
Antimicrobial Peptides SET-M33L and SET-M33L-PEG Are Promising Agents Against Strong Biofilm-Forming P. aeruginosa, Including Multidrug-Resistant Isolates

 ,
,  ,
,  ,
,  ,
,  ,
,  and
and
Abstract

1. Introduction
2. Results
2.1. Antibiotic Susceptibility Profiles of Clinical P. aeruginosa Isolates
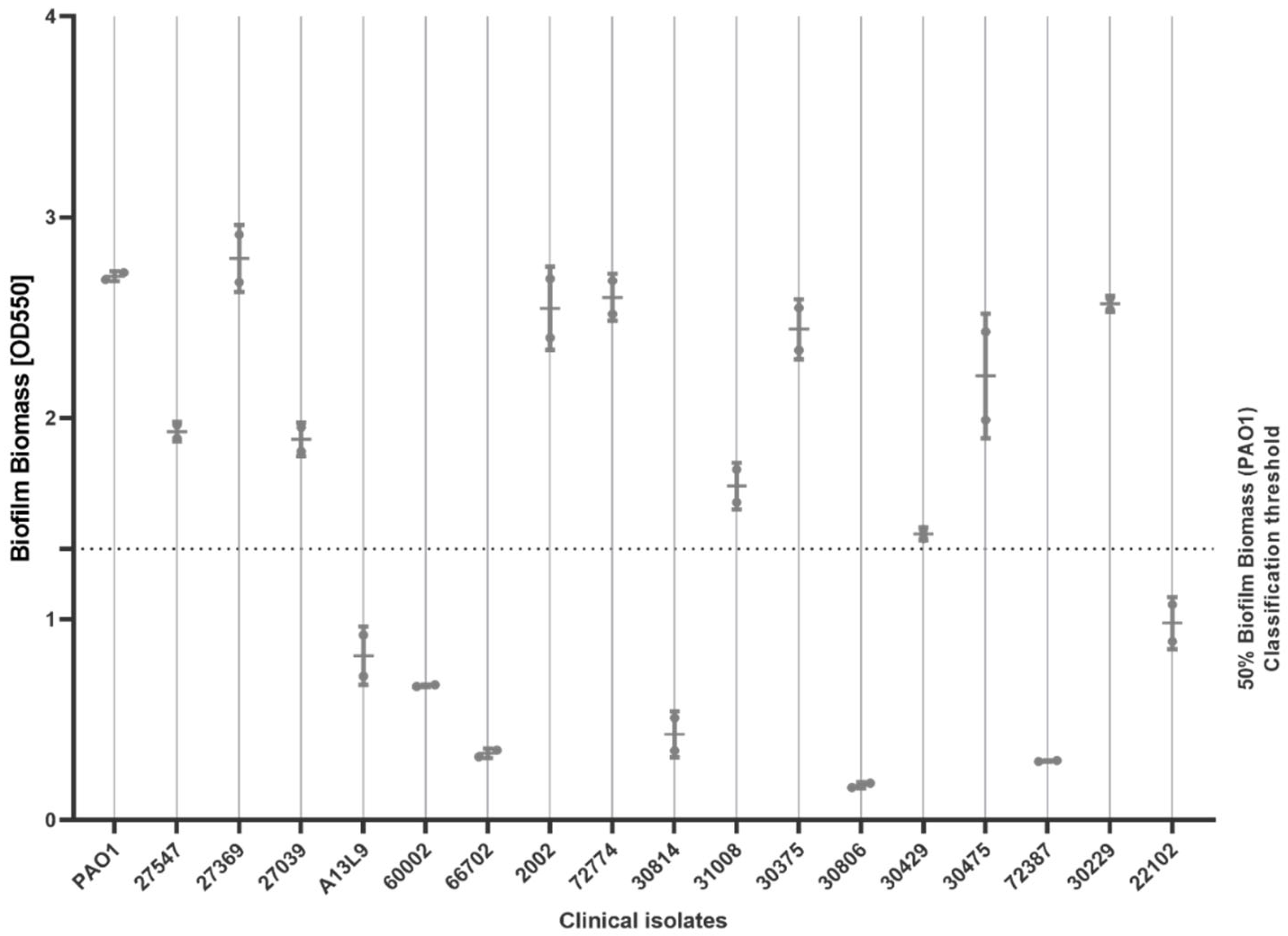
2.2. Biofilm-Forming Ability
2.3. Minimum Inhibitory Concentration (MIC) of AMPs
2.4. Minimum Biofilm Inhibitory Concentration (MBIC)
2.5. Bacterial Viability in Biofilms After MBIC Testing
2.6. Antimicrobial Synergy—Fractional Inhibitory Concentration (FIC) and Fractional Bactericidal Concentration (FBC)
3. Discussion
4. Materials and Methods
4.1. M33 Peptides Synthesis
4.2. Isolation and Characterization of Clinical Isolates of P. aeruginosa
4.3. Biofilm-Forming Ability
4.4. Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC) of AMPs
4.5. Minimum Biofilm Inhibitory Concentration (MBIC)
4.6. Bacterial Viability in Biofilms after MBIC Testing
4.7. Antimicrobial Synergy—Fractional Inhibitory Concentration (FIC) and Fractional Bactericidal Concentration (FBC)
- (1)
- FIC index ≤ 0.5: synergy (the drugs work better in combination than alone)
- (2)
- 0.5 < FIC index ≤ 1: additive (the drugs work equally well in combination and alone)
- (3)
- 1 < FIC index ≤ 4: indifference (the drugs do not significantly affect each other’s performance)
- (4)
- FIC index > 4: antagonism (the drugs work worse in combination than alone)
- (1)
- FBC index ≤ 0.5: synergy (the drugs work better in combination than alone)
- (2)
- 0.5 < FBC index ≤ 1: additive (the drugs work equally well in combination as alone)
- (3)
- 1 < FBC index ≤ 4: indifference (the drugs do not significantly affect each other’s performance)
- (4)
- FBC index > 4: antagonism (the drugs work worse in combination than alone)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef] [PubMed]
- Almughem, F.A.; Aldossary, A.M.; Tawfik, E.A.; Alomary, M.N.; Alharbi, W.S.; Alshahrani, M.Y.; Alshehri, A.A. Cystic Fibrosis: Overview of the Current Development Trends and Innovative Therapeutic Strategies. Pharmaceutics 2020, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Vidaillac, C.; Chotirmall, S.H. Pseudomonas aeruginosa in bronchiectasis: Infection, inflammation, and therapies. Expert Rev. Respir. Med. 2021, 15, 649–662. [Google Scholar] [CrossRef]
- Hall, C.W.; Mah, T.-F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef]
- Elfadadny, A.; Ragab, R.F.; AlHarbi, M.; Badshah, F.; Ibáñez-Arancibia, E.; Farag, A.; Hendawy, A.O.; De Los Ríos-Escalante, P.R.; Aboubakr, M.; Zakai, S.A.; et al. Antimicrobial resistance of Pseudomonas aeruginosa: Navigating clinical impacts, current resistance trends, and innovations in breaking therapies. Front. Microbiol. 2024, 15, 1374466. [Google Scholar] [CrossRef]
- Guillaume, O.; Butnarasu, C.; Visentin, S.; Reimhult, E. Interplay between biofilm microenvironment and pathogenicity of Pseudomonas aeruginosa in cystic fibrosis lung chronic infection. Biofilm 2022, 4, 100089. [Google Scholar] [CrossRef]
- Caldara, M.; Belgiovine, C.; Secchi, E.; Rusconi, R. Environmental, Microbiological, and Immunological Features of Bacterial Biofilms Associated with Implanted Medical Devices. Clin. Microbiol. Rev. 2022, 35, e00221-20. [Google Scholar] [CrossRef]
- Karygianni, L.; Ren, Z.; Koo, H.; Thurnheer, T. Biofilm Matrixome: Extracellular Components in Structured Microbial Communities. Trends Microbiol. 2020, 28, 668–681. [Google Scholar] [CrossRef]
- Miranda, S.W.; Asfahl, K.L.; Dandekar, A.A.; Greenberg, E.P. Pseudomonas aeruginosa Quorum Sensing. In Pseudomonas aeruginosa; Filloux, A., Ramos, J.-L., Eds.; Springer International Publishing: Cham, Switzerland, 2022; Volume 1386, pp. 95–115. [Google Scholar] [CrossRef]
- Xuan, J.; Feng, W.; Wang, J.; Wang, R.; Zhang, B.; Bo, L.; Chen, Z.-S.; Yang, H.; Sun, L. Antimicrobial peptides for combating drug-resistant bacterial infections. Drug Resist. Updates 2023, 68, 100954. [Google Scholar] [CrossRef]
- Matthyssen, T.; Li, W.; Holden, J.A.; Lenzo, J.C.; Hadjigol, S.; O’Brien-Simpson, N.M. The Potential of Modified and Multimeric Antimicrobial Peptide Materials as Superbug Killers. Front. Chem. 2022, 9, 795433. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Pini, A.; Lozzi, L.; Bernini, A.; Brunetti, J.; Falciani, C.; Scali, S.; Bindi, S.; Di Maggio, T.; Rossolini, G.M.; Niccolai, N.; et al. Efficacy and toxicity of the antimicrobial peptide M33 produced with different counter-ions. Amino Acids 2012, 43, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Castiglia, F.; Zevolini, F.; Riolo, G.; Brunetti, J.; De Lazzari, A.; Moretto, A.; Manetto, G.; Fragai, M.; Algotsson, J.; Evenäs, J.; et al. NMR Study of the Secondary Structure and Biopharmaceutical Formulation of an Active Branched Antimicrobial Peptide. Molecules 2019, 24, 4290. [Google Scholar] [CrossRef]
- Brunetti, J.; Falciani, C.; Roscia, G.; Pollini, S.; Bindi, S.; Scali, S.; Arrieta, U.C.; Gómez-Vallejo, V.; Quercini, L.; Ibba, E.; et al. In vitro and in vivo efficacy, toxicity, bio-distribution and resistance selection of a novel antibacterial drug candidate. Sci. Rep. 2016, 6, 26077. [Google Scholar] [CrossRef]
- Brunetti, J.; Carnicelli, V.; Ponzi, A.; Di Giulio, A.; Lizzi, A.R.; Cristiano, L.; Cresti, L.; Cappello, G.; Pollini, S.; Mosconi, L.; et al. Antibacterial and Anti-Inflammatory Activity of an Antimicrobial Peptide Synthesized with D Amino Acids. Antibiotics 2020, 9, 840. [Google Scholar] [CrossRef]
- Marianantoni, G.; Meogrossi, G.; Tollapi, E.; Rencinai, A.; Brunetti, J.; Marruganti, C.; Gaeta, C.; Pini, A.; Bracci, L.; Ferrari, M.; et al. Antimicrobial Peptides Active in In Vitro Models of Endodontic Bacterial Infections Modulate Inflammation in Human Cardiac Fibroblasts. Pharmaceutics 2022, 14, 2081. [Google Scholar] [CrossRef]
- Falciani, C.; Lozzi, L.; Scali, S.; Brunetti, J.; Bracci, L.; Pini, A. Site-specific pegylation of an antimicrobial peptide increases resistance to Pseudomonas aeruginosa elastase. Amino Acids 2014, 46, 1403–1407. [Google Scholar] [CrossRef]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Tait, J.R.; Bilal, H.; Kim, T.H.; Oh, A.; Peleg, A.Y.; Boyce, J.D.; Oliver, A.; Bergen, P.J.; Nation, R.L.; Landersdorfer, C.B. Pharmacodynamics of ceftazidime plus tobramycin combination dosage regimens against hypermutable Pseudomonas aeruginosa isolates at simulated epithelial lining fluid concentrations in a dynamic in vitro infection model. J. Glob. Antimicrob. Resist. 2021, 26, 55–63. [Google Scholar] [CrossRef]
- Karruli, A.; Catalini, C.; D’Amore, C.; Foglia, F.; Mari, F.; Harxhi, A.; Galdiero, M.; Durante-Mangoni, E. Evidence-Based Treatment of Pseudomonas aeruginosa Infections: A Critical Reappraisal. Antibiotics 2023, 12, 399. [Google Scholar] [CrossRef] [PubMed]
- Llor, C.; Bjerrum, L. Antimicrobial resistance: Risk associated with antibiotic overuse and initiatives to reduce the problem. Ther. Adv. Drug Saf. 2014, 5, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, T.; Tian, X.; An, W.; Wang, Z.; Han, B.; Tao, H.; Wang, J.; Wang, X. Research progress on the PEGylation of therapeutic proteins and peptides (TPPs). Front. Pharmacol. 2024, 15, 1353626. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M. Antimicrobial Peptides as Future Therapeutics: Challenges and Possibilities. In Evolution of Antimicrobial Peptides; Baindara, P., Mandal, S.M., Eds.; Springer Nature: Cham, Switzerland, 2024; pp. 1–21. [Google Scholar] [CrossRef]
- Girdhar, M.; Sen, A.; Nigam, A.; Oswalia, J.; Kumar, S.; Gupta, R. Antimicrobial peptide-based strategies to overcome antimicrobial resistance. Arch. Microbiol. 2024, 206, 411. [Google Scholar] [CrossRef]
- Van Der Weide, H.; Brunetti, J.; Pini, A.; Bracci, L.; Ambrosini, C.; Lupetti, P.; Paccagnini, E.; Gentile, M.; Bernini, A.; Niccolai, N.; et al. Investigations into the killing activity of an antimicrobial peptide active against extensively antibiotic-resistant K. pneumoniae and P. aeruginosa. Biochim. Biophys. Acta (BBA)—Biomembr. 2017, 1859, 1796–1804. [Google Scholar] [CrossRef]
- Fiel, S.B.; Roesch, E.A. The use of tobramycin for Pseudomonas aeruginosa: A review. Expert Rev. Respir. Med. 2022, 16, 503–509. [Google Scholar] [CrossRef]
- Yost, R.L.; Ramphal, R.; McLeod, D.C. Ceftazidime Review. Drug Intell. Clin. Pharm. 1985, 19, 509–513. [Google Scholar] [CrossRef]
- Grace, A.; Sahu, R.; Owen, D.R.; Dennis, V.A. Pseudomonas aeruginosa reference strains PAO1 and PA14: A genomic, phenotypic, and therapeutic review. Front. Microbiol. 2022, 13, 1023523. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, Y.; Liu, C.; Kudinha, T.; Liu, X.; Luo, Y.; Yang, Q.; Sun, H.; Hu, J.; Xu, Y.-C. Comparison of five commonly used automated susceptibility testing methods for accuracy in the China Antimicrobial Resistance Surveillance System (CARSS) hospitals. Infect. Drug Resist. 2018, 11, 1347–1358. [Google Scholar] [CrossRef]
- Matuschek, E.; Brown, D.F.J.; Kahlmeter, G. Development of the EUCAST disk diffusion antimicrobial susceptibility testing method and its implementation in routine microbiology laboratories. Clin. Microbiol. Infect. 2014, 20, O255–O266. [Google Scholar] [CrossRef]
- Nadir, Y.; Alga Batırel, A. Kolistin Duyarlılık Testlerinin Güncel Bilgiler Eşliğinde İrdelenmesi. Review of colistin susceptibility testing with current data. Mediterr. J. Infect. Microbes Antimicrob. 2024, 13, 3. [Google Scholar] [CrossRef]
- Kragh, K.N.; Alhede, M.; Kvich, L.; Bjarnsholt, T. Into the well—A close look at the complex structures of a microtiter biofilm and the crystal violet assay. Biofilm 2019, 1, 100006. [Google Scholar] [CrossRef] [PubMed]
- Rasamiravaka, T.; Labtani, Q.; Duez, P.; El Jaziri, M. The formation of biofilms by Pseudomonas aeruginosa: A review of the natural and synthetic compounds interfering with control mechanisms. Biomed Res. Int. 2015, 2015, 759348. [Google Scholar] [CrossRef]
- Vetrivel, A.; Ramasamy, M.; Vetrivel, P.; Natchimuthu, S.; Arunachalam, S.; Kim, G.-S.; Murugesan, R. Pseudomonas aeruginosa Biofilm Formation and Its Control. Biologics 2021, 1, 312–336. [Google Scholar] [CrossRef]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48 (Suppl. S1), 1049. [Google Scholar] [CrossRef]
- Mulkern, A.J.; Oyama, L.B.; Cookson, A.R.; Creevey, C.J.; Wilkinson, T.J.; Olleik, H.; Maresca, M.; Da Silva, G.C.; Fontes, P.P.; Bazzolli, D.M.S.; et al. Microbiome-derived antimicrobial peptides offer therapeutic solutions for the treatment of Pseudomonas aeruginosa infections. Npj Biofilms Microbiomes 2022, 8, 70. [Google Scholar] [CrossRef]
- Christofilogiannis, P. Current inoculation methods in MIC determination. Aquaculture 2001, 196, 297–302. [Google Scholar] [CrossRef]
- Wilson, C.; Lukowicz, R.; Merchant, S.; Valquier-Flynn, H.; Caballero, J.; Sandoval, J.; Okuom, M.; Huber, C.; Brooks, T.D.; Wilson, E.; et al. Quantitative and Qualitative Assessment Methods for Biofilm Growth: A Mini-review. Res. Rev. J. Eng. Technol. 2017, 6, 1–42. [Google Scholar]
- Haney, E.; Trimble, M.; Cheng, J.; Vallé, Q.; Hancock, R. Critical Assessment of Methods to Quantify Biofilm Growth and Evaluate Antibiofilm Activity of Host Defence Peptides. Biomolecules 2018, 8, 29. [Google Scholar] [CrossRef]
- Díez-Aguilar, M.; Ekkelenkamp, M.; Morosini, M.-I.; Huertas, N.; Del Campo, R.; Zamora, J.; Fluit, A.C.; Tunney, M.M.; Obrecht, D.; Bernardini, F.; et al. Anti-biofilm activity of murepavadin against cystic fibrosis Pseudomonas aeruginosa isolates. J. Antimicrob. Chemother. 2021, 76, 2578–2585. [Google Scholar] [CrossRef]
- Luciani, L.; Stefanetti, V.; Rampacci, E.; Gobbi, P.; Valentini, L.; Capuozzo, R.; Passamonti, F. Comparison between clinical evaluations and laboratory findings and the impact of biofilm on antimicrobial susceptibility in vitro in canine otitis externa. Vet. Dermatol. 2023, 34, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Bardbari, A.M.; Arabestani, M.R.; Karami, M.; Keramat, F.; Aghazadeh, H.; Alikhani, M.Y.; Bagheri, K.P. Highly synergistic activity of melittin with imipenem and colistin in biofilm inhibition against multidrug-resistant strong biofilm producer strains of Acinetobacter baumannii. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Pedreira, A.; Fernandes, S.; Simões, M.; García, M.R.; Vázquez, J.A. Synergistic Bactericidal Effects of Quaternary Ammonium Compounds with Essential Oil Constituents. Foods 2024, 13, 1831. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Isolate | AMG | BL | PMX | CPN | FQ | ||||
|---|---|---|---|---|---|---|---|---|---|
| Isolate | TOB | GEN | CAZ | PIP-TAZ | AZT | COL | MER | IMP | CIP |
| 27547 | S | IE | I | I | nt | S | S | I | I |
| A13L9 | S | nt | I | nt | nt | nt | nt | nt | nt |
| 60002 | S | nt | I | I | nt | S | S | I | I |
| 31008 | S | IE | I | I | nt | S | S | I | I |
| 22102 | S | nt | I | I | nt | S | S | I | I |
| 66702 | S | nt | I | I | nt | nt | S | R | I |
| 2002 | S | IE (R) | I | I | nt | S | S | I | I |
| 72774 | S | IE (R) | I | I | nt | S | S | I | I |
| 30814 | S | IE | I | I | nt | S | S | I | R |
| 30375 | S | IE | I | I | nt | R | S | I | I |
| 30229 | S | IE | I | I | nt | S | S | R | R |
| 27369 | S | IE (R) | R | R | nt | S | S | I | R |
| 30475 | S | nt | R | R | I | S | R | R | R |
| 27039 | R | nt | R | R | R | nt | R | R | I |
| 30806 | R | nt | R | R | R | nt | R | R | R |
| 30429 | R | IE (R) | I | I | nt | S | R | R | R |
| 72387 | R | nt | R | R | I | S | R | R | R |
| Isolate | Antimicrobial Compounds [μM] | |||||||
|---|---|---|---|---|---|---|---|---|
| Tobramycin | Ceftazidime | SET-M33L | SET-M33L-PEG | |||||
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| PAO1 | 4.3 | 8.5 | 3.7 | 3.7 | 2.7 | 10.9 | 2.7 | 10.6 |
| 27547 | 4.3 | 8.5 | 1.8 | 7.3 | 1.4 | 5.5 | 0.7 | 2.7 |
| 31008 | 4.3 | 17.1 | 3.7 | 14.6 | 10.9 | 21.9 | 5.3 | 21.2 |
| 2002 | 8.5 | 17.1 | 3.7 | 7.3 | 1.4 | 5.5 | 1.3 | 5.3 |
| 72774 | 8.5 | 17.1 | 3.7 | 7.3 | 1.4 | 5.5 | 1.3 | 5.3 |
| 30375 | 4.3 | 17.1 | 1.8 | 7.3 | 10.9 | 43.7 | 10.6 | 42.4 |
| 30229 | 4.3 | 17.1 | 7.3 | 29.3 | 5.5 | 21.9 | 1.3 | 5.3 |
| 27369 | 2.1 | 8.5 | 29.3 | 58.5 | 1.4 | 2.7 | 0.7 | 2.7 |
| 30475 | 4.3 | 17.1 | 58.5 | 234.2 | 0.3 | 1.4 | 0.3 | 0.7 |
| 27039 | 547.6 | 1095.2 | 58.5 | 117.1 | 5.5 | 10.9 | 10.6 | 21.2 |
| 30429 | 34.2 | 68.4 | 14.6 | 117.1 | 2.7 | 10.9 | 5.3 | 10.6 |
| Isolate | Antimicrobial Compounds [μM] | |||||||
|---|---|---|---|---|---|---|---|---|
| Tobramycin | Polymyxin B | SET-M33L | SET-M33L-PEG | |||||
| MBIC min | MBIC max | MBIC min | MBIC max | MBIC min | MBIC max | MBIC min | MBIC max | |
| PAO1 | 4.3 | 8.6 | 1.6 | 3.3 | 2.7 | 5.5 | 2.7 | 5.3 |
| 27547 | 2.1 | 4.3 | 0.4 | 0.8 | 1.4 | 2.7 | 0.6 | 1.3 |
| 31008 | 8.6 | 17.1 | 0.8 | 1.6 | 5.5 | 10.9 | 5.3 | 10.6 |
| 2002 | 17.1 | 34.2 | 0.8 | 1.6 | 5.5 | 10.9 | 5.3 | 10.6 |
| 72774 | 17.1 | 34.2 | 0.8 | 1.6 | 5.5 | 10.9 | 5.3 | 10.6 |
| 30375 | 8.6 | 17.1 | - | >106.3 | 10.9 | 21.8 | 10.6 | 21.2 |
| 30229 | 4.3 | 8.6 | 0.4 | 0.8 | 5.5 | 10.9 | 2.7 | 5.3 |
| 27369 | 4.3 | 8.6 | 0.4 | 0.8 | 2.7 | 5.5 | 1.3 | 2.6 |
| 30475 | 17.1 | 34.2 | 0.2 | 0.4 | 0.6 | 1.4 | 0.3 | 0.6 |
| 27039 | 136.8 | 273.8 | 1.6 | 3.3 | 5.5 | 10.9 | 10.6 | 21.2 |
| 30429 | 68.4 | 136.8 | 0.8 | 1.6 | 10.9 | 21.8 | 5.3 | 10.6 |
| Isolate | Untreated | Tobramycin | Polymyxin B | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PAO1 | 0 | 2.1 | 4.3 | 8.6 | 17.1 | 0.8 | 1.7 | 3.3 | 6.6 | μM |
| 1 × 108 | 1 × 109 | 1 × 107 | 1 × 104 | 0.0 | 1 × 108 | 1 × 108 | 1 × 104 | 1 × 103 | CFU/mL | |
| 27547 | 0 | 1.1 | 2.1 | 4.3 | 8.6 | 0.2 | 0.4 | 0.8 | 1.7 | μM |
| 1 × 107 | 1 × 107 | 1 × 106 | 1 × 105 | 0.0 | 1 × 107 | 1 × 106 | 1 × 103 | 0.0 | CFU/mL | |
| 31008 | 0 | 4.3 | 8.6 | 17.1 | 34.2 | 0.4 | 0.8 | 1.7 | 3.3 | μM |
| 1 × 109 | 1 × 108 | 1 × 108 | 1 × 106 | 1 × 103 | 1 × 109 | 1 × 109 | 1 × 109 | 1 × 108 | CFU/mL | |
| 2002 | 0 | 8.6 | 17.1 | 34.2 | 68.4 | 0.4 | 0.8 | 1.7 | 3.3 | μM |
| 1 × 108 | 1 × 107 | 1 × 106 | 0.0 | 0.0 | 1 × 108 | 1 × 107 | 1 × 104 | 1 × 103 | CFU/mL | |
| 72774 | 0 | 8.6 | 17.1 | 34.2 | 68.4 | 0.4 | 0.8 | 1.7 | 3.3 | μM |
| 1 × 108 | 1 × 107 | 1 × 106 | 0.0 | 0.0 | 1 × 108 | 1 × 107 | 1 × 104 | 1 × 103 | CFU/mL | |
| 30375 | 0 | 4.3 | 8.6 | 17.1 | 34.2 | 26.6 | 53.2 | 106.4 | 212.7 | μM |
| 1 × 107 | 1 × 106 | 1 × 104 | 1 × 104 | 0.0 | 1 × 107 | 1 × 107 | 1 × 106 | 1 × 105 | CFU/mL | |
| 30229 | 0 | 2.1 | 4.3 | 8.6 | 17.1 | 0.4 | 0.8 | 1.7 | 3.3 | μM |
| 1 × 108 | 1 × 107 | 1 × 107 | 1 × 105 | 1 × 104 | 1 × 107 | 1 × 107 | 1 × 107 | 1 × 105 | CFU/mL | |
| 27369 | 0 | 2.1 | 4.3 | 8.6 | 17.1 | 0.2 | 0.4 | 0.8 | 1.7 | μM |
| 1 × 107 | 1 × 107 | 1 × 107 | 1 × 105 | 1 × 102 | 1 × 106 | 1 × 106 | 1 × 106 | 1 × 103 | CFU/mL | |
| 30475 | 0 | 8.6 | 17.1 | 34.2 | 68.4 | 0.2 | 0.4 | 0.8 | 1.7 | μM |
| 1 × 107 | 1 × 106 | 0.0 | 0.0 | 0.0 | 1 × 107 | 1 × 106 | 1 × 102 | 0.0 | CFU/mL | |
| 27039 | 0 | 68.4 | 136.9 | 273.3 | 547.6 | 0.8 | 1.7 | 3.3 | 6.6 | μM |
| 1 × 106 | 1 × 107 | 1 × 106 | 1 × 104 | 0.0 | 1 × 106 | 1 × 105 | 1 × 102 | 0.0 | CFU/mL | |
| 30429 | 0 | 34.2 | 68.4 | 136.9 | 273.3 | 0.4 | 0.8 | 1.7 | 3.3 | μM |
| 1 × 108 | 1 × 107 | 1 × 105 | 0.0 | 0.0 | 1 × 108 | 1 × 107 | 1 × 107 | 1 × 105 | CFU/mL | |
| Isolate | Untreated | SET-M33L | SET-M33L-PEG | |||||||
| PAO1 | 0 | 1.4 | 2.7 | 5.5 | 10.9 | 1.3 | 2.7 | 5.3 | 10.6 | μM |
| 1 × 108 | 1 × 108 | 1 × 108 | 1 × 105 | 0.0 | 1 × 108 | 1 × 108 | 1 × 105 | 0.0 | CFU/mL | |
| 27547 | 0 | 0.7 | 1.4 | 2.7 | 5.5 | 0.3 | 0.7 | 1.3 | 2.7 | μM |
| 1 × 107 | 1 × 108 | 1 × 106 | 1 × 104 | 1 × 103 | 1 × 107 | 1 × 107 | 1 × 104 | 1 × 103 | CFU/mL | |
| 31008 | 0 | 2.7 | 5.5 | 10.9 | 21.8 | 2.7 | 5.3 | 10.6 | 21.2 | μM |
| 1 × 109 | 1 × 108 | 1 × 108 | 1 × 106 | 1 × 103 | 1 × 108 | 1 × 108 | 1 × 105 | 1 × 103 | CFU/mL | |
| 2002 | 0 | 2.7 | 5.5 | 10.9 | 21.8 | 2.7 | 5.3 | 10.6 | 21.2 | μM |
| 1 × 108 | 1 × 108 | 1 × 108 | 1 × 105 | 0.0 | 1 × 109 | 1 × 108 | 1 × 104 | 0.0 | CFU/mL | |
| 72774 | 0 | 2.7 | 5.5 | 10.9 | 21.8 | 2.7 | 5.3 | 10.6 | 21.2 | μM |
| 1 × 108 | 1 × 108 | 1 × 108 | 1 × 105 | 0.0 | 1 × 109 | 1 × 108 | 1 × 104 | 0.0 | CFU/mL | |
| 30375 | 0 | 5.5 | 10.9 | 21.8 | 43.7 | 5.3 | 10.6 | 21.2 | 43.7 | μM |
| 1 × 107 | 1 × 107 | 1 × 106 | 1 × 103 | 0.0 | 1 × 107 | 1 × 107 | 1 × 105 | 0.0 | CFU/mL | |
| 30229 | 0 | 2.7 | 5.5 | 10.9 | 21.8 | 1.3 | 2.7 | 5.3 | 10.6 | μM |
| 1 × 108 | 1 × 108 | 1 × 108 | 1 × 107 | 1 × 104 | 1 × 108 | 1 × 108 | 1 × 108 | 1 × 105 | CFU/mL | |
| 27369 | 0 | 1.4 | 2.7 | 5.5 | 10.9 | 0.7 | 1.3 | 2.7 | 5.3 | μM |
| 1 × 107 | 1 × 108 | 1 × 108 | 1 × 107 | 1 × 102 | 1 × 107 | 1 × 107 | 1 × 104 | 1 × 103 | CFU/mL | |
| 30475 | 0 | 0.3 | 0.7 | 1.4 | 2.7 | 0.2 | 0.3 | 0.7 | 1.3 | μM |
| 1 × 107 | 1 × 107 | 1 × 105 | 1 × 103 | 1 × 102 | 1 × 107 | 1 × 106 | 1 × 105 | 1 × 103 | CFU/mL | |
| 27039 | 0 | 2.7 | 5.5 | 10.9 | 21.8 | 5.3 | 10.6 | 21.2 | 43.7 | μM |
| 1 × 106 | 1 × 106 | 1 × 106 | 1 × 106 | 1 × 103 | 1 × 106 | 1 × 106 | 1 × 104 | 0.0 | CFU/mL | |
| 30429 | 0 | 5.5 | 10.9 | 21.8 | 43.7 | 2.7 | 5.3 | 10.6 | 21.2 | μM |
| 1 × 108 | 1 × 108 | 1 × 105 | 1 × 103 | 1 × 103 | 1 × 108 | 1 × 107 | 1 × 102 | 0.0 | CFU/mL | |
| Isolate | SET-M33L | SET-M33L-PEG | ||
|---|---|---|---|---|
| Tobramycin | Ceftazidime | Tobramycin | Ceftazidime | |
| PAO1 | 1 | 0.8 | 1 | 0.8 |
| 27547 | 1 | 1.5 | 1 | 1.5 |
| 31008 | 1 | 0.8 | 1 | 0.8 |
| 2002 | 1 | 1 | 0.8 | 1 |
| 72774 | 1 | 1 | 0.8 | 1 |
| 30375 | 1.3 | 1.5 | 1.3 | 1 |
| 30229 | 0.8 | 0.4 | 1.3 | 1.1 |
| 27369 | 1 | 1.1 | 1.5 | 1.1 |
| 30475 | 2.3 | 2.1 | 2.3 | 2.1 |
| 27039 | 1.1 | 0.6 | 1.1 | 0.4 |
| 30429 | 1 | 0.8 | 0.8 | 0.6 |
| Isolate | SET-M33L | SET-M33L-PEG | ||
|---|---|---|---|---|
| Tobramycin | Ceftazidime | Tobramycin | Ceftazidime | |
| PAO1 | 0.8 | 0.8 | 0.8 | 0.8 |
| 27547 | 0.5 | 0.6 | 0.6 | 0.5 |
| 31008 | 0.6 | 0.4 | 0.5 | 0.4 |
| 2002 | 0.6 | 0.6 | 0.6 | 0.6 |
| 72774 | 0.6 | 0.6 | 0.6 | 0.6 |
| 30375 | 0.3 | 0.3 | 0.3 | 0.4 |
| 30229 | 0.3 | 0.1 | 0.6 | 0.4 |
| 27369 | 0.8 | 1.1 | 0.5 | 0.6 |
| 30475 | 0.6 | 0.6 | 1.1 | 1.1 |
| 27039 | 0.6 | 0.6 | 0.6 | 0.4 |
| 30429 | 0.6 | 0.2 | 0.8 | 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontanot, A.; Croughs, P.D.; Cortese, C.; de Bruijn, A.C.J.M.; Falciani, C.; Pini, A.; Ellinger, I.; Unger, W.W.J.; Hays, J.P. Antimicrobial Peptides SET-M33L and SET-M33L-PEG Are Promising Agents Against Strong Biofilm-Forming P. aeruginosa, Including Multidrug-Resistant Isolates. Antibiotics 2025, 14, 699. https://doi.org/10.3390/antibiotics14070699
Fontanot A, Croughs PD, Cortese C, de Bruijn ACJM, Falciani C, Pini A, Ellinger I, Unger WWJ, Hays JP. Antimicrobial Peptides SET-M33L and SET-M33L-PEG Are Promising Agents Against Strong Biofilm-Forming P. aeruginosa, Including Multidrug-Resistant Isolates. Antibiotics. 2025; 14(7):699. https://doi.org/10.3390/antibiotics14070699
Chicago/Turabian StyleFontanot, Alessio, Peter D. Croughs, Clelia Cortese, Adrianus C. J. M. de Bruijn, Chiara Falciani, Alessandro Pini, Isabella Ellinger, Wendy W. J. Unger, and John P. Hays. 2025. "Antimicrobial Peptides SET-M33L and SET-M33L-PEG Are Promising Agents Against Strong Biofilm-Forming P. aeruginosa, Including Multidrug-Resistant Isolates" Antibiotics 14, no. 7: 699. https://doi.org/10.3390/antibiotics14070699
APA StyleFontanot, A., Croughs, P. D., Cortese, C., de Bruijn, A. C. J. M., Falciani, C., Pini, A., Ellinger, I., Unger, W. W. J., & Hays, J. P. (2025). Antimicrobial Peptides SET-M33L and SET-M33L-PEG Are Promising Agents Against Strong Biofilm-Forming P. aeruginosa, Including Multidrug-Resistant Isolates. Antibiotics, 14(7), 699. https://doi.org/10.3390/antibiotics14070699