1. Introduction
DNA-binding proteins that regulate expression of efflux pump-encoding genes have significance in controlling levels of bacterial resistance to clinically important antibiotics and other antimicrobials [
1,
2,
3,
4,
5]. A well-established, and often repeated, observation is that mutations that abrogate the DNA-binding ability of these regulators or expression of their respective gene can, depending on the nature of the protein, increase or decrease efflux pump gene expression and levels of bacterial resistance to antimicrobials recognized by the cognate efflux pump [
6,
7,
8,
9,
10,
11,
12]. Results from recent studies suggest, however, that such DNA-binding proteins also participate in the regulation of other genes that are important in the overall biology of the bacterium [
7,
13,
14]; we have termed such regulated genes as being “off-target” [
14].
While a considerable amount of information is available regarding how DNA-binding proteins control efflux pump gene expression, less is known about how they influence the expression of “off-target” genes and the impact such regulation has for bacterial physiology and pathogenesis. For instance, results from a transcriptional profiling study performed by us [
7] defined the MtrR regulon of
N. gonorrhoeae strain FA19 as consisting of at least 47 repressed and 22 activated genes. MtrR is a member of the TetR/QacR family of DNA-binding proteins that are known for their capacity to repress bacterial genes encoding drug efflux proteins [
2,
8,
15,
16]. MtrR represses transcription of the
mtrCDE efflux pump operon [
15,
16] by binding as two homodimers to a DNA sequence containing two pseudo-direct repeats within the
mtrCDE promoter region [
8,
10]. Missense mutations that cause radical amino changes (e.g., A39T or G45D) in the helix-turn-helix motif of MtrR significantly decrease its DNA-binding activity resulting in increased expression of
mtrCDE and decreased susceptibility of gonococci to hydrophobic drugs, dyes, and detergents [
11]. We hypothesize that such regulation (and loss thereof) is important
in vivo, as null mutations in
mtrR increase fitness of gonococci
in vivo when assessed in a female mouse model of lower genital tract infection [
17,
18]. Expression of
mtrR is controlled by a
cis-acting 13 bp inverted repeat sequence within the
mtrR promoter, which overlaps the adjacent but divergent
mtrCDE promoter; a single bp deletion within this sequence abolishes
mtrR expression [
15,
19]. Expression of
mtrR is also subject to repression by the product of the
mpeR gene [
20], which is in-turn repressed by Fur (Ferric Uptake Regulator) in the presence of iron [
21] (
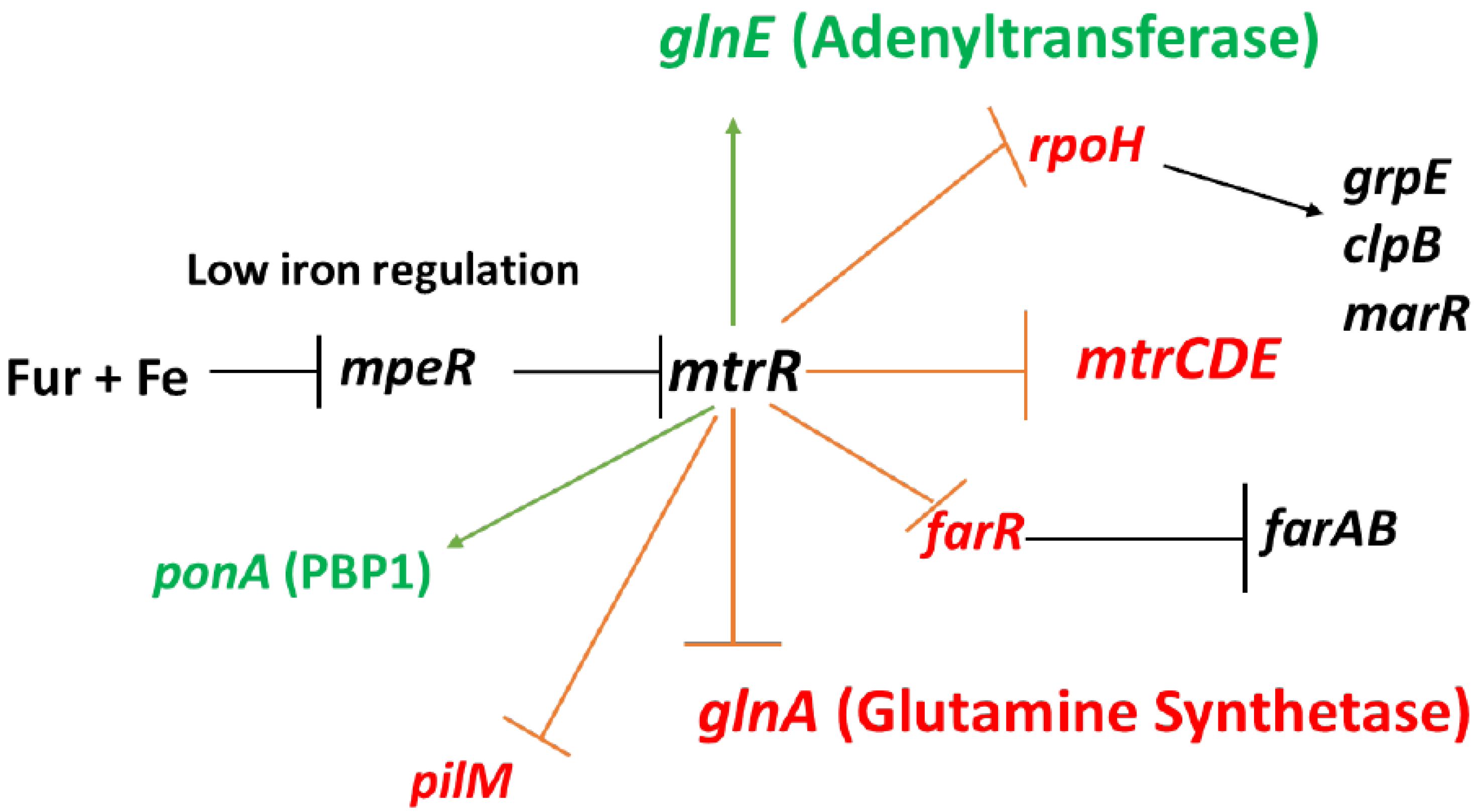
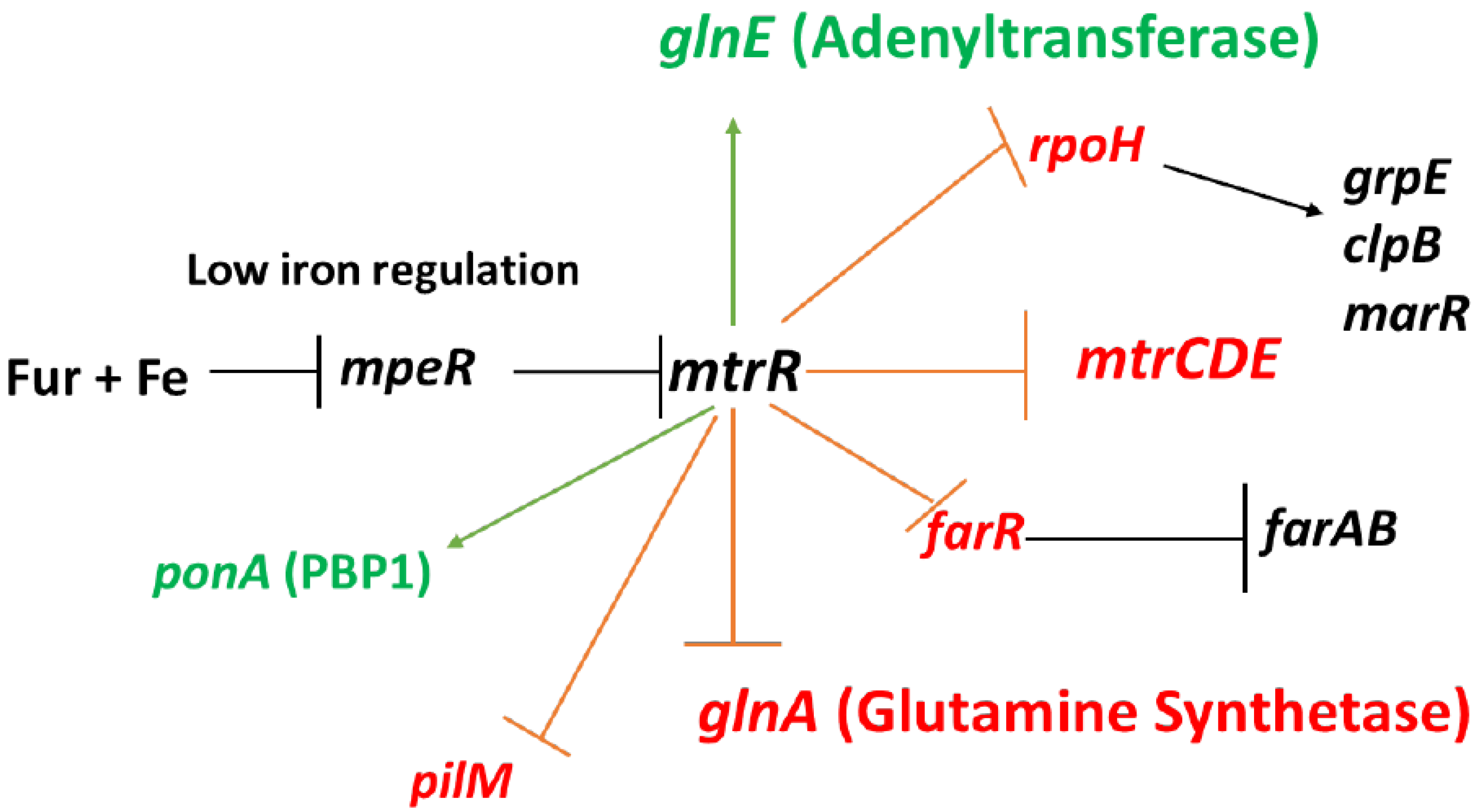
Figure 1).
In addition to repressing expression of
mtrCDE, MtrR also directly down-regulates expression of other genes such as:
farR (encodes the repressor of the
farAB efflux pump operon) [
9],
rpoH (encodes the major stress-related sigma factor produced by gonococci) [
7],
ponA (encodes penicillin-binding protein 1) [
13], and
glnA (encoding glutamine synthetase) [
7,
14]; these regulatory schemes are summarized in
Figure 1. MtrR also indirectly activates gonococcal genes such as
pilM [
7] and
farAB [
9], but evidence for its capacity to directly activate genes has heretofore been lacking. In order to test if MtrR can serve as a direct activator of gonococcal genes, we studied its regulation of
glnE since its expression was significantly decreased in an MtrR-negative mutant compared to the wild-type parent strain [
7].
Figure 1.
MtrR regulation of gonococcal genes. Shown are genes subject to direct or indirect regulation by MtrR (see text for details). Genes under negative control by MtrR are indicated by a red barred line extending from mtrR while those activated by MtrR are indicated by a green arrow extending from mtrR; other important regulated genes are shown with black arrowed or barred lines to signify transcriptional activation or repression of expression, respectively. Repression of mtrR expression by the product of the mpeR gene which is in-turn negatively controlled by Fur + iron are also shown.
Figure 1.
MtrR regulation of gonococcal genes. Shown are genes subject to direct or indirect regulation by MtrR (see text for details). Genes under negative control by MtrR are indicated by a red barred line extending from mtrR while those activated by MtrR are indicated by a green arrow extending from mtrR; other important regulated genes are shown with black arrowed or barred lines to signify transcriptional activation or repression of expression, respectively. Repression of mtrR expression by the product of the mpeR gene which is in-turn negatively controlled by Fur + iron are also shown.
We hypothesize that the fidelity of the glutamine biosynthesis pathway is important for optimal physiology and metabolism of gonococci during infection since the levels of this amino acid are very low in mucosal fluids and within phagocytes compared to the levels found in blood [
22]. Further, since
glnE has been reported to be an essential gene in other pathogens such as
Mycobacterium tuberculosis [
23,
24] and we have been unable to construct a gonococcal
glnE null mutant (data not presented), we posit that the opposing regulatory properties of MtrR on
glnA and
glnE likely serves to modulate the fidelity of glutamine biosynthesis during infection.
2. Results and Discussion
Given our inability (see above) to isolate a
glnE null mutant of gonococci (data not presented), which suggests that this gene is essential gene in this pathogen), and the results from transcriptional profiling studies that suggested it is an MtrR-activated gene [
7], we hypothesize that MtrR regulation of
glnE is of importance for fidelity of glutamine biosynthesis and the overall physiology of gonococci. Accordingly, to test this possibility we examined if such regulation is direct or indirect and used DNA-binding assays to distinguish these possibilities. This is an important regulatory issue to resolve as MtrR can indirectly activate other genes (e.g., the
farAB efflux operon) by its ability to repress expression of genes encoding other transcriptional repressors (e.g.,
farR) [
9]. In contrast to its previously reported ability to repress the expression of
mtrCDE [
8,
10,
11] and other gonococcal genes [
7,
12,
13,
14], microarray work by Folster
et al. [
7] indicated that MtrR significantly (seven-fold;
p ≤ 0.05) enhances
glnE expression in strain FA19. In confirmation of this report, using qRT-PCR with RNA extracted from MtrR-positive strain FA19 and its MtrR-negative deletion mutant strain JF1 [
7,
17], we found a nine-fold reduction in the
glnE transcript level in strain JF1 compared to parental strain FA19 (data not presented).
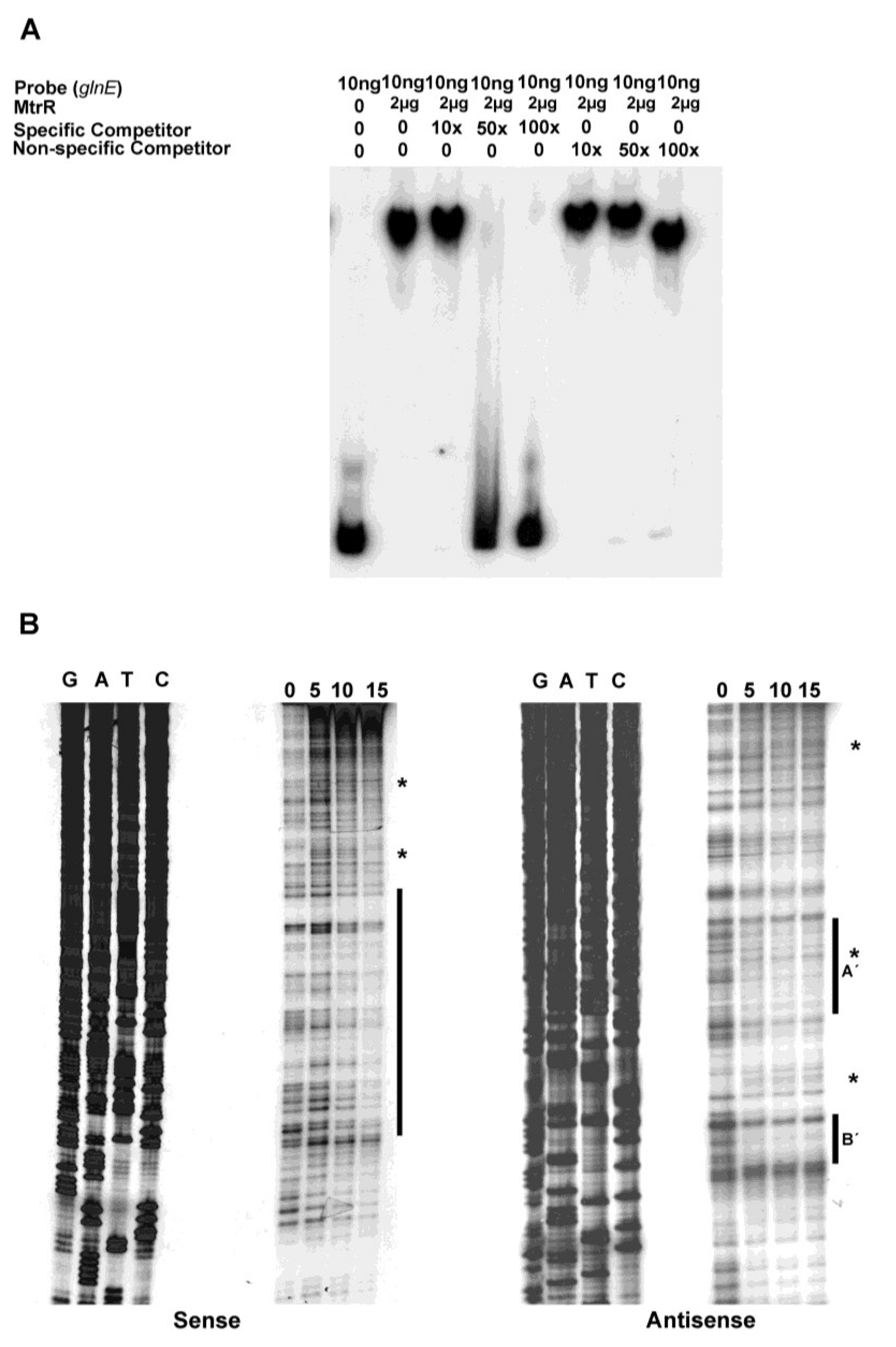
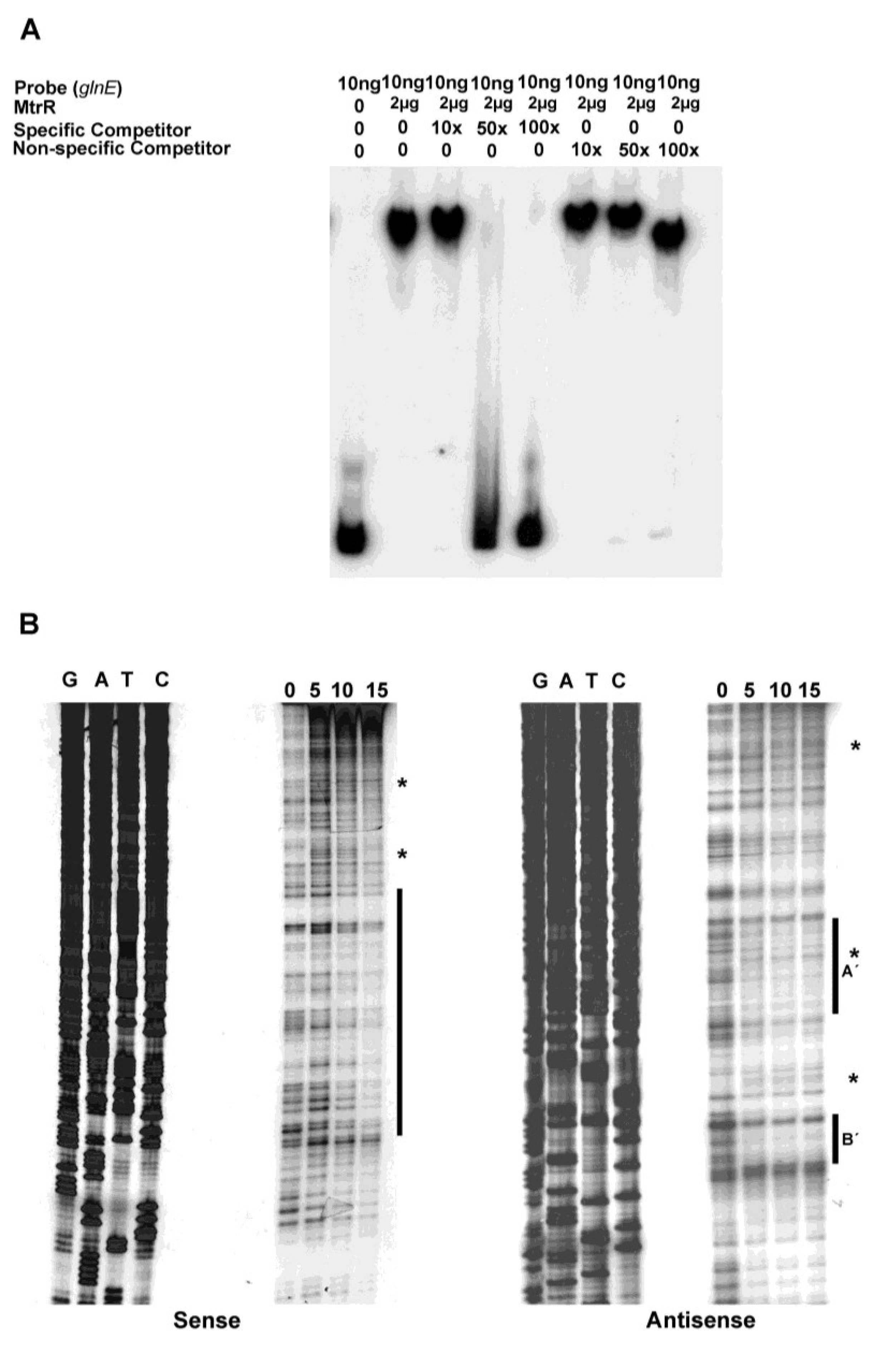
In order to determine if MtrR activation of
glnE is by a direct mechanism, we tested if it can bind to the sequence upstream of
glnE (
Figure 2). Based on the published MtrR-binding site sequences upstream of
mtrCDE [
7,
10],
farR [
9], and
rpoH [
7], we detected four potential MtrR-binding sites on the sense and anti-sense strands in this sequence, which ranged in sequence identity from 67% to 52% for regions on the sense and anti-sense strands, respectively. Using competitive EMSA, we confirmed that the DNA sequence upstream of
glnE could bind MtrR (
Figure 3A) in a specific manner as such binding was competed by unlabeled specific DNA. The nonspecific DNA failed to show such competition, although at its highest level (100× excess) the electrophoretic mobility of the MtrR::
glnE DNA complex was slightly faster than at lower concentrations of competing DNA; this suggested that some MtrR::
glnE DNA complexes may be weak or non-specific.
Figure 2.
The nucleotide sequence upstream of
glnE and MtrR-binding sites. The 301 bp sequence of the DNA upstream of
glnE and the first two codons (encoding M and S, respectively) is shown with the annotated −10 and −35 hexamer sequences of the
glnE promoter identified by a line under the sequences. The putative extended −10 element is shown in blue. The alternative −35 hexamer is shown by the dashed line above the sequence. The boxed regions represent predicted MtrR binding sites that were identified based on sequence similarity to that of a site upstream of
mtrCDE [
8,
10] or
rpoH [
7]. The grey box represents a sequence with 53% identity to the region upstream of
rpoH [
7] while the yellow, black, and red boxes represent sequences with 55%, 67%, and 52%, respectively, identity to regions upstream of
mtrCDE [
8,
10]. The MtrR-binding sites identified by DNase I protection (
Figure 3) are noted by the solid line above the sense strand or below the anti-sense strand; the two sites on the anti-sense strand are denoted as A′ and B′ with the DNase I hypersensitive site in A′ shown by an
*. The adjacent seven nucleotide imperfect inverted element is shown in green and red.
Figure 2.
The nucleotide sequence upstream of
glnE and MtrR-binding sites. The 301 bp sequence of the DNA upstream of
glnE and the first two codons (encoding M and S, respectively) is shown with the annotated −10 and −35 hexamer sequences of the
glnE promoter identified by a line under the sequences. The putative extended −10 element is shown in blue. The alternative −35 hexamer is shown by the dashed line above the sequence. The boxed regions represent predicted MtrR binding sites that were identified based on sequence similarity to that of a site upstream of
mtrCDE [
8,
10] or
rpoH [
7]. The grey box represents a sequence with 53% identity to the region upstream of
rpoH [
7] while the yellow, black, and red boxes represent sequences with 55%, 67%, and 52%, respectively, identity to regions upstream of
mtrCDE [
8,
10]. The MtrR-binding sites identified by DNase I protection (
Figure 3) are noted by the solid line above the sense strand or below the anti-sense strand; the two sites on the anti-sense strand are denoted as A′ and B′ with the DNase I hypersensitive site in A′ shown by an
*. The adjacent seven nucleotide imperfect inverted element is shown in green and red.
![Antibiotics 04 00188 g002]()
MtrR-binding sites upstream of
glnE on both the sense and anti-sense strands were detected by DNase I protection. On the sense strand, we detected a relatively long sequence (82 nucleotides) that had regions protected by MtrR (
Figure 3B). This region of protection encompassed two of the predicted MtrR binding sites and overlapped (16 nucleotides) the predicted MtrR-binding site that had the highest identity to previously identified binding sites [
8,
9,
10] (
Figure 2). Additionally, we detected DNase I hypersensitive regions on the sense strand, which were near the annotated
glnE promoter. While no clear areas of protection were evident at these DNase I hypersensitive sites, their presence suggested that DNA-protein interactions occurred in or near these regions and resulted in alterations in the local DNA structure. On the anti-sense strand, we observed two protected sites (labeled as A′ and B′ in
Figure 2 and
Figure 3B), one of which straddles the annotated
glnE promoter (site A′), while the other was located further downstream (site B′). Site A′ is notable as it overlaps the 27 bp region that was identified as being a possible MtrR-binding site. Of interest, we also noted a DNase I hypersensitive C residue in site A′ (see asterisk in
Figure 2 and
Figure 3B) that became apparent in the presence of MtrR; this site overlaps the annotated promoter region on the complementary strand.
Figure 3.
Identification of the MtrR-binding site in the
glnE upstream DNA. (
A) The binding specificity of MtrR for the DNA shown in
Figure 1 was determined by competitive EMSA; (
B) The MtrR-binding sites within this sequence were identified by DNase I protection assays that employed increasing amounts of purified MtrR-MBP (0, 5, 10, and 15 μg) with both sense and anti-sense probes. The protected regions on each probe are identified by the black bars and the two sites on the anti-sense strand are labeled as A′ and B′. Regions containing DNase I hypersensitive sites, which could contain more than one nucleotide, on the sense and antisense strands are denoted by
*. The sequencing reactions for each probe are adjacent to the DNase I protection reactions and oriented G, A, T, C.
Figure 3.
Identification of the MtrR-binding site in the
glnE upstream DNA. (
A) The binding specificity of MtrR for the DNA shown in
Figure 1 was determined by competitive EMSA; (
B) The MtrR-binding sites within this sequence were identified by DNase I protection assays that employed increasing amounts of purified MtrR-MBP (0, 5, 10, and 15 μg) with both sense and anti-sense probes. The protected regions on each probe are identified by the black bars and the two sites on the anti-sense strand are labeled as A′ and B′. Regions containing DNase I hypersensitive sites, which could contain more than one nucleotide, on the sense and antisense strands are denoted by
*. The sequencing reactions for each probe are adjacent to the DNase I protection reactions and oriented G, A, T, C.
Examination of the nucleotide sequence surrounding this DNase I hypersensitive C nucleotide revealed the presence of an imperfect seven nucleotide indirect repeat element (TAGCTTC/CAACGAT) that intervenes the annotated −35 and −10 hexamers. Examination of the MtrR-protected region identified two interesting features regarding the putative
glnE promoter region. First, we noted the presence of a potential extended −10 motif (TGATATAGT) [
25], previously observed for other −10 sequences in
N. gonorrhoeae [
26] that could mitigate the impact of a poor −35 element that was identified in the annotated sequence. Second, we identified an alternative −35 hexamer sequence (TTCAGA) that is more consistent with the sigma-70 consensus −35 hexamer sequence (TTGACA) in
E. coli. This alternative −35 sequence would change the spacing between the annotated −10 hexamer from an optimal 17 nucleotides to a suboptimal 21 nucleotides or 18 nucleotides from the proposed extended −10 element. For either −10 element, the binding of MtrR to this region could enhance interaction of the promoter with RNA polymerase leading to increased transcription of
glnE. In this respect, it is of interest that BmrR of
Bacillus subtilis activates transcription of the
bmr-encoded efflux transporter gene by binding to an imperfect inverted repeat element within the respective promoter that also has sub-optimal spacing (19 nucleotides) between the −10 and −35 hexamers [
1,
2].
In the case of MtrR activation of
glnE, we do not yet know if all of the binding sites identified by DNase I protection are important in such regulation. However, given the gene activation model for BmrR [
1], as a model to help explain the data we hypothesize that binding Site A′, which contains the imperfect indirect repeat element (
Figure 2) between the −35 and −10 domains, is important in
glnE activation by MtrR. Moreover, since the −10 element of the
glnE promoter is similar to other extended −10 element bearing promoters in gonococci [
26] and
E.
coli [
25], the ability of MtrR to bind to this region could facilitate interactions of RNA polymerase with the promoter.
Our past and current studies on MtrR control of gonococcal genes, especially its opposing action on
glnA and
glnE (
Figure 1), emphasize that this member of the TetR family of DNA-binding proteins, which are known [
2] for their ability to transcriptionally repress efflux pump-encoding genes, can also function as a repressor or activator of “off-target” genes. Is there data to believe that the ability of MtrR to repress its main target, the
mtrCDE efflux pump operon, as well as its capacity to activate expression of “off target” genes (e.g.,
glnE) is important
in vivo? Based on the findings of Warner
et al. [
17,
18] that employed an experimental murine model of lower genital tract infection in females, we posit that the answer is yes. In this infection model, which measured fitness during a dual infection by MtrR-positive and MtrR-negative isogenic gonococci, loss of MtrR was found to enhance fitness of gonococci during the first 4–5 days, but this advantage waned at later time points. The early competitive advantage due to loss of MtrR was linked to enhanced expression of the
mtrCDE-encoded efflux pump, which exports host-derived antimicrobials such as antimicrobial peptides [
27] and progesterone [
18]. As the infection progressed, the observed diminution of the competitive advantage afforded by loss of MtrR was suggested to be due to the inability of gonococci to activate certain genes. Based on this proposal, we hypothesize that MtrR activation of
glnE expression could contribute to maximal gonococcal fitness and growth during late stages of infection at mucosal surfaces where levels of glutamine are often low [
22]. This hypothesis is consistent with the notion that glutamine synthesis is important for virulence in other bacterial pathogens (e.g.,
Salmonella typhimurium) [
22].
The results presented herein are, to our knowledge, the first report of a repressor of drug efflux genes that can also directly activate a gene involved in basic metabolism. Taken together, we suggest that our work highlights the diverse global regulatory properties that could be displayed by DNA-binding proteins, such as MtrR, known for their capacity to negatively regulate drug efflux genes. Such regulation of “off target” genes and the consequences for bacterial physiology should be considered in research studies dealing with transcriptional regulation of drug efflux pump-encoding genes and bacterial resistance to antimicrobials.




{kind=link}
{kind=link}
{kind=link}
{kind=link}