

Substitution of Proline Residues by 4-Fluoro-l-Proline Affects the Mechanism of the Proline-Rich Antimicrobial Peptide Api137 †

 , , , , , and
, , , , , and
Abstract

1. Introduction
2. Results
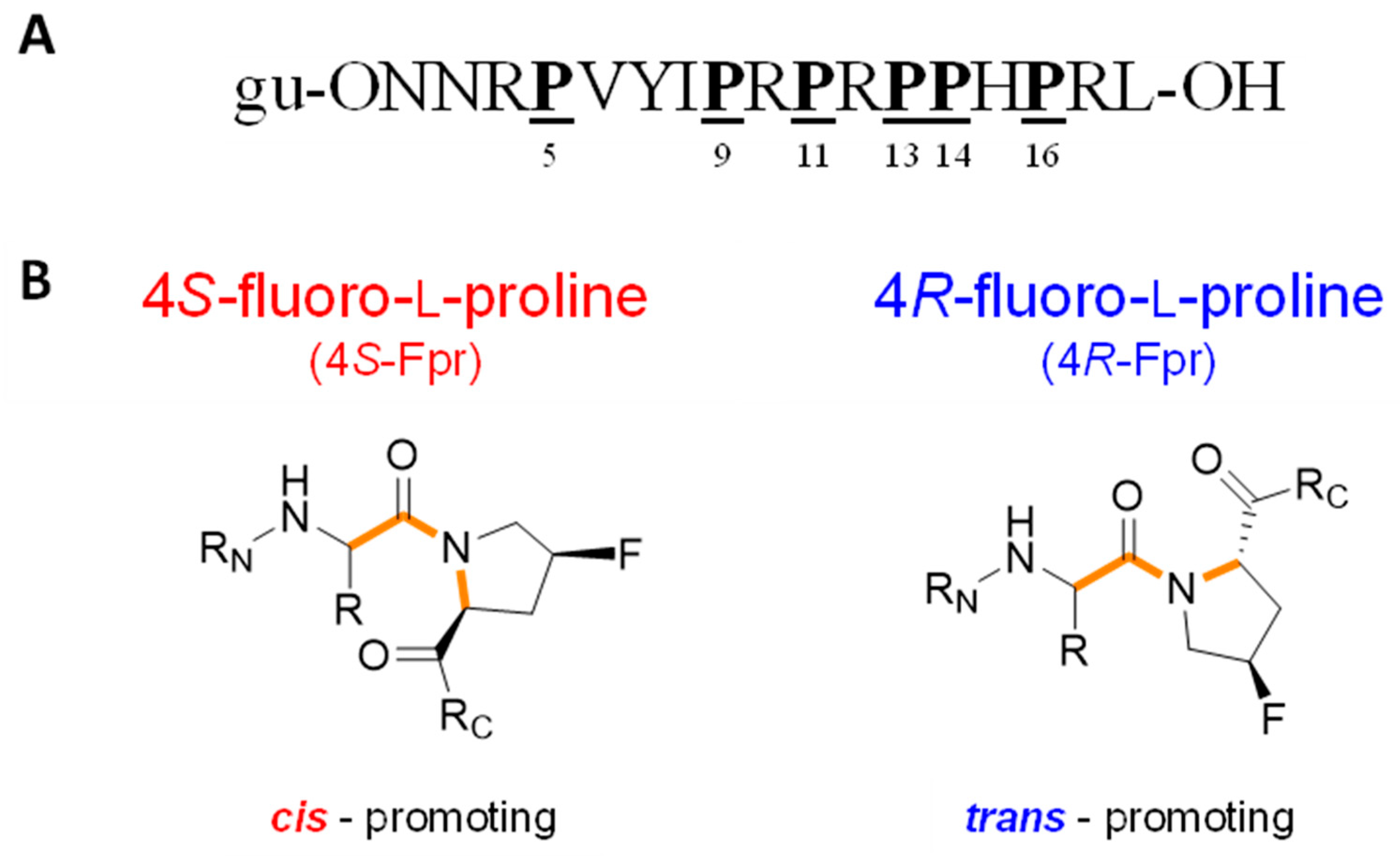
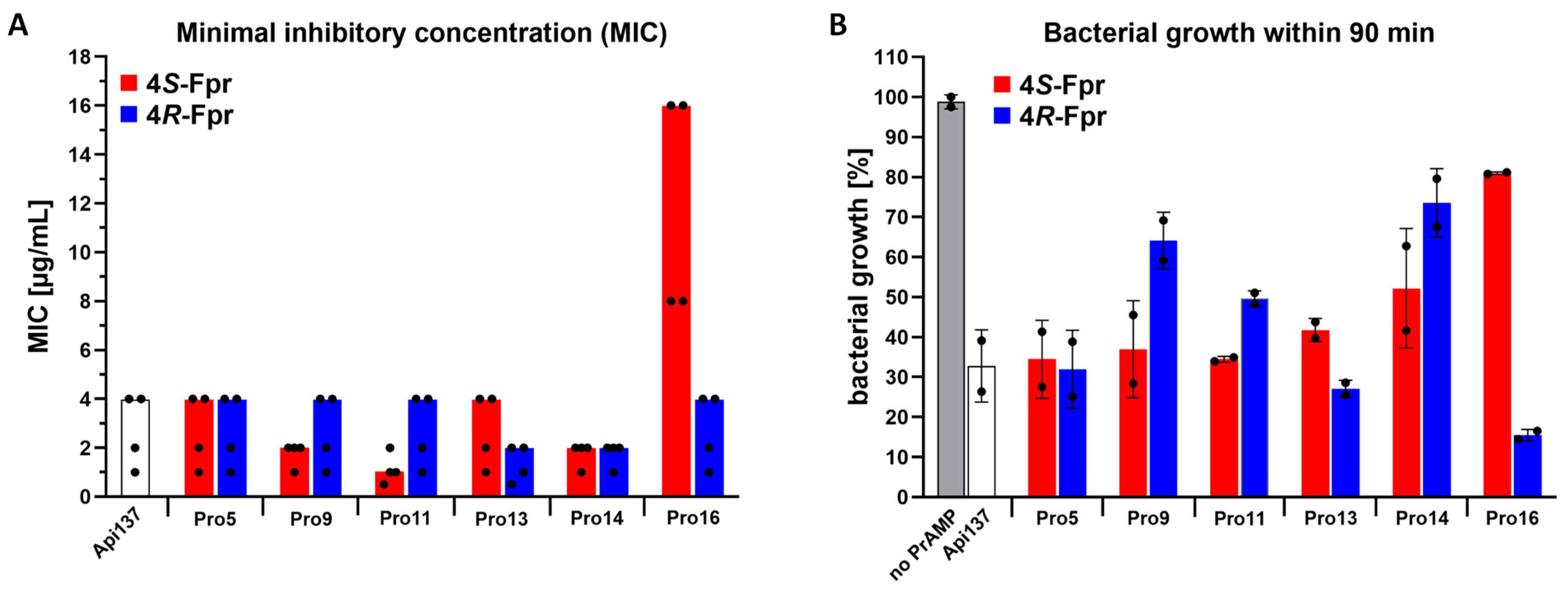
2.1. Antimicrobial Activity Against Escherichia coli
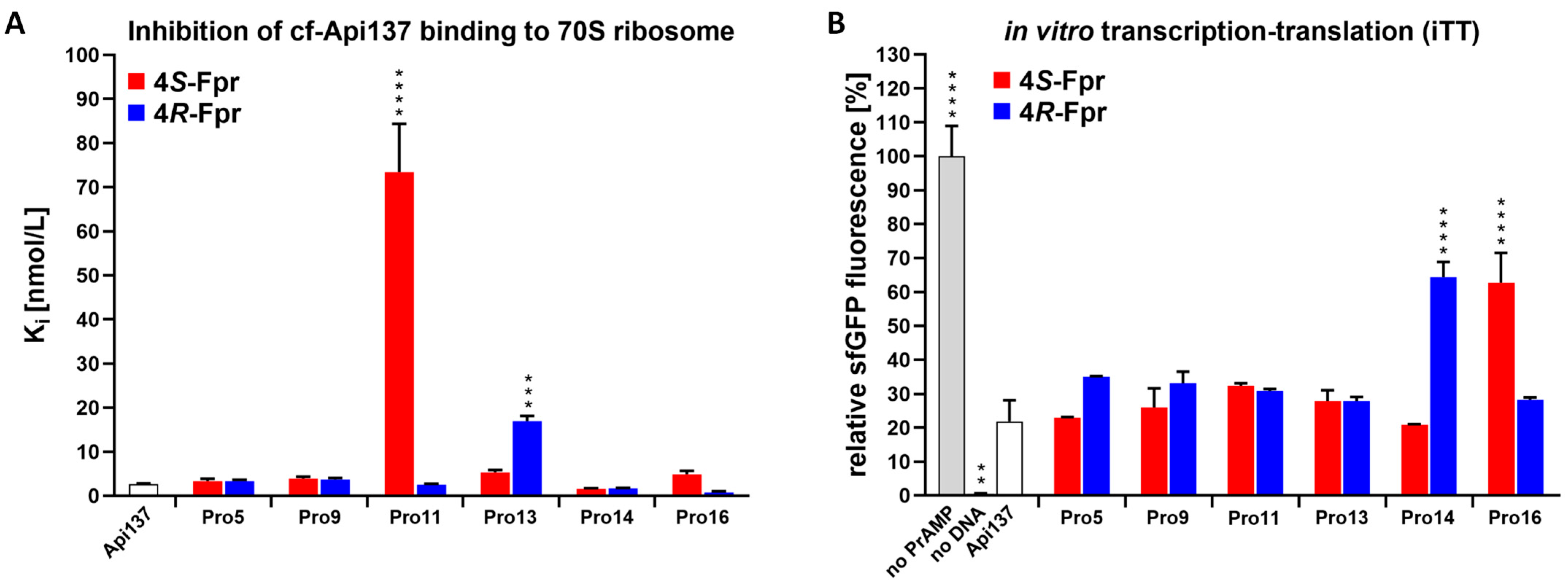
2.2. Ribosome Binding and Inhibition of In Vitro Translation
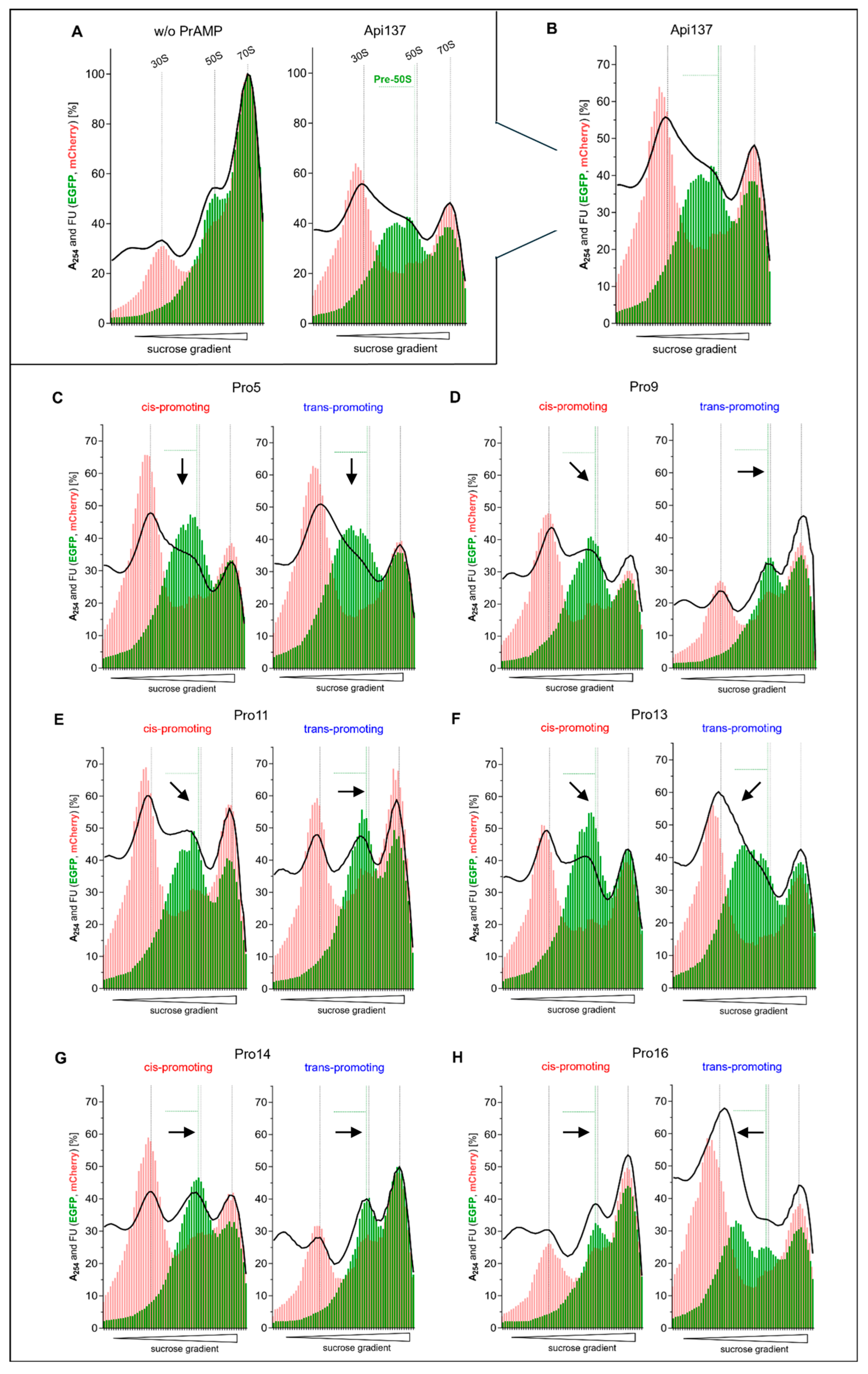
2.3. Ribosome Profile Analysis and 50S Subunit Assembly Disruption
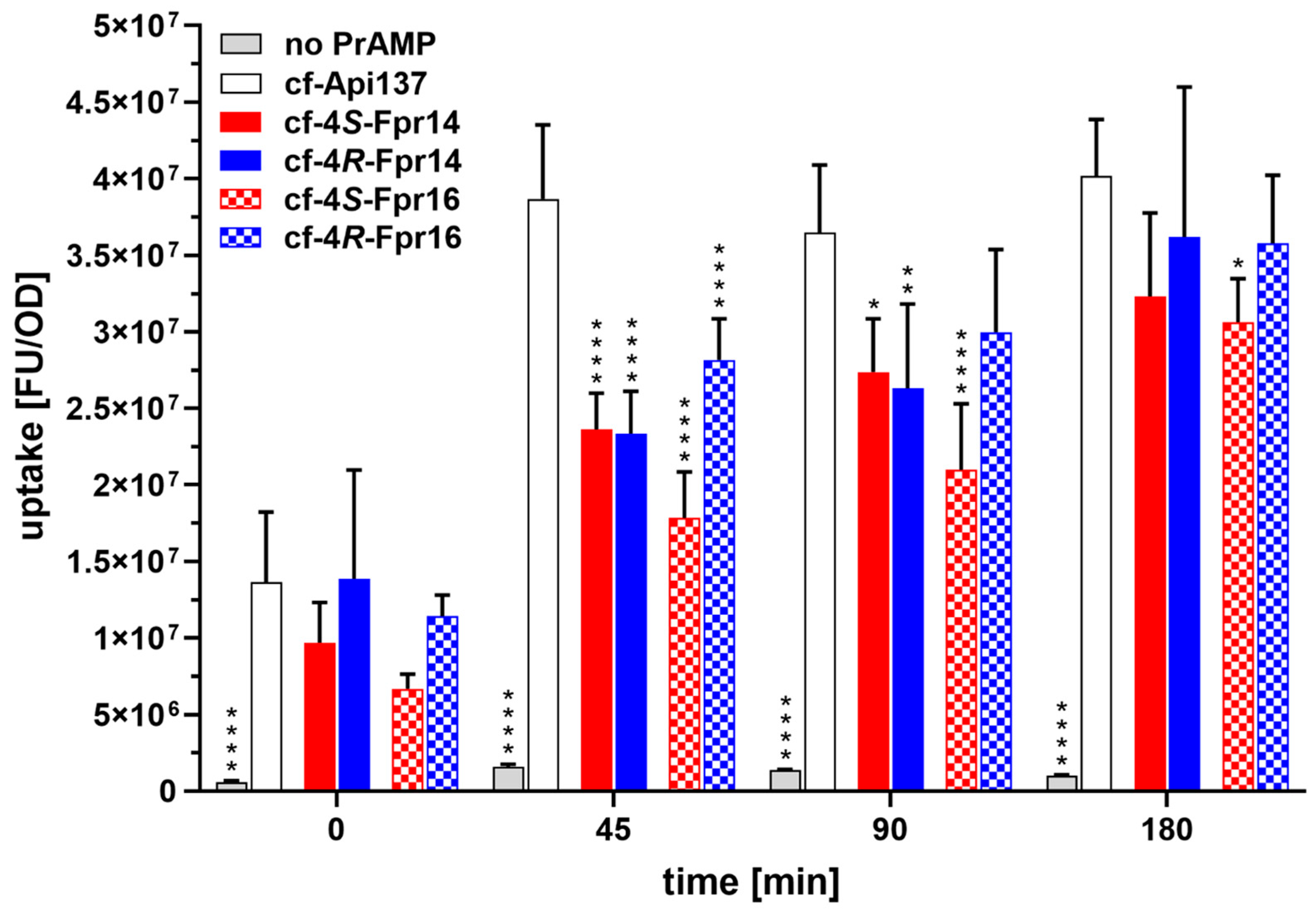
2.4. Further Analysis of the C-Terminal Proline Substitutions
3. Discussion
4. Materials and Methods
4.1. Materials and Chemicals
4.2. Peptide Synthesis
4.3. Ribosome Preparation
4.4. Fluorescence Polarization
4.5. Antibacterial Activity
4.6. In Vitro Transcription/Translation (iTT) Assay
4.7. Cultivation of E. coli RN31 for Ribosomal Profile Analysis
4.8. Ribosomal Profile Analysis
4.9. Cellular Uptake
4.10. Bacterial Growth Rate
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antimicrobial Peptides Database. Available online: https://aps.unmc.edu/home (accessed on 18 March 2025).
- Bakare, O.O.; Gokul, A.; Fadaka, A.O.; Wu, R.; Niekerk, L.-A.; Barker, A.M.; Keyster, M.; Klein, A. Plant Antimicrobial Peptides (PAMPs): Features, Applications, Production, Expression, and Challenges. Molecules 2022, 27, 3703. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zietz, C.M.; Mudgapalli, A.; Wang, S.; Wang, Z. The Evolution of the Antimicrobial Peptide Database over 18 Years: Milestones and New Features. Protein Sci. 2022, 31, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Boman, H.G. Peptide Antibiotics and Their Role in Innate Immunity. Annu. Rev. Immunol. 1995, 13, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Castle, M.; Nazarian, A.; Yi, S.S.; Tempst, P. Lethal Effects of Apidaecin on Escherichia coliInvolve Sequential Molecular Interactions with Diverse Targets. J. Biol. Chem. 1999, 274, 32555–32564. [Google Scholar] [CrossRef]
- Krizsan, A.; Prahl, C.; Goldbach, T.; Knappe, D.; Hoffmann, R. Short Proline-Rich Antimicrobial Peptides Inhibit Either the Bacterial 70S Ribosome or the Assembly of Its Large 50S Subunit. ChemBioChem 2015, 16, 2304–2308. [Google Scholar] [CrossRef]
- Mattiuzzo, M.; Bandiera, A.; Gennaro, R.; Benincasa, M.; Pacor, S.; Antcheva, N.; Scocchi, M. Role of the Escherichia Coli SbmA in the Antimicrobial Activity of Proline-rich Peptides. Mol. Microbiol. 2007, 66, 151–163. [Google Scholar] [CrossRef]
- Krizsan, A.; Volke, D.; Weinert, S.; Sträter, N.; Knappe, D.; Hoffmann, R. Insect-Derived Proline-Rich Antimicrobial Peptides Kill Bacteria by Inhibiting Bacterial Protein Translation at the 70 S Ribosome. Angew. Chem. Int. Ed. 2014, 53, 12236–12239. [Google Scholar] [CrossRef]
- Benincasa, M.; Pelillo, C.; Zorzet, S.; Garrovo, C.; Biffi, S.; Gennaro, R.; Scocchi, M. The Proline-Rich Peptide Bac7(1-35) Reduces Mortality from Salmonella Typhimurium in a Mouse Model of Infection. BMC Microbiol. 2010, 10, 178. [Google Scholar] [CrossRef]
- Czihal, P.; Knappe, D.; Fritsche, S.; Zahn, M.; Berthold, N.; Piantavigna, S.; Müller, U.; Van Dorpe, S.; Herth, N.; Binas, A.; et al. Api88 Is a Novel Antibacterial Designer Peptide To Treat Systemic Infections with Multidrug-Resistant Gram-Negative Pathogens. ACS Chem. Biol. 2012, 7, 1281–1291. [Google Scholar] [CrossRef]
- Boman, H.G.; Agerberth, B.; Boman, A. Mechanisms of Action on Escherichia Coli of Cecropin P1 and PR-39, Two Antibacterial Peptides from Pig Intestine. Infect. Immun. 1993, 61, 2978–2984. [Google Scholar] [CrossRef]
- Selmer, M.; Dunham, C.M.; Murphy, F.V.; Weixlbaumer, A.; Petry, S.; Kelley, A.C.; Weir, J.R.; Ramakrishnan, V. Structure of the 70 S Ribosome Complexed with mRNA and tRNA. Science 2006, 313, 1935–1942. [Google Scholar] [CrossRef]
- Bashan, A.; Yonath, A. Correlating Ribosome Function with High-Resolution Structures. Trends Microbiol. 2008, 16, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Sievers, A.; Beringer, M.; Rodnina, M.V.; Wolfenden, R. The Ribosome as an Entropy Trap. Proc. Natl. Acad. Sci. USA 2004, 101, 7897–7901. [Google Scholar] [CrossRef]
- Rodnina, M.V. Translation in Prokaryotes. Cold Spring Harb. Perspect. Biol. 2018, 10, a032664. [Google Scholar] [CrossRef]
- Nierhaus, K.H. The Assembly of the Prokaryotic Ribosome. Biosystems 1980, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Williamson, J.R. Structure and Dynamics of Bacterial Ribosome Biogenesis. Philos. Trans. R. Soc. B 2017, 372, 20160181. [Google Scholar] [CrossRef]
- Loveland, A.B.; Demo, G.; Grigorieff, N.; Korostelev, A.A. Ensemble Cryo-EM Elucidates the Mechanism of Translation Fidelity. Nature 2017, 546, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.N.; Rodnina, M.V.; et al. An Antimicrobial Peptide That Inhibits Translation by Trapping Release Factors on the Ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757. [Google Scholar] [CrossRef]
- Berthold, N.; Czihal, P.; Fritsche, S.; Sauer, U.; Schiffer, G.; Knappe, D.; Alber, G.; Hoffmann, R. Novel Apidaecin 1b Analogs with Superior Serum Stabilities for Treatment of Infections by Gram-Negative Pathogens. Antimicrob. Agents Chemother. 2013, 57, 402–409. [Google Scholar] [CrossRef]
- Casteels, P.; Ampe, C.; Jacobs, F.; Vaeck, M.; Tempst, P. Apidaecins: Antibacterial Peptides from Honeybees. EMBO J. 1989, 8, 2387–2391. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Huter, P.; Maracci, C.; Peterek, M.; Rodnina, M.V.; Wilson, D.N. Visualization of Translation Termination Intermediates Trapped by the Apidaecin 137 Peptide during RF3-Mediated Recycling of RF1. Nat. Commun. 2018, 9, 3053. [Google Scholar] [CrossRef]
- Ludwig, T.; Krizsan, A.; Mohammed, G.K.; Hoffmann, R. Antimicrobial Activity and 70S Ribosome Binding of Apidaecin-Derived Api805 with Increased Bacterial Uptake Rate. Antibiotics 2022, 11, 430. [Google Scholar] [CrossRef] [PubMed]
- Baliga, C.; Brown, T.J.; Florin, T.; Colon, S.; Shah, V.; Skowron, K.J.; Kefi, A.; Szal, T.; Klepacki, D.; Moore, T.W.; et al. Charting the Sequence-Activity Landscape of Peptide Inhibitors of Translation Termination. Proc. Natl. Acad. Sci. USA 2021, 118, e2026465118. [Google Scholar] [CrossRef]
- Lauer, S.M.; Reepmeyer, M.; Berendes, O.; Klepacki, D.; Gasse, J.; Gabrielli, S.; Grubmüller, H.; Bock, L.V.; Krizsan, A.; Nikolay, R.; et al. Multimodal Binding and Inhibition of Bacterial Ribosomes by the Antimicrobial Peptides Api137 and Api88. Nat. Commun. 2024, 15, 3945. [Google Scholar] [CrossRef]
- Lauer, S.M.; Gasse, J.; Krizsan, A.; Reepmeyer, M.; Sprink, T.; Nikolay, R.; Spahn, C.M.T.; Hoffmann, R. The Proline-Rich Antimicrobial Peptide Api137 Disrupts Large Ribosomal Subunit Assembly and Induces Misfolding. Nat. Commun. 2025, 16, 567. [Google Scholar] [CrossRef] [PubMed]
- Nikolay, R.; Schmidt, S.; Schlömer, R.; Deuerling, E.; Nierhaus, K. Ribosome Assembly as Antimicrobial Target. Antibiotics 2016, 5, 18. [Google Scholar] [CrossRef]
- Qin, B.; Lauer, S.M.; Balke, A.; Vieira-Vieira, C.H.; Bürger, J.; Mielke, T.; Selbach, M.; Scheerer, P.; Spahn, C.M.T.; Nikolay, R. Cryo-EM Captures Early Ribosome Assembly in Action. Nat. Commun. 2023, 14, 898. [Google Scholar] [CrossRef]
- Skowron, K.J.; Baliga, C.; Johnson, T.; Kremiller, K.M.; Castroverde, A.; Dean, T.T.; Allen, A.C.; Lopez-Hernandez, A.M.; Aleksandrova, E.V.; Klepacki, D.; et al. Structure–Activity Relationships of the Antimicrobial Peptide Natural Product Apidaecin. J. Med. Chem. 2023, 66, 11831–11842. [Google Scholar] [CrossRef]
- Knappe, D.; Cassone, M.; Nollmann, F.; Otvos, L.; Hoffmann, R. Hydroxyproline Substitutions Stabilize Non-Glycosylated Drosocin Against Serum Proteases Without Challenging Its Antibacterial Activity. Protein Pept. Lett. 2014, 21, 321–329. [Google Scholar] [CrossRef]
- Thomas, K.M.; Naduthambi, D.; Zondlo, N.J. Electronic Control of Amide Cis−trans Isomerism via the Aromatic−Prolyl Interaction. J. Am. Chem. Soc. 2006, 128, 2216–2217. [Google Scholar] [CrossRef] [PubMed]
- Newberry, R.W.; Raines, R.T. 4-Fluoroprolines: Conformational Analysis and Effects on the Stability and Folding of Peptides and Proteins. In Peptidomimetics I; Lubell, W.D., Ed.; Topics in Heterocyclic Chemistry; Springer International Publishing: Cham, Switzerland, 2016; Volume 48, pp. 1–25. ISBN 978-3-319-49117-2. [Google Scholar]
- Dunitz, J.D.; Taylor, R. Organic Fluorine Hardly Ever Accepts Hydrogen Bonds. Chem. A Eur. J. 1997, 3, 89–98. [Google Scholar] [CrossRef]
- Verhoork, S.J.M.; Killoran, P.M.; Coxon, C.R. Fluorinated Prolines as Conformational Tools and Reporters for Peptide and Protein Chemistry. Biochemistry 2018, 57, 6132–6143. [Google Scholar] [CrossRef]
- Huang, W.; Baliga, C.; Aleksandrova, E.V.; Atkinson, G.; Polikanov, Y.S.; Vázquez-Laslop, N.; Mankin, A.S. Activity, Structure, and Diversity of Type II Proline-Rich Antimicrobial Peptides from Insects. EMBO Rep. 2024, 25, 5194–5211. [Google Scholar] [CrossRef]
- Huang, W.; Baliga, C.; Vázquez-Laslop, N.; Mankin, A.S. Sequence Diversity of Apidaecin-like Peptides Arresting the Terminating Ribosome. Nucleic Acids Res. 2024, 52, 8967–8978. [Google Scholar] [CrossRef]
- Knappe, D.; Schmidt, R.; Adermann, K.; Hoffmann, R. Continuous Subcutaneous Delivery of Proline-Rich Antimicrobial Peptide Api137 Provides Superior Efficacy to Intravenous Administration in a Mouse Infection Model. Front. Microbiol. 2019, 10, 2283. [Google Scholar] [CrossRef]
- Schmidt, R.; Knappe, D.; Wende, E.; Ostorházi, E.; Hoffmann, R. In Vivo Efficacy and Pharmacokinetics of Optimized Apidaecin Analogs. Front. Chem. 2017, 5, 15. [Google Scholar] [CrossRef]
- Berthold, N.; Hoffmann, R. Cellular Uptake of Apidaecin 1b and Related Analogs in Gram-Negative Bacteria Reveals Novel Antibacterial Mechanism for Proline-Rich Antimicrobial Peptides. Protein Pept. Lett. 2014, 21, 391–398. [Google Scholar] [CrossRef]
- Kolano, L.; Knappe, D.; Volke, D.; Sträter, N.; Hoffmann, R. Ribosomal Target-Binding Sites of Antimicrobial Peptides Api137 and Onc112 Are Conserved among Pathogens Indicating New Lead Structures To Develop Novel Broad-Spectrum Antibiotics. ChemBioChem 2020, 21, 2628–2634. [Google Scholar] [CrossRef]
- Corbalan, N.; Runti, G.; Adler, C.; Covaceuszach, S.; Ford, R.C.; Lamba, D.; Beis, K.; Scocchi, M.; Vincent, P.A. Functional and Structural Study of the Dimeric Inner Membrane Protein SbmA. J. Bacteriol. 2013, 195, 5352–5361. [Google Scholar] [CrossRef]
- Siibak, T.; Peil, L.; Xiong, L.; Mankin, A.; Remme, J.; Tenson, T. Erythromycin- and Chloramphenicol-Induced Ribosomal Assembly Defects Are Secondary Effects of Protein Synthesis Inhibition. Antimicrob. Agents Chemother. 2009, 53, 563–571. [Google Scholar] [CrossRef]
- Siibak, T.; Peil, L.; Dönhöfer, A.; Tats, A.; Remm, M.; Wilson, D.N.; Tenson, T.; Remme, J. Antibiotic-induced Ribosomal Assembly Defects Result from Changes in the Synthesis of Ribosomal Proteins. Mol. Microbiol. 2011, 80, 54–67. [Google Scholar] [CrossRef]
- Champney, W. Bacterial Ribosomal Subunit Synthesis A Novel Antibiotic Target. Curr. Drug Targets-Infect. Disord. 2001, 1, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Champney, W.S. Antibiotics Targeting Bacterial Ribosomal Subunit Biogenesis. J. Antimicrob. Chemother. 2020, 75, 787–806. [Google Scholar] [CrossRef]
- Demange, L.; Ménez, A.; Dugave, C. Practical Synthesis of Boc and Fmoc Protected 4-Fluoro and 4-Difluoroprolines from Trans-4-Hydroxyproline. Tetrahedron Lett. 1998, 39, 1169–1172. [Google Scholar] [CrossRef]
- Chorghade, M.S.; Mohapatra, D.K.; Sahoo, G.; Gurjar, M.K.; Mandlecha, M.V.; Bhoite, N.; Moghe, S.; Raines, R.T. Practical Syntheses of 4-Fluoroprolines. J. Fluor. Chem. 2008, 129, 781–784. [Google Scholar] [CrossRef]
- Mathias, U.; Jung, M. Determination of Drug–Serum Protein Interactions via Fluorescence Polarization Measurements. Anal. Bioanal. Chem. 2007, 388, 1147–1156. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PrAMP | Substitution (Position) | Sequence |
|---|---|---|
| Api137 | None | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api839 | 4S-Fpr (5) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api840 | 4R-Fpr (5) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api845 | 4S-Fpr (9) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api846 | 4R-Fpr (9) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api841 | 4S-Fpr (11) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api842 | 4R-Fpr (11) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api843 | 4S-Fpr (13) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api844 | 4R-Fpr (13) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api847 | 4S-Fpr (14) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api848 | 4R-Fpr (14) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api849 | 4S-Fpr (16) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Api850 | 4R-Fpr (16) | gu-ONNRPVYIPRPRPPHPRL-OH |
| Substitution (Promoting Conformer) | MIC [µg/mL] | Ki [nM] | Residual iTT [%] | RPA [pre-50S Formation] | |
|---|---|---|---|---|---|
| Api137 | None | 4 | 2.6 ± 0.2 | 22 ± 6.2 | yes |
| Pro5 | 4S-Fpr (cis) | 4 | 3.3 ± 0.5 | 23 ± 0.2 | yes |
| 4R-Fpr (trans) | 4 | 3.3 ± 0.3 | 35 ± 0.1 | yes | |
| Pro9 | 4S-Fpr (cis) | 2 | 3.9 ± 0.4 | 26 ± 5.7 | yes |
| 4R-Fpr (trans) | 4 | 3.7 ± 0.4 | 33 ± 3.5 | no | |
| Pro11 | 4S-Fpr (cis) | 1 | 73.4 ± 10.9 | 32 ± 0.9 | yes |
| 4R-Fpr (trans) | 4 | 2.5 ± 0.2 | 31 ± 0.7 | reduced | |
| Pro13 | 4S-Fpr (cis) | 4 | 5.3 ± 0.6 | 28 ± 3.2 | yes |
| 4R-Fpr (trans) | 2 | 16.9 ± 1.2 | 28 ± 1.2 | yes | |
| Pro14 | 4S-Fpr (cis) | 2 | 1.6 ± 0.1 | 21 ± 0.2 | reduced |
| 4R-Fpr (trans) | 2 | 1.6± 0.1 | 64 ± 4.6 | no | |
| Pro16 | 4S-Fpr (cis) | 16 | 4.8 ± 0.8 | 63 ± 8.9 | no |
| 4R-Fpr (trans) | 4 | 0.8 ± 0.2 | 28 ± 0.7 | yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reepmeyer, M.; Krizsan, A.; Brakel, A.; Kolano, L.; Gasse, J.; Husselbee, B.W.; Robinson, A.J.; Hoffmann, R. Substitution of Proline Residues by 4-Fluoro-l-Proline Affects the Mechanism of the Proline-Rich Antimicrobial Peptide Api137. Antibiotics 2025, 14, 566. https://doi.org/10.3390/antibiotics14060566
Reepmeyer M, Krizsan A, Brakel A, Kolano L, Gasse J, Husselbee BW, Robinson AJ, Hoffmann R. Substitution of Proline Residues by 4-Fluoro-l-Proline Affects the Mechanism of the Proline-Rich Antimicrobial Peptide Api137. Antibiotics. 2025; 14(6):566. https://doi.org/10.3390/antibiotics14060566
Chicago/Turabian StyleReepmeyer, Maren, Andor Krizsan, Alexandra Brakel, Lisa Kolano, Jakob Gasse, Benjamin W. Husselbee, Andrea J. Robinson, and Ralf Hoffmann. 2025. "Substitution of Proline Residues by 4-Fluoro-l-Proline Affects the Mechanism of the Proline-Rich Antimicrobial Peptide Api137" Antibiotics 14, no. 6: 566. https://doi.org/10.3390/antibiotics14060566
APA StyleReepmeyer, M., Krizsan, A., Brakel, A., Kolano, L., Gasse, J., Husselbee, B. W., Robinson, A. J., & Hoffmann, R. (2025). Substitution of Proline Residues by 4-Fluoro-l-Proline Affects the Mechanism of the Proline-Rich Antimicrobial Peptide Api137. Antibiotics, 14(6), 566. https://doi.org/10.3390/antibiotics14060566