

Integrating Nanopore MinION Sequencing into National Animal Health AMR Surveillance Programs: An Indonesian Pilot Study of Chicken Slaughterhouse Effluent and Rivers

 ,
,  ,
,
Abstract

1. Introduction
- What types and concentrations of E. coli are present in the effluent of chicken slaughterhouses and the receiving rivers?
- Can the Oxford Nanopore MinION sequencing technology provide valid and valuable data considering costs within regional Indonesian AMR monitoring systems currently deploying the Tricycle Protocol?
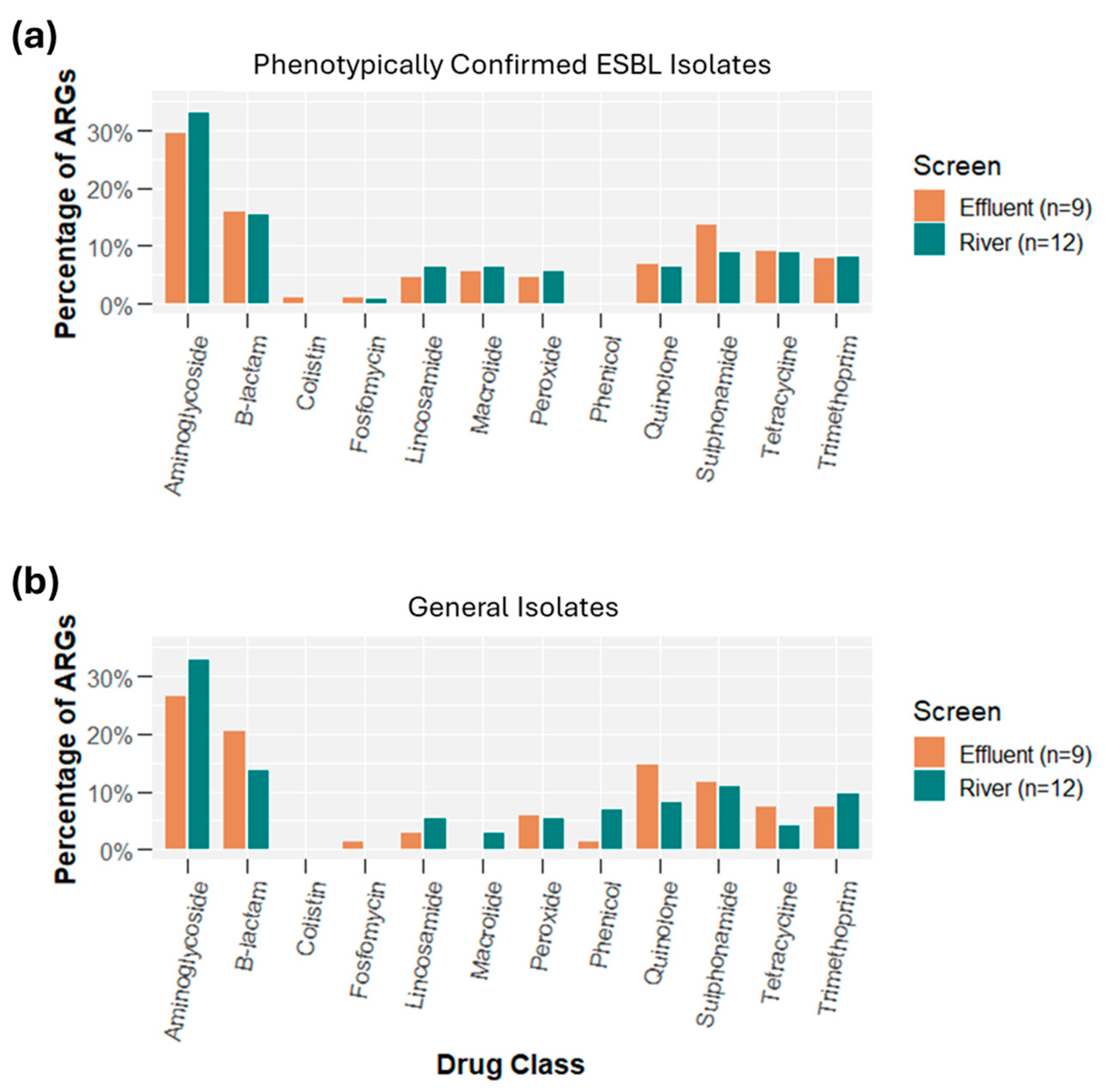
2. Results
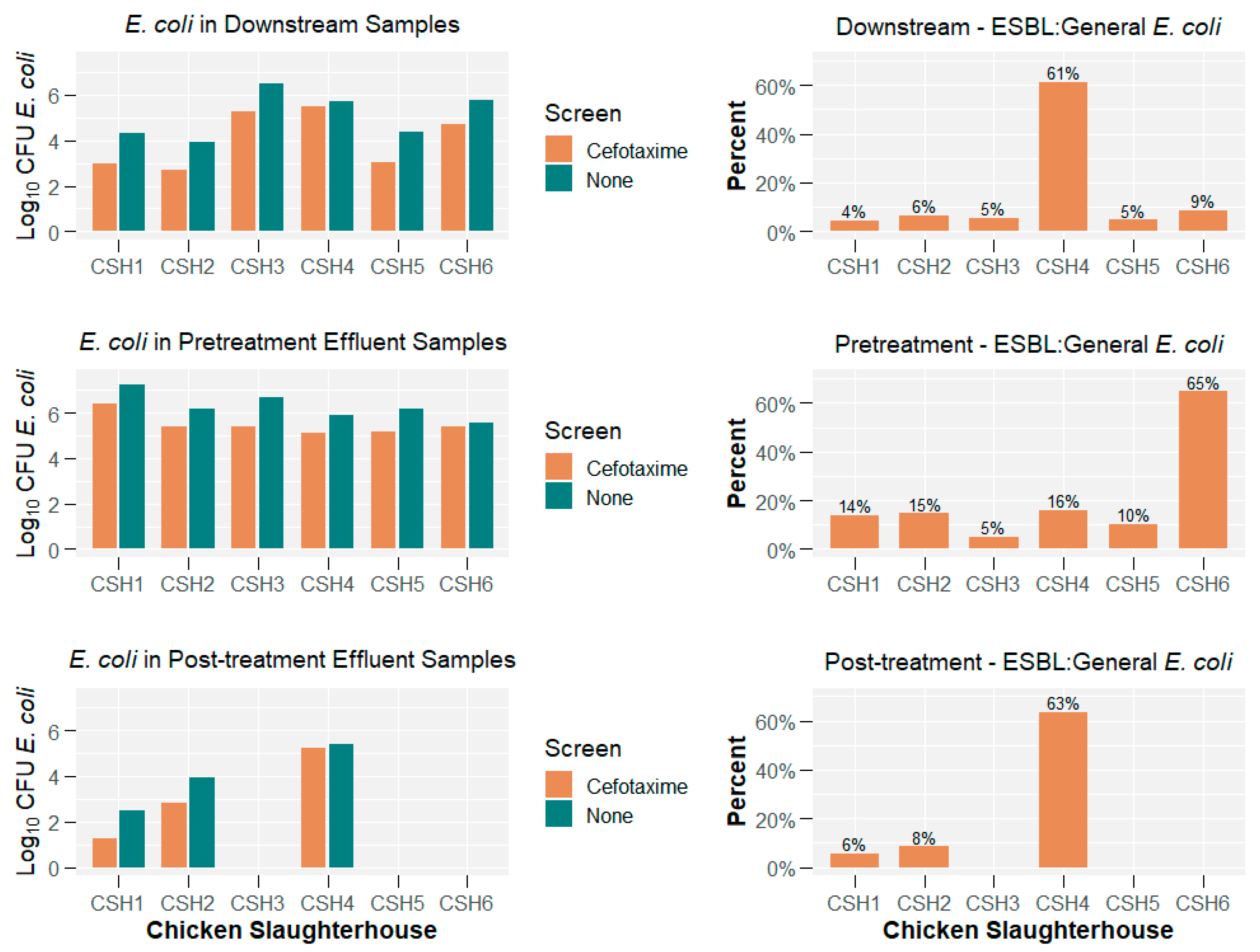
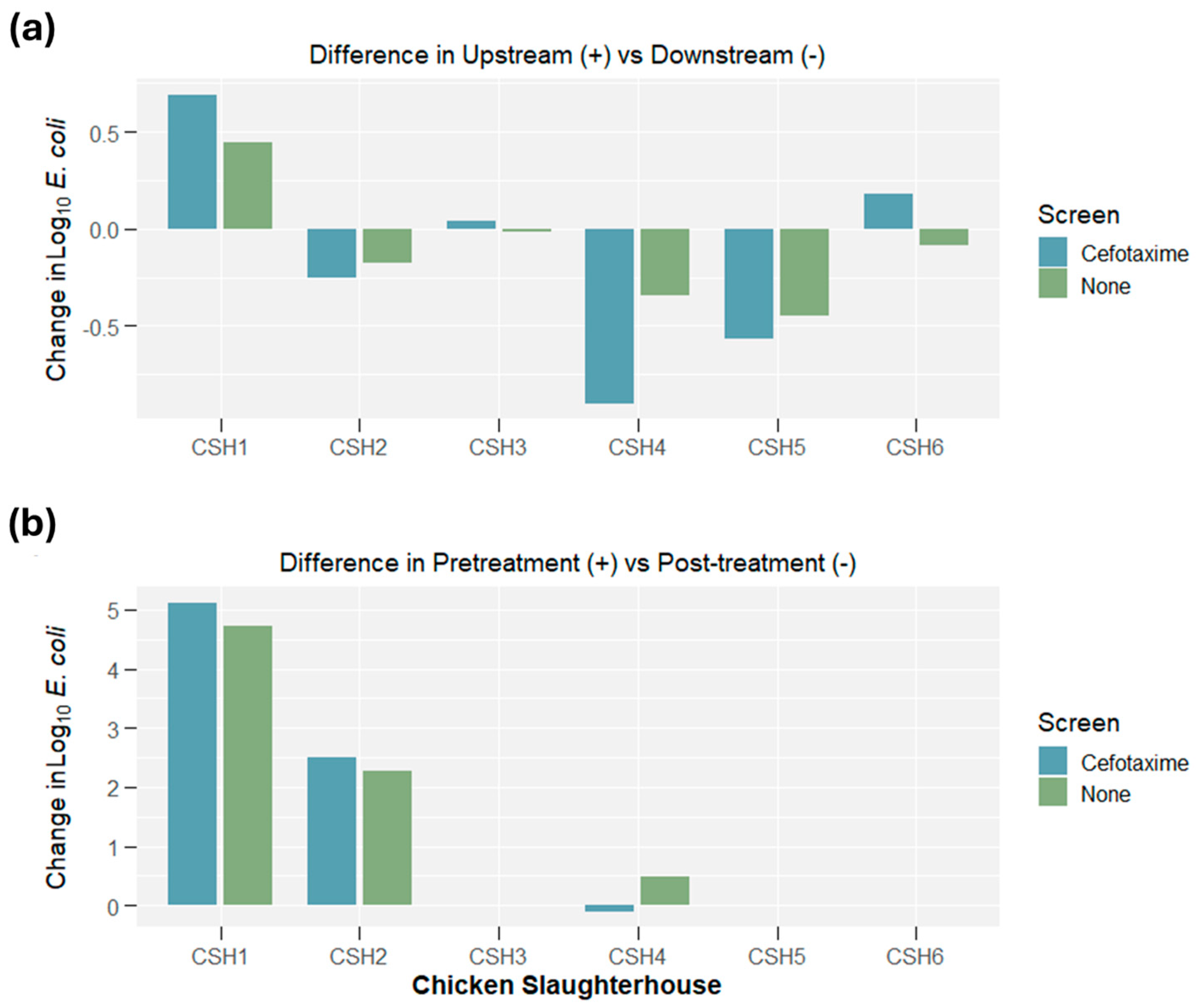
2.1. E. coli Concentrations
2.2. Whole Genome Sequencing: Oxford Nanopore vs. Illumina
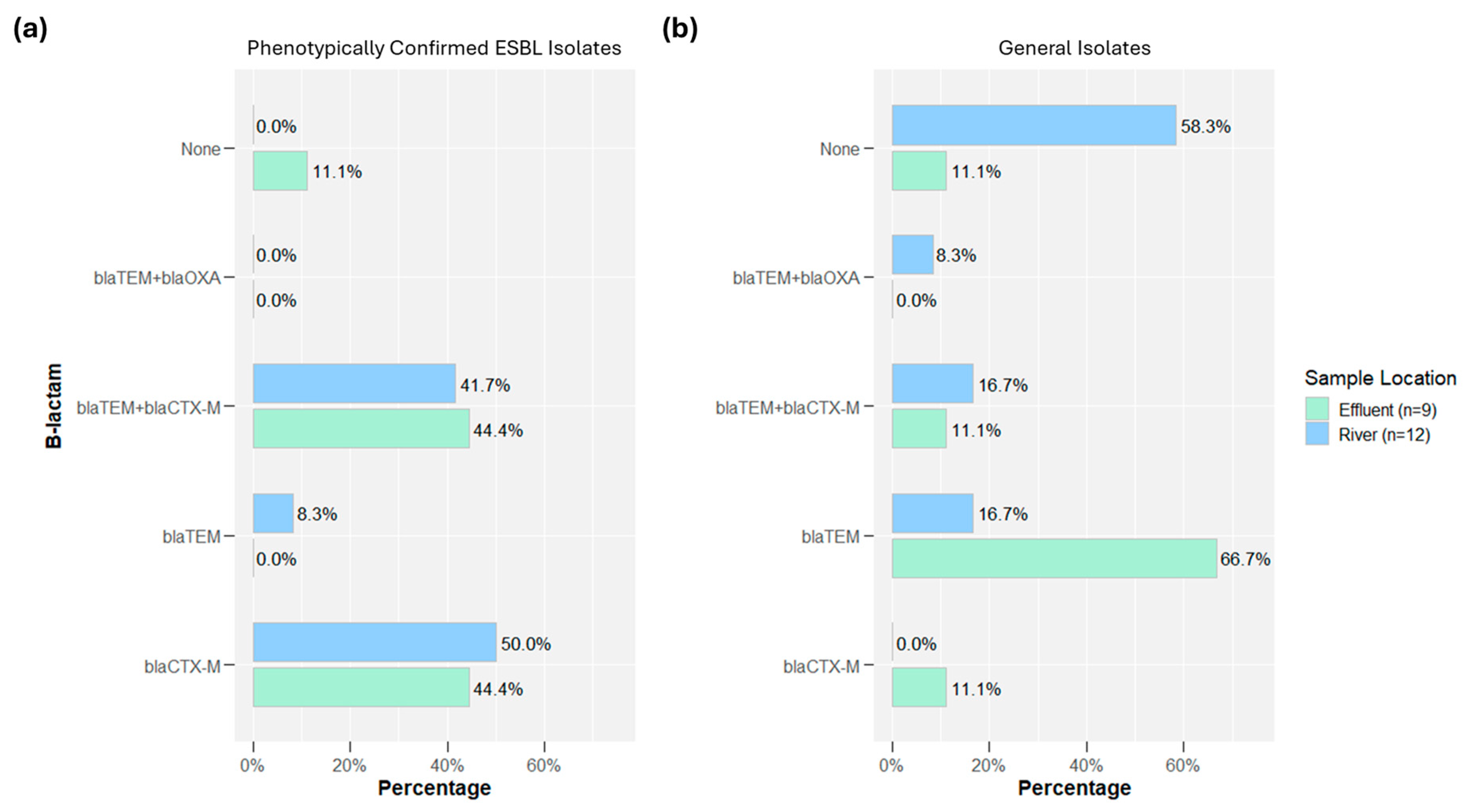
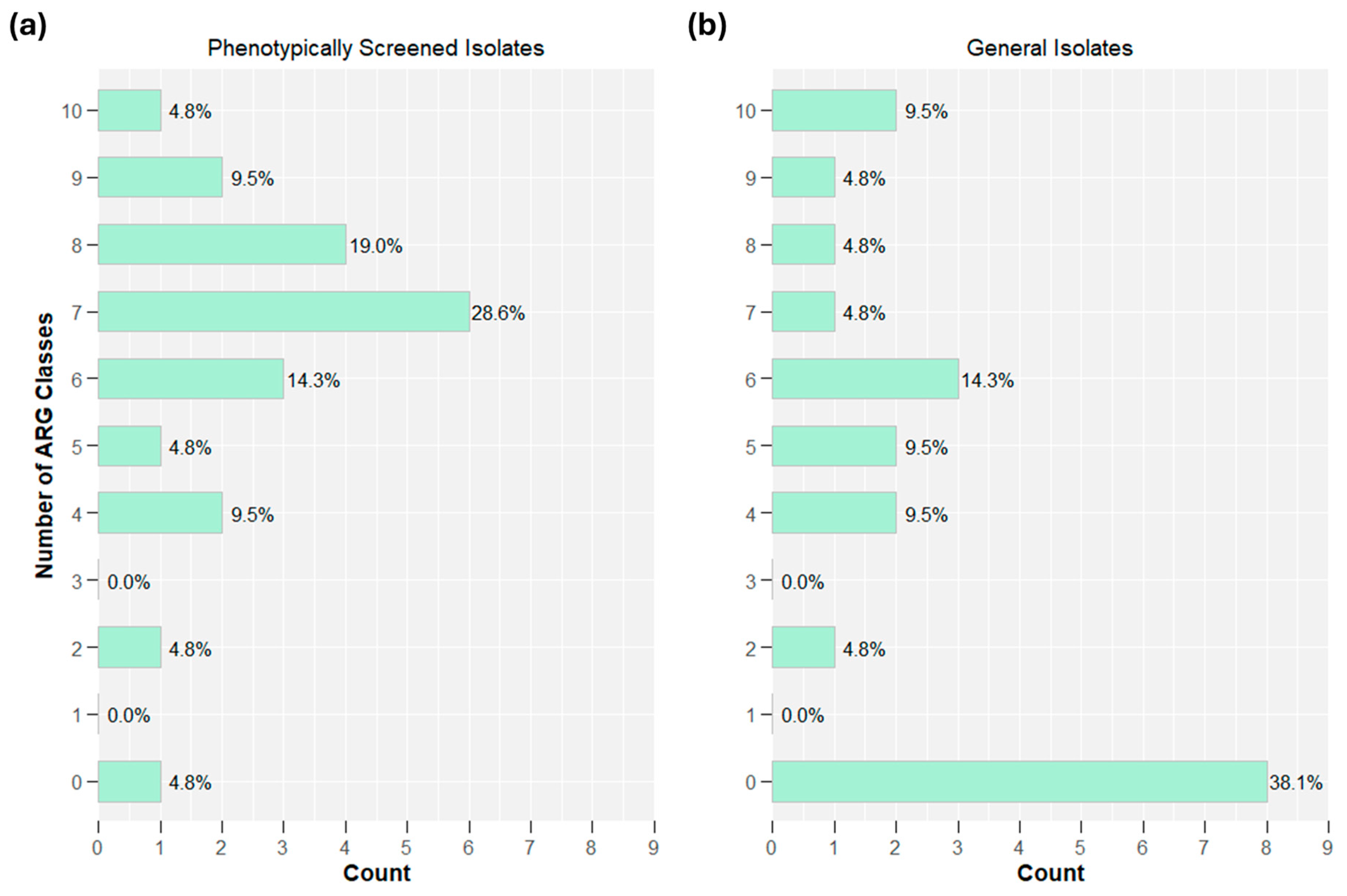
2.3. Whole Genome Sequencing: Oxford Nanopore
2.4. Cost Analysis
3. Discussion
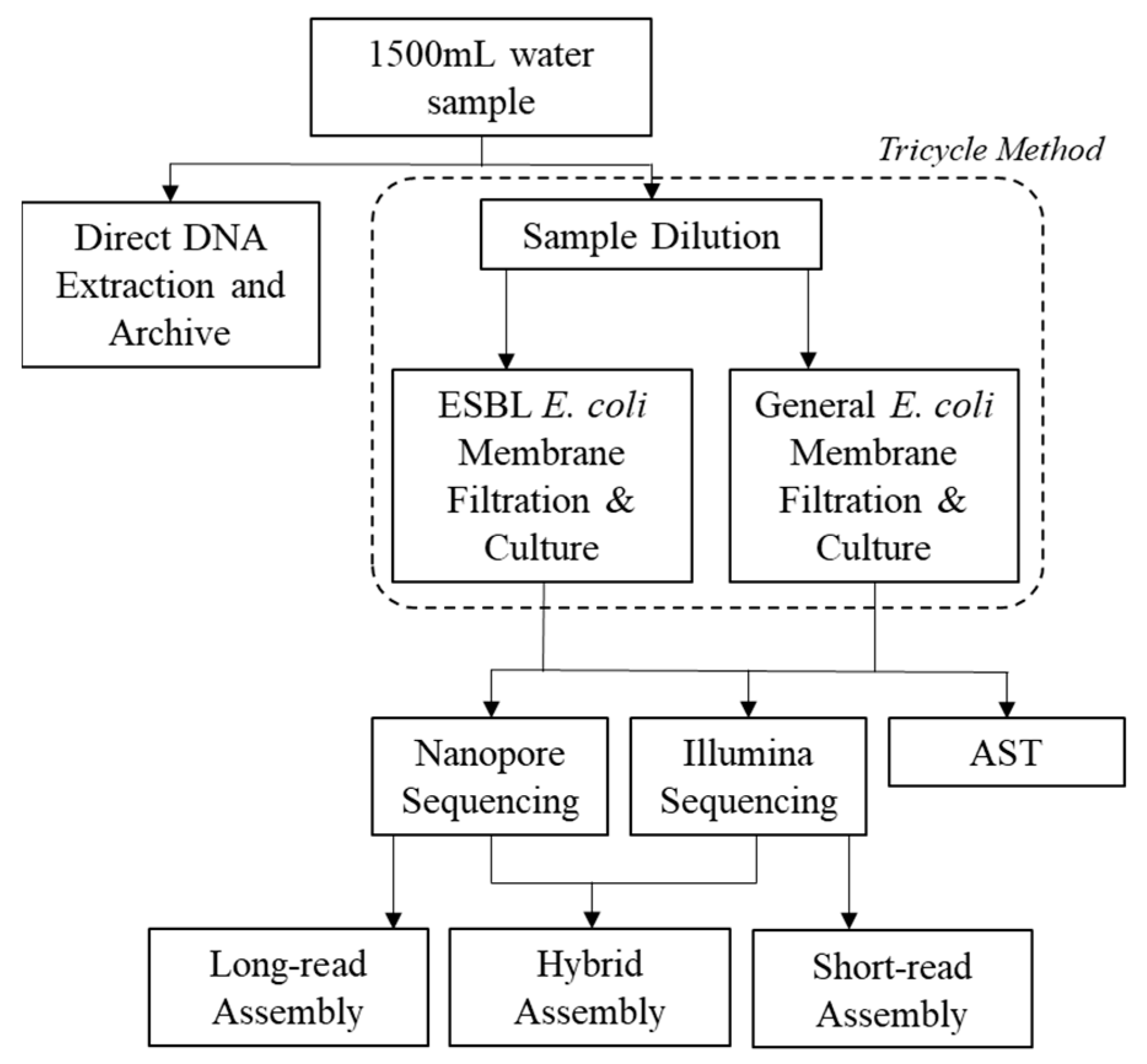
4. Materials and Methods
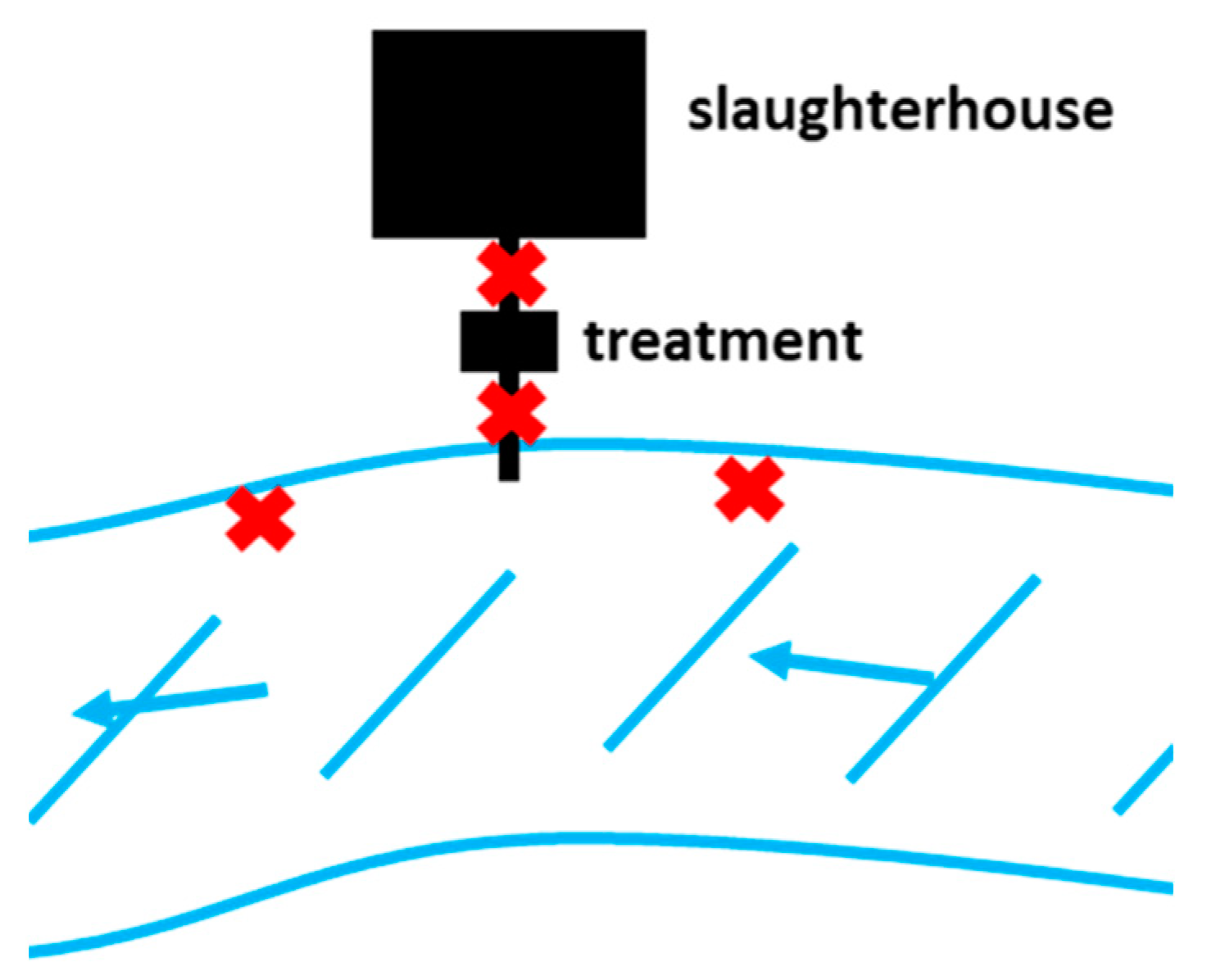
4.1. Study Site and Sample Collection
4.2. Tricycle Protocol and Antimicrobial Susceptibility Testing (AST)
4.3. Whole Genome Sequencing: Oxford Nanopore and Illumina
4.4. Quality Control
4.5. Bioinformatic Analyses
4.6. Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berendonk, T.U.; Manaia, C.M.; Merlin, C.; Fatta-Kassinos, D.; Cytryn, E.; Walsh, F.; Burgmann, H.; Sorum, H.; Norstrom, M.; Pons, M.-N.; et al. Tackling antibiotic resistance: The environmental framework. Nat. Rev. Microbiol. 2015, 13, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.; Vollset, S.E.; Ikuta, K.S.; Swetschinski, L.R.; Gray, A.P.; Wool, E.E.; Aguilar, G.R.; Mestrovic, T.; Smith, G.; Han, C.; et al. Global burden of bacterial antimicrobial resistance 1990–2021: A systematic analysis with forecasts to 2050. Lancet 2024, 404, 1199–1226. [Google Scholar] [CrossRef] [PubMed]
- McEwen, S.A.; Collignon, P. Antimicrobial resistance: A One Health perspective. Microbiol. Spectr. 2017, 6, 255–260. [Google Scholar] [CrossRef]
- Goulas, A.; Belhadi, D.; Descamps, A.; Andremont, A.; Benoit, P.; Courtois, S.; Dagot, C.; Grall, N.; Makowski, D.; Nazaret, S.; et al. How effective are strategies to control the dissemination of antibiotic resistance in the environment? A systematic review. Environ. Evid. 2020, 9, 4. [Google Scholar] [CrossRef]
- Keenum, I.; Liguori, K.; Calarco, J.; Davis, B.C.; Milligan, E.; Harwood, V.J.; Pruden, A. A framework for standardized qPCR-targets and protocols for quantifying antibiotic resistance in surface water, recycled water and wastewater. Crit. Rev. Environ. Sci. Technol. 2022, 52, 4395–4419. [Google Scholar] [CrossRef]
- Liguori, K.; Keenum, I.; Davis, B.C.; Calarco, J.; Milligan, E.; Harwood, V.J.; Pruden, A. Antimicrobial Resistance Monitoring of Water Environments: A Framework for Standardized Methods and Quality Control. Environ. Sci. Technol. 2022, 2022, 9160. [Google Scholar] [CrossRef] [PubMed]
- Sano, D.; Wester, A.L.; Schmitt, H.; Amarasiri, M.; Kirby, A.; Medlicott, K.; de Roda Husman, A.M. Updated research agenda for water, sanitation and antimicrobial resistance. J. Water Health 2020, 18, 858–866. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tricycle Surveillance: ESBL E. coli; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- World Health Organization. WHO Integrated Global Surveillance on ESBL-Producing E. coli Using a “One Health” Approach: Implementation and Opportunities; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Nadon, C.; Van Walle, I.; Gerner-Smidt, P.; Campos, J.; Chinen, I.; Concepcion-Acevedo, J.; Gilpin, B.; Smith, A.M.; Kam, K.M.; Perez, E.; et al. PulseNet International: Vision for the implementation of whole genome sequencing (WGS) for global food-borne disease surveillance. Eurosurveillance 2017, 22, 30544. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019. [Google Scholar]
- Day, M.J.; Hopkins, K.L.; Wareham, D.W.; Toleman, M.A.; Elviss, N.; Randall, L.; Teale, C.; Cleary, P.; Wiuff, C.; Doumith, M.; et al. Extended-spectrum β-lactamase-producing Escherichia coli in human-derived and foodchain-derived samples from England, Wales, and Scotland: An epidemiological surveillance and typing study. Lancet Infect. Dis. 2019, 19, 1325–1335. [Google Scholar] [CrossRef]
- Blaak, H.; Lynch, G.; Italiaander, R.; Hamidjaja, R.A.; Schets, F.M.; De Husman, A.M.R. Multidrug-resistant and extended spectrum beta-lactamase-producing Escherichia coli in dutch surface water and wastewater. PLoS ONE 2015, 10, e0127752. [Google Scholar] [CrossRef]
- Jorgensen, S.B.; Soraas, A.V.; Arnesen, L.S.; Leegaard, T.M.; Sundsfjord, A.; Jenum, P.A. A comparison of extended spectrum β-lactamase producing Escherichia coli from clinical, recreational water and wastewater samples associated in time and location. PLoS ONE 2017, 12, e0186576. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmeister, E.R.; Voth-Gaeddert, L.E.; Metilda, A.; Tai, A.; Batorsky, R.E.; Veeraraghavan, B.; Ward, H.D.; Kang, G.; Pickering, A.J. Surveillance of potential pathogens and antibiotic resistance in wastewater and surface water from Boston, USA and Vellore, India using long-read metagenomic sequencing. medRxiv 2021. [Google Scholar] [CrossRef]
- Berendes, D.M.; de Mondesert, L.; Kirby, A.E.; Yakubu, H.; Adomako, L.; Michiel, J.; Raj, S.; Robb, K.; Wang, Y.; Doe, B.; et al. Variation in E. coli concentrations in open drains across neighborhoods in Accra, Ghana: The influence of onsite sanitation coverage and interconnectedness of urban environments. Int. J. Hyg. Environ. Health 2020, 224, 113433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mairinger, W.; Raj, S.J.; Yakubu, H.; Siesel, C.; Green, J.; Durry, S.; Joseph, G.; Rahman, M.; Amin, N.; et al. Quantitative assessment of exposure to fecal contamination in urban environment across nine cities in low-income and lower-middle-income countries and a city in the United States. Sci. Total Environ. 2022, 806, 151273. [Google Scholar] [CrossRef] [PubMed]
- US Environmental Protection Agency. Recreational Water Quality Criteria 820-F-12-058; US Environmental Protection Agency: Washington, DC, USA, 2012. [Google Scholar]
- Nguyen, A.Q.; Vu, H.P.; Nguyen, L.N.; Wang, Q.; Djordjevic, S.P.; Donner, E.; Yin, H.; Nghiem, L.D. Monitoring antibiotic resistance genes in wastewater treatment: Current strategies and future challenges. Sci. Total Environ. 2021, 783, 146964. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.-T.; Su, Z.-X.; Lai, W.-X.; Zhang, Y.-B.; Liu, Y.-W. Insights into the fate and removal of antibiotics and antibiotic resistance genes using biological wastewater treatment technology. Sci. Total Environ. 2021, 776, 145906. [Google Scholar] [CrossRef]
- Uluseker, C.; Kaster, K.M.; Thorsen, K.; Basiry, D.; Shobana, S.; Jain, M.; Kumar, G.; Kommedal, R.; Pala-Ozkok, I. A Review on Occurrence and Spread of Antibiotic Resistance in Wastewaters and in Wastewater Treatment Plants: Mechanisms and Perspectives. Front. Microbiol. 2021, 12, 3003. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, X. Removal of antibiotic resistance genes (ARGs) in various wastewater treatment processes: An overview. Crit. Rev. Environ. Sci. Technol. 2022, 52, 571–630. [Google Scholar] [CrossRef]
- Anwar Sani, R.; Sunandar, S.; Rachmawati, A.; Pertela, G.; Susanti, O.; Rahayu, K.P.; Allamanda, P.; Suandy, I.; Nurbiyanti, N.; Jahja, E.J.; et al. Antimicrobial Usage and Antimicrobial Resistance in Commensal Escherichia coli from Broiler Farms: A Farm-Level Analysis in West Java, Indonesia. Antibiotics 2024, 13, 1181. [Google Scholar] [CrossRef]
- FAO in Indonesia. Preserving Critically Important Antibiotics for Humans, by Banning Their Use in Animals; Food and Agriculture Organization of the United Nations: Jakarta, Indonesia, 2019. [Google Scholar]
- Bessa, L.J.; Barbosa-Vasconcelos, A.; Mendes, Â.; Vaz-Pires, P.; Martins da Costa, P. High prevalence of multidrug-resistant Escherichia coli and Enterococcus spp. in river water, upstream and downstream of a wastewater treatment plant. J. Water Health 2014, 12, 426–435. [Google Scholar] [CrossRef]
- Agusi, E.R.; Kabantiyok, D.; Mkpuma, N.; Atai, R.B.; Okongwu-Ejike, C.; Bakare, E.L.; Budaye, J.; Sule, K.G.; Rindaps, R.J.; James, G.K.; et al. Prevalence of multidrug-resistant Escherichia coli isolates and virulence gene expression in poultry farms in Jos, Nigeria. Front. Microbiol. 2024, 15, 1298582. [Google Scholar] [CrossRef] [PubMed]
- Saliu, E.-M.; Vahjen, W.; Zentek, J. Types and prevalence of extended-spectrum beta-lactamase producing Enterobacteriaceae in poultry. Anim. Health Res. Rev. 2017, 18, 46–57. [Google Scholar] [CrossRef]
- Nhung, N.T.; Chansiripornchai, N.; Carrique-Mas, J.J. Antimicrobial Resistance in Bacterial Poultry Pathogens: A Review. Front. Vet. Sci. 2017, 4, 126. [Google Scholar] [CrossRef] [PubMed]
- Stromberg, Z.R.; Johnson, J.R.; Fairbrother, J.M.; Kilbourne, J.; Van Goor, A.; Curtiss, R.; Mellata, M. Evaluation of Escherichia coli isolates from healthy chickens to determine their potential risk to poultry and human health. PLoS ONE 2017, 12, e0180599. [Google Scholar] [CrossRef]
- Hamelin, K.; Bruant, G.; El-Shaarawi, A.; Hill, S.; Edge, T.A.; Fairbrother, J.; Harel, J.; Maynard, C.; Masson, L.; Brousseau, R. Occurrence of Virulence and Antimicrobial Resistance Genes in Escherichia coli Isolates from Different Aquatic Ecosystems within the St. Clair River and Detroit River Areas. Appl. Environ. Microbiol. 2007, 73, 477–484. [Google Scholar] [CrossRef]
- Foster-Nyarko, E.; Cottingham, H.; Wick, R.R.; Judd, L.M.; Lam, M.M.C.; Wyres, K.L.; Stanton, T.D.; Tsang, K.K.; David, S.; Aanensen, D.M.; et al. Nanopore-only assemblies for genomic surveillance of the global priority drug-resistant pathogen, Klebsiella pneumoniae. Microb. Genom. 2023, 9, 000936. [Google Scholar] [CrossRef]
- Neal-McKinney, J.M.; Liu, K.C.; Lock, C.M.; Wu, W.H.; Hu, J. Comparison of MiSeq, MinION, and hybrid genome sequencing for analysis of Campylobacter jejuni. Sci. Rep. 2021, 11, 5676. [Google Scholar] [CrossRef]
- Oh, S.; Nam, S.K.; Chang, H.E.; Park, K.U. Comparative Analysis of Short- and Long-Read Sequencing of Vancomycin-Resistant Enterococci for Application to Molecular Epidemiology. Front. Cell. Infect. Microbiol. 2022, 12, 857801. [Google Scholar] [CrossRef] [PubMed]
- Griswold, T.; Kapsak, C.; Chen, J.C.; Den Bakker, H.C.; Williams, G.; Kelley, A.; Vidyaprakash, E.; Katz, L.S. SneakerNet: A modular quality assurance and quality check workflow for primary genomic and metagenomic read data. J. Open Source Softw. 2021, 6, 2334. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. “Laboratory Standard Operating Procedure for Whole Genome Sequencing On Miseq.” Protocol PNL38. US Centers for Disease Control and Prevention. 2019. Available online: https://www.aphl.org/programs/global_health/Documents/PNL38_WGS%20on%20MiSeq%20SOP_v4.pdf (accessed on 15 December 2024).
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Kapsak, C.; Katz, L. NanoporeWorkflow v0.4.4. Available online: https://github.com/kapsakcj/nanoporeWorkflow (accessed on 1 June 2020).
- Wick, R. Filtlong. Available online: https://github.com/rrwick/Filtlong (accessed on 6 July 2020).
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Vaser, R.; Sovic, I.; Nagarajan, N.; Mile, Š. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies LLC Medaka v1.2.3. Available online: https://github.com/nanoporetech/medaka (accessed on 4 August 2020).
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: Predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef]
- Katz, L. SneakerNet. Available online: https://github.com/lskatz/SneakerNet (accessed on 12 December 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Seemann, T. Shovill. Available online: https://github.com/tseemann/shovill (accessed on 6 September 2020).
- Souvorov, A.; Agarwala, R.; Lipman, D.J. SKESA: Strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018, 19, 153. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Lukjancenko, O.; Saputra, D.; Rasmussen, S.; Hasman, H.; Sicheritz-Pontén, T.; Aarestrup, F.M.; Ussery, D.W.; Lund, O. Benchmarking of methods for genomic taxonomy. J. Clin. Microbiol. 2014, 52, 1529–1539. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; Cattoir, V.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, M. Real-Time Whole-Genome Sequencing for Routine Typing, Surveillance, and Outbreak Detection of Verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; García-fernández, A.; Larsen, V.; Lund, O.; Villa, L.; Aarestrup, M.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Franz, E.; Veenman, C.; van Hoek, A.H.A.M.; Husman, A.D.R.; Blaak, H. Pathogenic Escherichia coli producing Extended-Spectrum β-Lactamases isolated from surface water and wastewater. Sci. Rep. 2015, 5, 14372. [Google Scholar] [CrossRef] [PubMed]
- Sarowska, J.; Koloch, B.F.; Kmiecik, A.J.; Madrzak, M.F.; Ksiazczyk, M.; Ploskonska, G.B.; Krol, I.C. Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: Recent reports. Gut Pathog. 2019, 11, 10. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Sequence analysis Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Results per Run | Oxford Nanopore MinION Mean (Range) | Illumina MiSeq Mean (Range) |
|---|---|---|
| QC | ||
| Total Mbases | 479.6 (54.5–1400) | - |
| Total Mbases (post QC) | 391.4 (59.4–600 ^) | 273 (110–404) |
| Total reads (post QC) | 82,432 (14,790–167,621) | 1,215,360 (555,750–1,606,430) |
| Read quality (post QC) | Q9.91 (8.8–11.2) | Q36 (35.28–36.09) |
| Read length (post QC) | 4810 bp (2691–10,511) | 236 bp (215–247) |
| N50 (post QC) | 6819 bp (4031–11,748) | - |
| Contamination * | ||
| Per run (%) | Classified reads: (0.97–3.55) Total reads: (1.10–3.75) | - |
| Per isolate (%) | Classified reads: (0.22–5.79) Total reads: (0.25–6.25) | Classified reads: (0.36–4.87) ^ Total reads: (0.58–5.84) |
| Assembly ** | ||
| Estimated coverage; mean (median) | 96x (84x) | 109x (113x) |
| Total chromosomal contigs; mean (median) | 3.5 (1) | 54.7 (51.5) |
| Total plasmid contigs; mean (median) | 5.4 (4) | 39.1 (38.5) |
| Tricycle Protocol | MinION Sequencing | MiSeq Sequencing | Hybrid Approach | Senstitire AST | |
|---|---|---|---|---|---|
| Pilot Study Cost per Sample | USD $10 | USD $292 | USD $225 | USD $517 | USD $20 |
| Projected Cost per Sample at Scale * | USD $5 | USD $70–100 | USD $80–150 | USD $150–250 | USD $12 |
| ESBL E. coli Concentration | X | ||||
| Total E. coli Concentration | X | ||||
| ESBL to Total E. coli Ratio | X | ||||
| ARG Identification | X | x | X | X | |
| ARG Location | X | x | X | ||
| Virulence Factor Identification | X | x | X | ||
| Virulence Factor Location | X | x | X | ||
| Serotype | X | x | X | ||
| Phylogenetic Relatedness | x | X | X |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telussa, R.; Rahayu, P.; Yunindika, T.; Kapsak, C.J.; Rahayu, K.P.; Susanti, O.; Suandy, I.; Triwijayanti, N.; Niasono, A.B.; Ma’arif, S.; et al. Integrating Nanopore MinION Sequencing into National Animal Health AMR Surveillance Programs: An Indonesian Pilot Study of Chicken Slaughterhouse Effluent and Rivers. Antibiotics 2025, 14, 624. https://doi.org/10.3390/antibiotics14070624
Telussa R, Rahayu P, Yunindika T, Kapsak CJ, Rahayu KP, Susanti O, Suandy I, Triwijayanti N, Niasono AB, Ma’arif S, et al. Integrating Nanopore MinION Sequencing into National Animal Health AMR Surveillance Programs: An Indonesian Pilot Study of Chicken Slaughterhouse Effluent and Rivers. Antibiotics. 2025; 14(7):624. https://doi.org/10.3390/antibiotics14070624
Chicago/Turabian StyleTelussa, Rallya, Puji Rahayu, Thufeil Yunindika, Curtis J. Kapsak, Kanti Puji Rahayu, Oli Susanti, Imron Suandy, Nuraini Triwijayanti, Aji B. Niasono, Syamsul Ma’arif, and et al. 2025. "Integrating Nanopore MinION Sequencing into National Animal Health AMR Surveillance Programs: An Indonesian Pilot Study of Chicken Slaughterhouse Effluent and Rivers" Antibiotics 14, no. 7: 624. https://doi.org/10.3390/antibiotics14070624
APA StyleTelussa, R., Rahayu, P., Yunindika, T., Kapsak, C. J., Rahayu, K. P., Susanti, O., Suandy, I., Triwijayanti, N., Niasono, A. B., Ma’arif, S., Wibawa, H., Lestari, L., Utomo, G. B., Zenal, F. C., Schoonman, L., & Voth-Gaeddert, L. E. (2025). Integrating Nanopore MinION Sequencing into National Animal Health AMR Surveillance Programs: An Indonesian Pilot Study of Chicken Slaughterhouse Effluent and Rivers. Antibiotics, 14(7), 624. https://doi.org/10.3390/antibiotics14070624