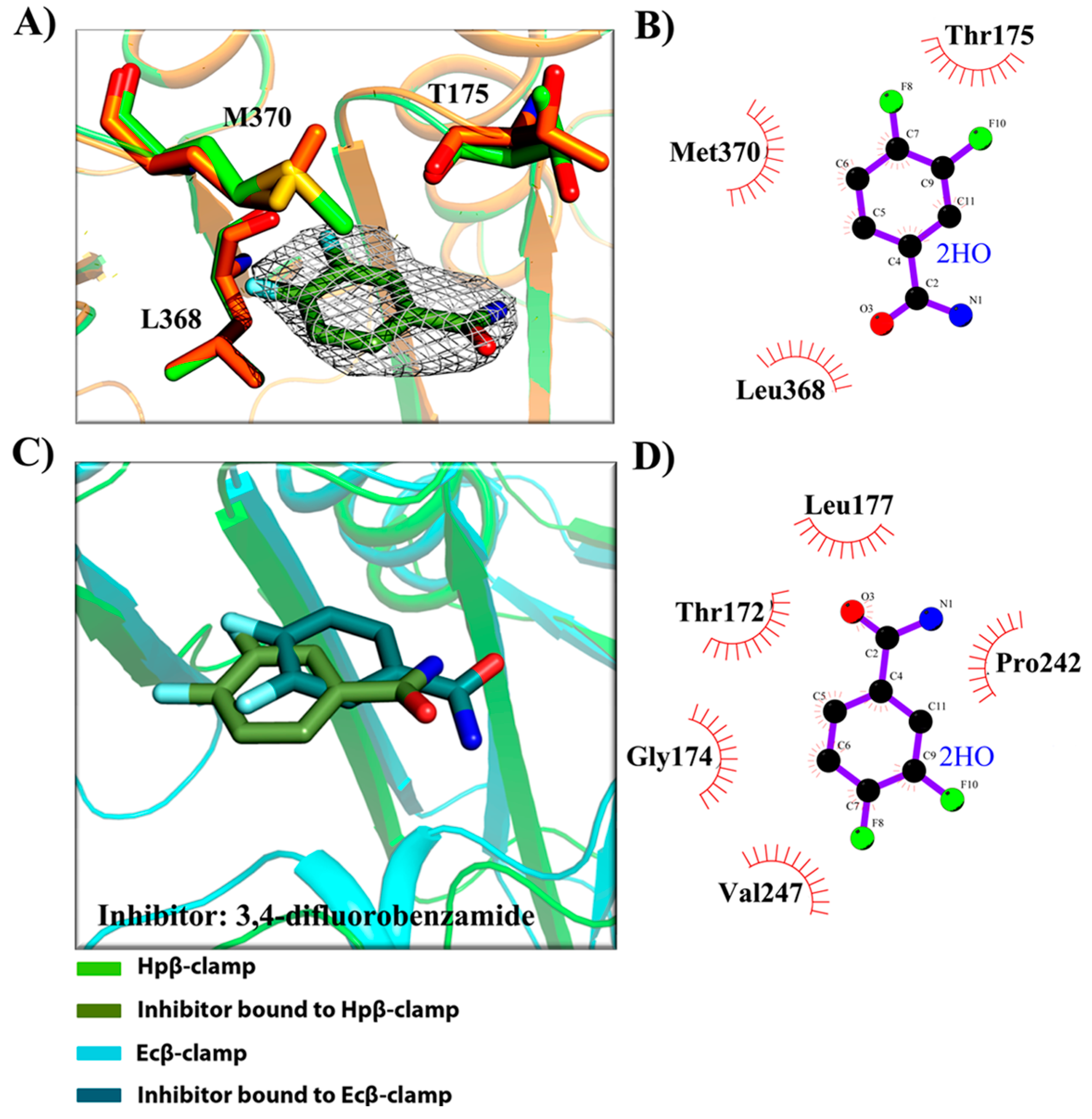
2.3. Binding Pattern Analysis in Complex Crystal Structures
- (a)
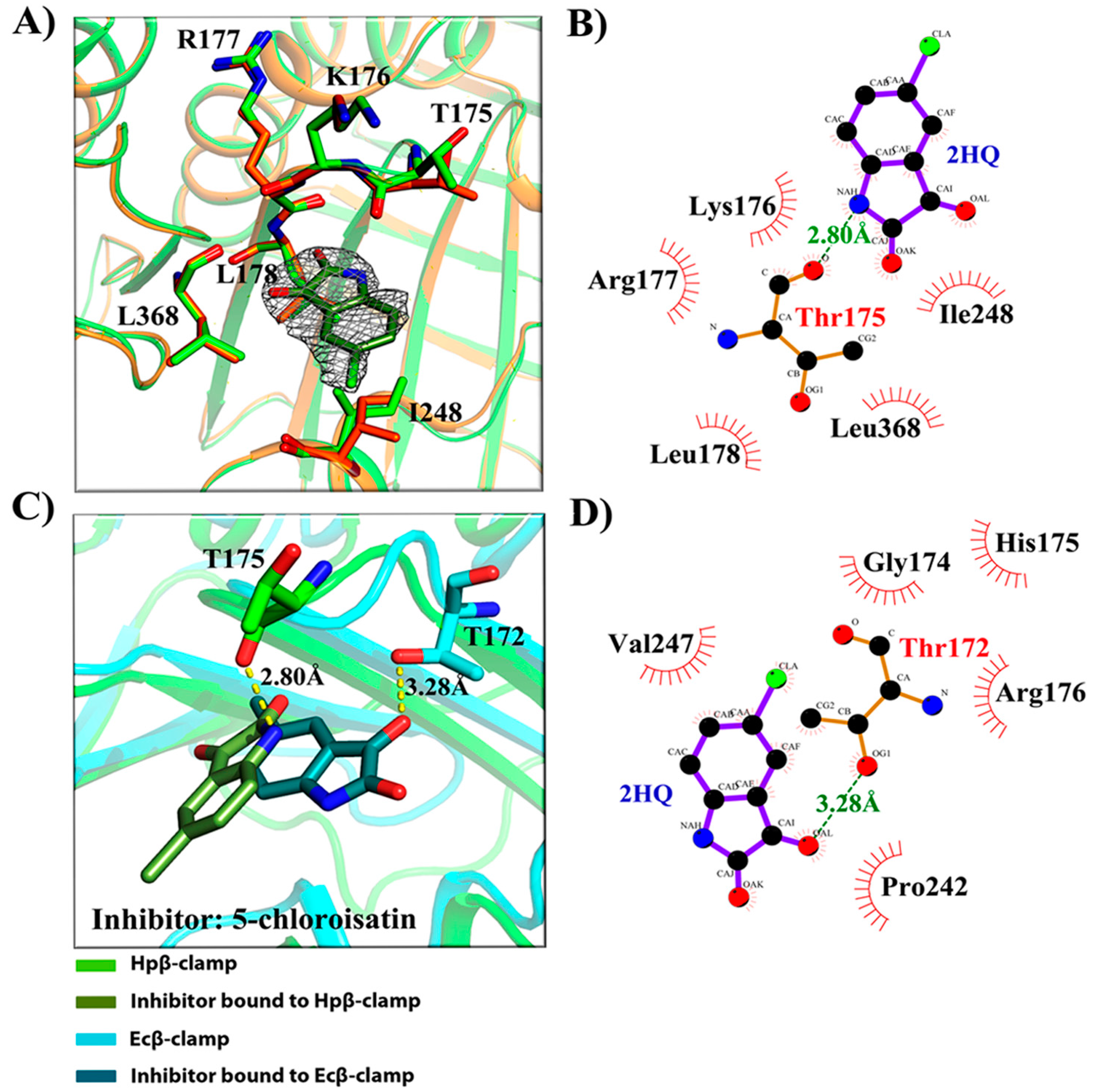
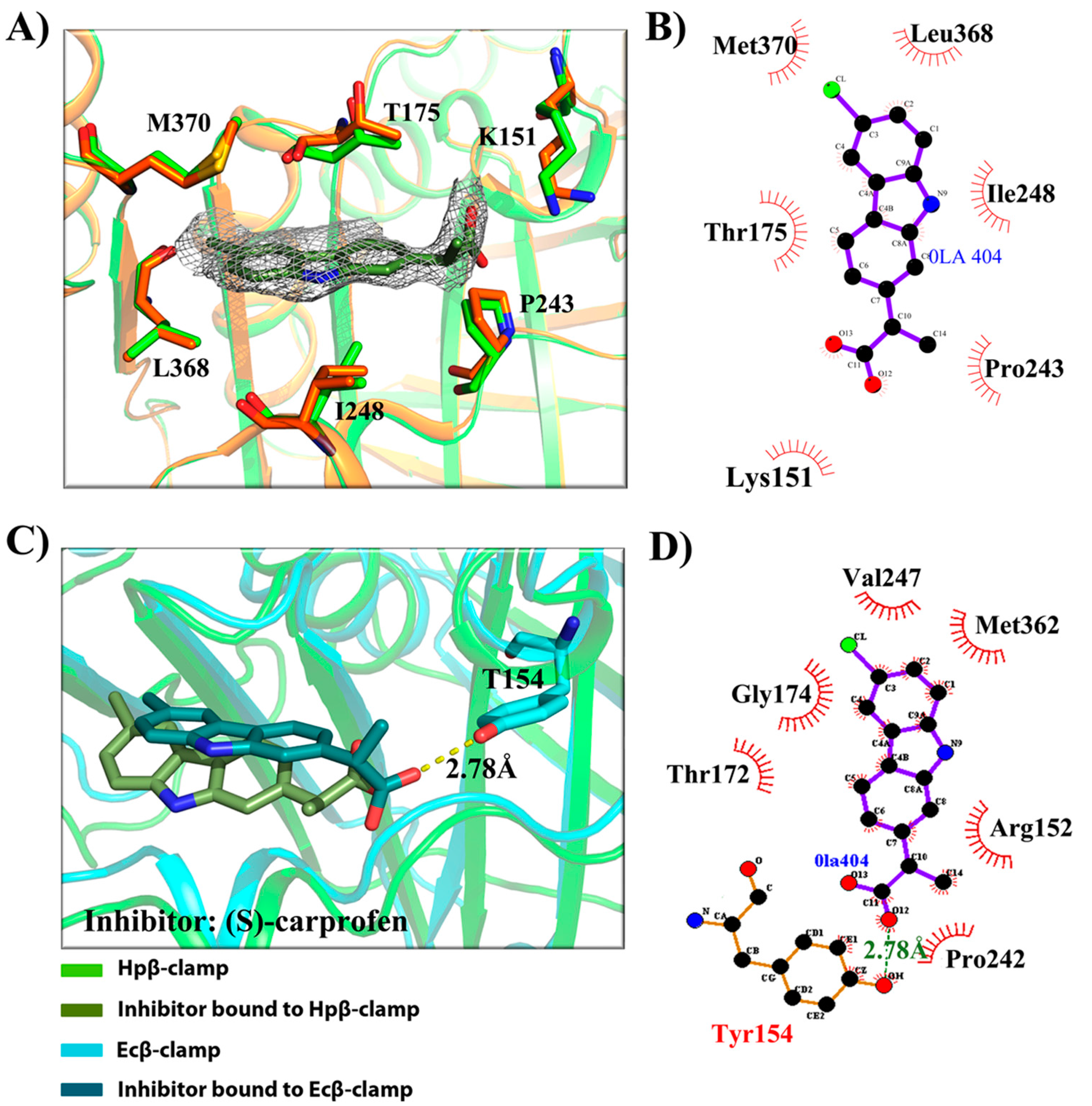
- Binding site of β-clamp is conserved in all bacterial species: The ligand-binding site of β-clamp consists of hydrophobic amino acid residues that have been found to be fairly conserved but not identical in the β-clamps of the various species reported so far. As described in introduction, two protein-binding subsites located near each other, subsite I and subsite II, have been identified in β-clamp (Figure S1). All of the proteins and peptides that have been observed to interact with β-clamp in structures that are deposited in the PDB have been shown to bind to the region of β-clamp encompassing subsites I and II as seen in Ecβ-clamp [9,24,28] and in our earlier studies on ligase peptide-bound Hpβ-clamp [17]. Inspection of the crystal structures of the inhibitor-Hpβ-clamp complexes (PDB id: 5g4q, 5FXT, 5FVE) and inhibitor-Ecβ-clamp complexes (PDB id: 4N95, 4MJR, 4N94) revealed the inhibitor-interacting residues of Hpβ-clamp correspond to those of Ecβ-clamp [9,24,28], suggesting the importance of these residues in the protein-binding cleft (Table S2) (Figure 2, Figure 3 and Figure 4). These residues, especially those corresponding to Thr173, Thr175, Pro243, Ile248, Met370 of subsite I, are quite conserved in β-clamp and are ligand binding residues. Most of these residues also observed to interact with the ligase peptide, clearly indicating that these small molecules will compete with other proteins, like ligase, to bind to β-clamp.
- (b)
- All of the inhibitors occupy subsite I of the protein interaction site: In the crystal structure of Hpβ-clamp complexed with the FIRSLF peptide from HpDNA ligase (PDB ID: 5FRQ), the peptide was observed to occupy both subsites I and II, like other clamp-interacting proteins/peptides from E. coli [16]. Residues Leu360 and Phe361 of this ligase peptide were observed to be buried deep in the Hpβ-clamp cleft and to make hydrophobic contacts with Hpβ-clamp subsite I residues Thr173, Lys176, Ile248, Pro347, Leu368, and Met370 (PDB ID: 5FRQ). Moreover, all of the inhibitors we crystallized with Hpβ-clamp also bound to subsite I. The ligplots of the Hpβ-clamp residues contacting each inhibitor are shown in Figure 2, Figure 3 and Figure 4. The inhibitor-interacting residues were found to be the same as those that were observed to interact with HpDNA ligase (Table S2). All of these inhibitors were observed to interact with most of the residues of subsite I that formed contacts with the ligase peptide. A comparison of the ligand-bound Hpβ-clamp structure with the native Hpβ-clamp structure yielded no significant difference in the positions of the ligand-interacting residues (Figure 2, Figure 3 and Figure 4) except for Lys176 and Met370, which moved a little bit from their native positions to firmly bind the inhibitor. Thr175 of Hpβ-clamp was also observed to make a hydrogen bond with the inhibitor 5-chloroisatin.
- (c)
- Comparison of co-crystal structures of H. pylori and E. coli β-clamp: The comparison of co-complex structures of H. pylori and E. coli with various inhibitors showed some critical differences in the binding pattern. In case of 5-chloroisatin (Figure 2), the Thr175 of Hpβ-clamp makes hydrogen bond with ligand however that in Ecβ-clamp is made by Thr172 in the neighborhood. Out of various hydrophobic contacts between the β-clamp and inhibitor, three of them are common in both H. pylori and E. coli β-clamp. These residues include Ile248, Lys176, Arg177 in Hpβ-clamp, and Val247, His175, Arg176 in Ecβ-clamp. In case of (S)-carprofen (Figure 3), Thr154 of Ecβ-clamp makes H-bond with the ligand along with other hydrophobic contacts however in H. pylori the contact between β-clamp and ligand is favored by only hydrophobic interactions. Five of these interactions are common in both, which includes residues Lys151, Thr175, Pro243, Ile248, Met370 in Hpβ-clamp and Arg152, Gly174, Pro242, Val247, Met362 in Ecβ-clamp. In case of 3, 4-difluorobenzamide (Figure 4), the interaction between β-clamp and ligand in both H. pylori and E. coli is dominated by hydrophobic interactions. Among them, three of these interactions are common in both the clamps, Thr175, Pro243, Ile248 in Hpβ-clamp and Gly174, Pro242, Val247 in Ecβ-clamp, respectively. All of the ligand-interacting residues of Hpβ-clamp as well as Ecβ-clamp are tabulated (Table S2). The structure-based sequence alignment of Hpβ-clamp and Ecβ-clamp is shown in Figure 5 with highlighted ligand-interacting residues, which shows that the mode of interaction between inhibitor/ligand and β-clamp of both organisms are almost same.
- (d)
- Comparison of the co-crystal Hpβ-clamp structures with docked structure: In all of the docked structures, like in crystal structure, the inhibitors were found to be present in the same pocket (Figure S2). The orientation of the 5-chloroisatin inhibitor in the docked structure did differ from that in the crystal structure (Figure S2), but this difference did not affect much the structure of the binding site. Hpβ-clamp residue Thr173 form hydrogen bond with every docked inhibitor, while residue Lys247 hydrogen bonded the docked inhibitors (S)-carprofen. In contrast, in the actual crystal structure, only 5-chloroisatin was observed to make a hydrogen bond with Thr175 (Figure 2B). In every case, hydrophobic contact dominates the interactions between the protein and inhibitor. Interestingly, all of the docked ligands and ligands in co-crystal structures bound in the same pocket with similar hydrophobic interactions, but the hydrogen bond pattern did not match.
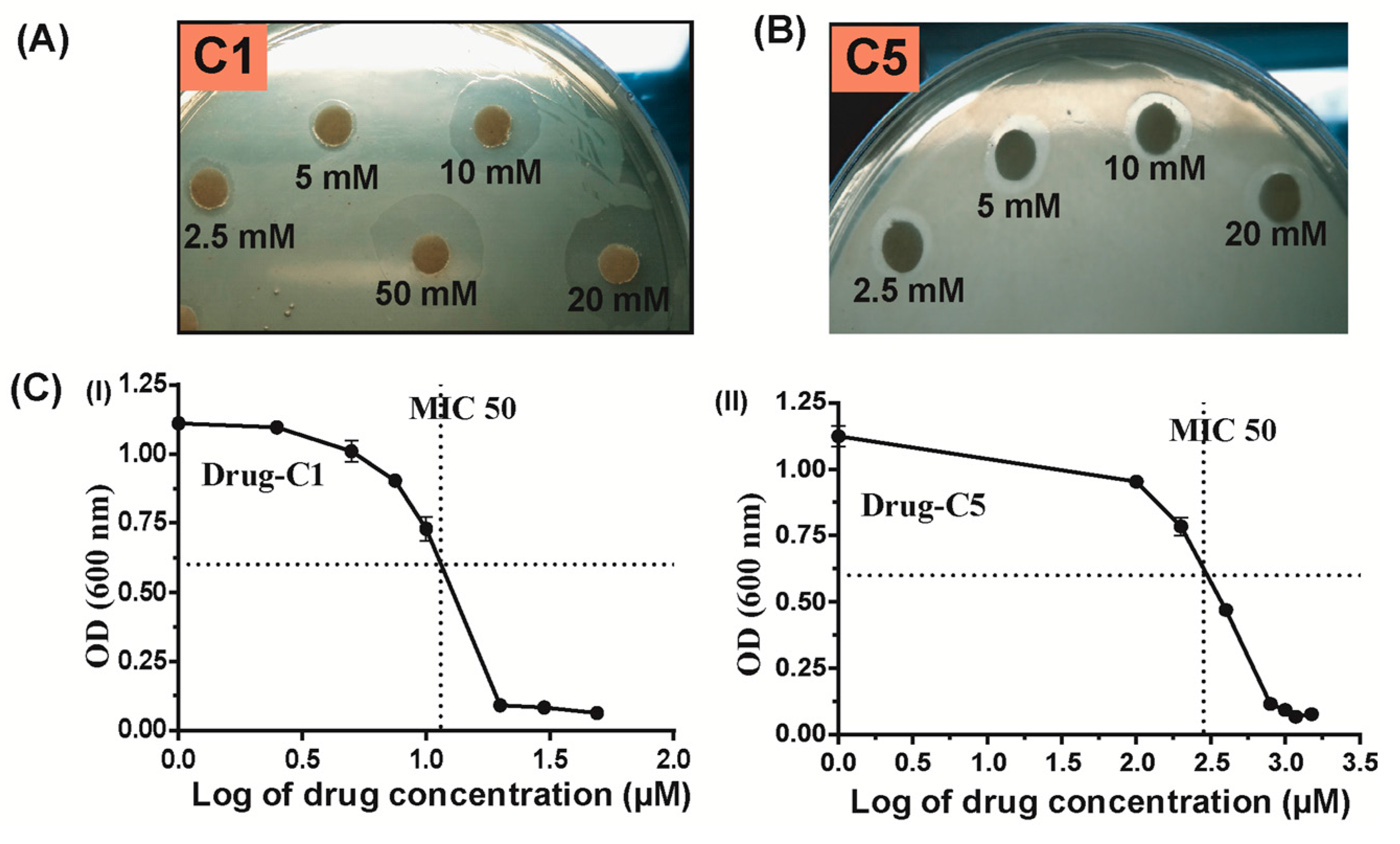
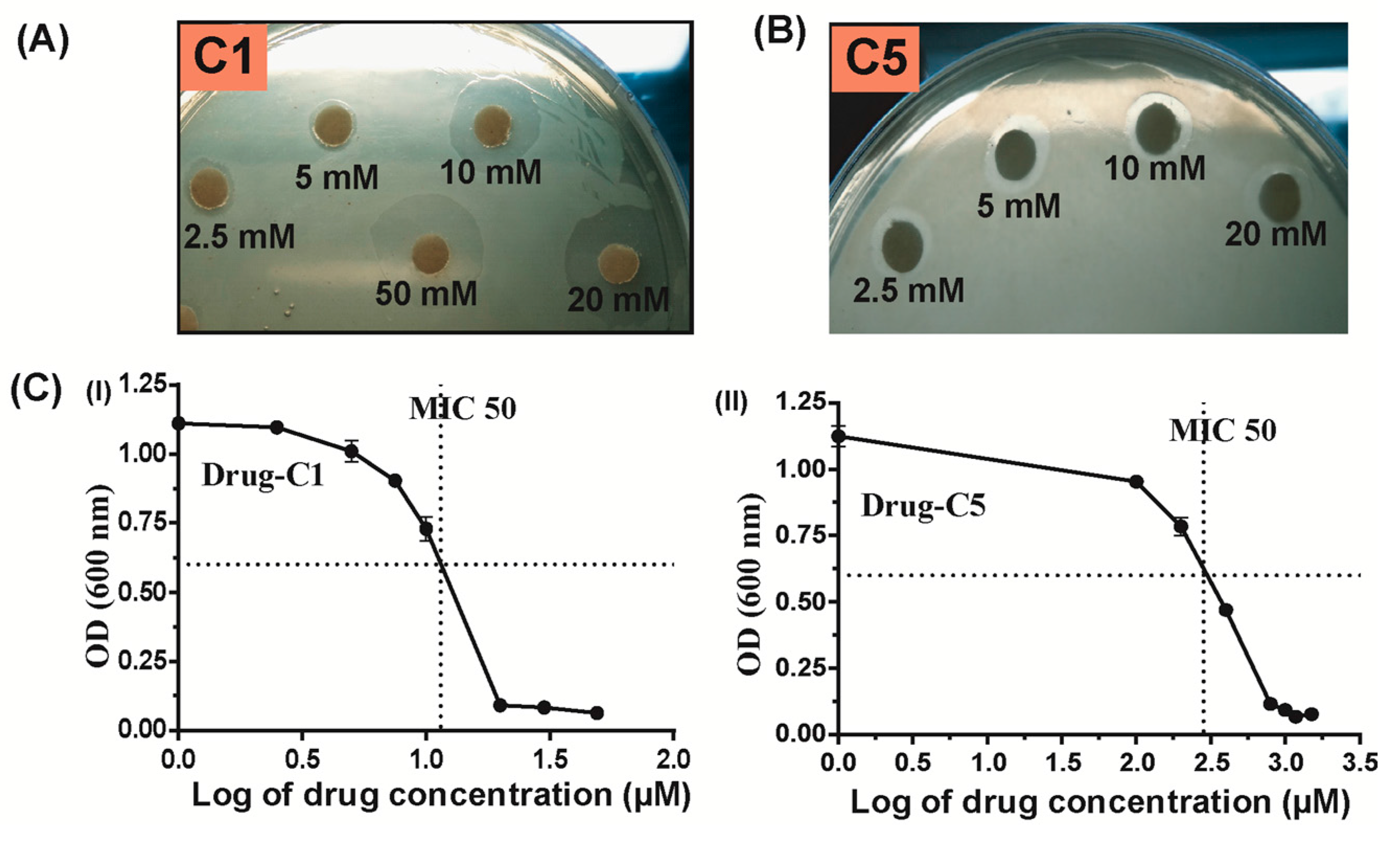
2.4. Antibacterial Activity of Hpβ-clamp Inhibitors
3. Material and Methods
3.1. Overexpression and Purification of Hpβ-clamp
3.2. In Silico Screening of Inhibitors
3.3. Surface Competition Assay
3.4. Crystallization, Structure Determination and Analysis of the Complex Structures
3.5. Antimicrobial Test
- (a)
- H. pylori strain and culture conditions: The H. pylori strain and culture conditions were used, as mentioned in our previous work [17]. In brief, Brain heart infusion (BHI) agar/broth medium was used to culture the H. pylori strain 26695. The agar/broth medium contains the antibiotics amphotericin B (8 mg/mL), trimethoprim (5 mg/mL), and vancomycin (6 mg/mL), 8% horse blood serum and 0.4% IsoVitaleX. The bacterial plates were incubated under microaerobic conditions (5% O2, 10% CO2) at 37 °C for 36 h and the broth cultures were incubated at 37 °C on a shaker operating at 110 rpm under microaerobic conditions (5% O2, 10% CO2).
- (b)
- Anti-H. pylori susceptibility tests: To screen the susceptibility of H. pylori against drugs the Kirby-Bauer disk diffusion susceptibility test was used, as mentioned earlier [17,29]. In brief, 200 μL of the H. pylori suspension was spread evenly on 90 mm BHI-agar plates. Because of the slow growth of H. pylori, high inoculum was used for the disk diffusion method. Different concentrations of inhibitors (Sigma, St Louis, MI, USA) were inoculated (10 μL of each drug or DMSO as control) on 6 mm disks, which are placed on bacterial lawn surface. The disks were dried and incubated at 37 °C under micro-aerophilic conditions for about 3–5 days.
- (c)
- Minimum inhibitory concentrations (MICs): The dilution method was applied to determine the MICs of drugs described earlier [17]. In brief, different concentrations of the drugs were used in 2 mL BHI broth that was inoculated with fresh H. pylori suspensions (20 µL; 1:100 dilution; initial optical density OD600 < 0.1). Further, the tubes containing broth and different concentration of drugs were incubated under microaerobic condition for 20 h at 37 °C on a shaker (110 rpm). After 20 h, the OD600 were taken and GraphPad software was used for analysis of MICs. OD600 measurements were also taken for the H. pylori suspension without drugs or with kanamycin (positive control) and with/without bacteria. All of the experiments were done in triplicate.
4. Conclusions
Supplementary Materials
Acknowledgements and Funding
Author Contributions
Conflicts of Interest
References
- Khatoon, J.; Rai, R.P.; Prasad, K.N. Role of Helicobacter pylori in gastric cancer: Updates. World J. Gastrointest. Oncol. 2016, 8, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, M.D.; Duckworth, C.A.; Williams, J.M.; Pritchard, D.M. Helicobacter pylori-induced gastric pathology: Insights from in vivo and ex vivo models. Dis. Model. Mech. 2017, 10, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, F.; Campbell, B.J.; Alfizah, H.; Varro, A.; Zahra, R.; Yamaoka, Y.; Pritchard, D.M. Analysis of clinical isolates of Helicobacter pylori in pakistan reveals high degrees of pathogenicity and high frequencies of antibiotic resistance. Helicobacter 2014, 19, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Wang, Y.; Whittell, L.R.; Jergic, S.; Liu, M.; Harry, E.; Dixon, N.E.; Kelso, M.J.; Beck, J.L.; Oakley, A.J. DNA replication is the target for the antibacterial effects of nonsteroidal anti-inflammatory drugs. Chem. Biol. 2014, 21, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Danks, M.K. Topoisomerase enzymes as drug targets. Curr. Opin. Oncol. 1999, 11, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Saro, F.J.; O’Donnell, M. Interaction of the beta sliding clamp with muts, ligase, and DNA polymerase I. Proc. Natl. Acad. Sci. USA 2001, 98, 8376–8380. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J., Jr.; Bryan, S.K.; Chen, H.; Moses, R.E.; McHenry, C.S. Escherichia coli DNA polymerase II is stimulated by DNA polymerase III holoenzyme auxiliary subunits. J. Biol. Chem. 1991, 266, 4568–4573. [Google Scholar] [PubMed]
- Bonner, C.A.; Stukenberg, P.T.; Rajagopalan, M.; Eritja, R.; O’Donnell, M.; McEntee, K.; Echols, H.; Goodman, M.F. Processive DNA synthesis by DNA polymerase II mediated by DNA polymerase III accessory proteins. J. Biol. Chem. 1992, 267, 11431–11438. [Google Scholar] [PubMed]
- Bunting, K.A.; Roe, S.M.; Pearl, L.H. Structural basis for recruitment of translesion DNA polymerase pol IV/DinB to the beta-clamp. EMBO J. 2003, 22, 5883–5892. [Google Scholar] [CrossRef] [PubMed]
- Patoli, A.A.; Winter, J.A.; Bunting, K.A. The umuc subunit of the E. Coli DNA polymerase V shows a unique interaction with the beta-clamp processivity factor. BMC Struct. Biol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Kurz, M.; Dalrymple, B.; Wijffels, G.; Kongsuwan, K. Interaction of the sliding clamp beta-subunit and Hda, a DnaA-related protein. J. Bacteriol. 2004, 186, 3508–3515. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, B.P.; Kongsuwan, K.; Wijffels, G.; Dixon, N.E.; Jennings, P.A. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc. Natl. Acad. Sci. USA 2001, 98, 11627–11632. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Saro, F.J.; Georgescu, R.E.; O’Donnell, M. A peptide switch regulates DNA polymerase processivity. Proc. Natl. Acad. Sci. USA 2003, 100, 14689–14694. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Kelso, M.J.; Beck, J.L.; Oakley, A.J. Structural and thermodynamic dissection of linear motif recognition by the E. coli sliding clamp. J. Med. Chem. 2013, 56, 8665–8673. [Google Scholar] [CrossRef] [PubMed]
- Wolff, P.; Amal, I.; Olieric, V.; Chaloin, O.; Gygli, G.; Ennifar, E.; Lorber, B.; Guichard, G.; Wagner, J.; Dejaegere, A.; et al. Differential modes of peptide binding onto replicative sliding clamps from various bacterial origins. J. Med. Chem. 2014, 57, 7565–7576. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Tarique, K.F.; Mazumder, M.; Rehman, S.A.A.; kumari, N.; Gourinath, S. Structural insight into β-clamp and its interaction with DNA ligase in Helicobacter pylori. Sci. Rep. 2016, 6, 31181. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Verma, V.; Gautam, G.; Kumari, N.; Dhar, S.K.; Gourinath, S. Targeting the beta-clamp in Helicobacter pylori with FDA-approved drugs reveals micromolar inhibition by diflunisal. FEBS Lett. 2017, 591, 2311–2322. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Whittell, L.R.; Wang, Y.; Jergic, S.; Liu, M.; Harry, E.J.; Dixon, N.E.; Beck, J.L.; Kelso, M.J.; Oakley, A.J. Discovery of lead compounds targeting the bacterial sliding clamp using a fragment-based approach. J. Med. Chem. 2014, 57, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Furuta, Y.; Yahara, K.; Tsuru, T.; Oshima, K.; Handa, N.; Takahashi, N.; Yoshida, M.; Azuma, T.; Hattori, M.; et al. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiol. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Abdul Rehman, S.A.; Verma, V.; Mazumder, M.; Dhar, S.K.; Gourinath, S. Crystal structure and mode of helicase binding of the C-terminal domain of primase from Helicobacter pylori. J. Bacteriol. 2013, 195, 2826–2838. [Google Scholar] [CrossRef] [PubMed]
- Kashav, T.; Nitharwal, R.; Abdulrehman, S.A.; Gabdoulkhakov, A.; Saenger, W.; Dhar, S.K.; Gourinath, S. Three-dimensional structure of N-terminal domain of dnab helicase and helicase-primase interactions in Helicobacter pylori. PLoS ONE 2009, 4, e7515. [Google Scholar] [CrossRef] [PubMed]
- Nitharwal, R.G.; Verma, V.; Subbarao, N.; Dasgupta, S.; Choudhury, N.R.; Dhar, S.K. DNA binding activity of Helicobacter pylori dnab helicase: The role of the N-terminal domain in modulating DNA binding activities. FEBS J. 2012, 279, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Kumar, A.; Nitharwal, R.G.; Alam, J.; Mukhopadhyay, A.K.; Dasgupta, S.; Dhar, S.K. Modulation of the enzymatic activities of replicative helicase (DnaB) by interaction with Hp0897: A possible mechanism for helicase loading in Helicobacter pylori. Nucleic Acids Res. 2016, 44, 3288–3303. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, R.E.; Yurieva, O.; Kim, S.S.; Kuriyan, J.; Kong, X.P.; O’Donnell, M. Structure of a small-molecule inhibitor of a DNA polymerase sliding clamp. Proc. Natl. Acad. Sci. USA 2008, 105, 11116–11121. [Google Scholar] [CrossRef] [PubMed]
- Wolff, P.; Olieric, V.; Briand, J.P.; Chaloin, O.; Dejaegere, A.; Dumas, P.; Ennifar, E.; Guichard, G.; Wagner, J.; Burnouf, D.Y. Structure-based design of short peptide ligands binding onto the E. Coli processivity ring. J. Med. Chem. 2011, 54, 4627–4637. [Google Scholar] [CrossRef] [PubMed]
- Wijffels, G.; Johnson, W.M.; Oakley, A.J.; Turner, K.; Epa, V.C.; Briscoe, S.J.; Polley, M.; Liepa, A.J.; Hofmann, A.; Buchardt, J.; et al. Binding inhibitors of the bacterial sliding clamp by design. J. Med. Chem. 2011, 54, 4831–4838. [Google Scholar] [CrossRef] [PubMed]
- Biacore. Biacore Concentration Analysis Handbook; Biacore: Žilina Region, Slovakia, 2001. [Google Scholar]
- Jeruzalmi, D.; Yurieva, O.; Zhao, Y.; Young, M.; Stewart, J.; Hingorani, M.; O’Donnell, M.; Kuriyan, J. Mechanism of processivity clamp opening by the delta subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell 2001, 106, 417–428. [Google Scholar] [CrossRef]
- Nariman, F.; Eftekhar, F.; Habibi, Z.; Falsafi, T. Anti-helicobacter pylori activities of six iranian plants. Helicobacter 2004, 9, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2016-1: Maestro, version 10.5; Schrödinger, LLC: New York, NY, USA, 2016.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Minor, Z.O.A.W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | |||
|---|---|---|---|
| β-clamp Complexed with 5-chloroisatin | β-clamp Complexed with Carprofen | β-clamp Complexed with 3,4-difluorobenzamide | |
| Space group | P21 | C2 | C2 |
| Cell dimensions | |||
| a,b,c (Å) | 82.1, 65.4, 88.8 | 89.9, 66.4, 82.8 | 90.0, 66.3, 82.7 |
| α,β,γ (deg.) | 90.0, 115.7, 90.0 | 90.0, 115.5, 90.0 | 90.0, 115.4, 90.0 |
| Rsym (highest resolution range) | 5.0 (43.8) | 5.1 (21.6) | 5.5 (42.1) |
| Completeness (highest resolution range) | 91.1 (89.0) | 98.5 (79.7) | 98.8 (90.9) |
| Mean I/σ | 20.2 | 38.8 | 33.17 |
| Refinement | |||
| Resolution range (Å) | 50.0–2.3 | 50.0–1.97 | 50.0–2.07 |
| Rwork/Rfree | 22.7/28.5 | 20.6/24.3 | 21.3/25.2 |
| Number of atoms | |||
| Protein | 5588 | 3021 | 2883 |
| Water | 57 | 55 | 92 |
| R.m.s. deviation | |||
| Bond angles (deg.) | 1.78 | 1.16 | 1.98 |
| Bond lengths (Å) | 0.015 | 0.005 | 0.007 |
| Mean B value (Å2) | 50.3 | 50.8 | 44.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, P.; Verma, V.; Dhar, S.K.; Gourinath, S. Screening of E. coli β-clamp Inhibitors Revealed that Few Inhibit Helicobacter pylori More Effectively: Structural and Functional Characterization. Antibiotics 2018, 7, 5. https://doi.org/10.3390/antibiotics7010005
Pandey P, Verma V, Dhar SK, Gourinath S. Screening of E. coli β-clamp Inhibitors Revealed that Few Inhibit Helicobacter pylori More Effectively: Structural and Functional Characterization. Antibiotics. 2018; 7(1):5. https://doi.org/10.3390/antibiotics7010005
Chicago/Turabian StylePandey, Preeti, Vijay Verma, Suman Kumar Dhar, and Samudrala Gourinath. 2018. "Screening of E. coli β-clamp Inhibitors Revealed that Few Inhibit Helicobacter pylori More Effectively: Structural and Functional Characterization" Antibiotics 7, no. 1: 5. https://doi.org/10.3390/antibiotics7010005
APA StylePandey, P., Verma, V., Dhar, S. K., & Gourinath, S. (2018). Screening of E. coli β-clamp Inhibitors Revealed that Few Inhibit Helicobacter pylori More Effectively: Structural and Functional Characterization. Antibiotics, 7(1), 5. https://doi.org/10.3390/antibiotics7010005