
The Current Landscape of Phage–Antibiotic Synergistic (PAS) Interactions



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Antimicrobial Resistance
1.2. Bacteriophage Therapy
1.3. Phage–Antibiotic Synergy (PAS)
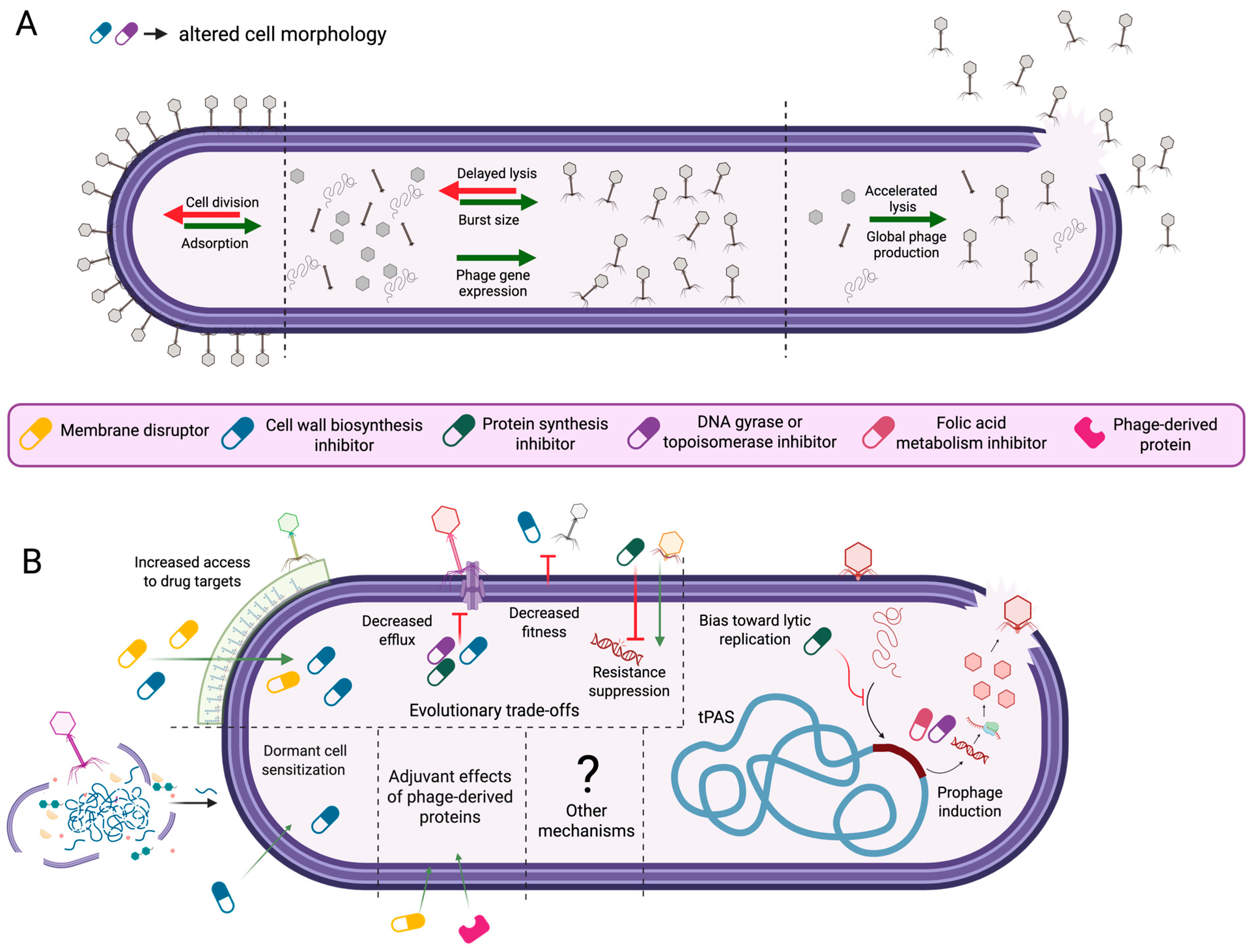
2. Potential Mechanisms of PAS
2.1. Changes to Cell Morphology
2.1.1. Antibiotic-Induced Changes to Host Cell Morphology and Phage Infection Dynamics
2.1.2. Morphology-Driven Modulation of Phage–Host Interactions
2.1.3. Increased Phage Production as a Function of Lysis Rate and Burst Size
2.2. Temperate Phage–Antibiotic Synergy and Resistance Trade-Offs
2.2.1. Temperate Phage–Antibiotic Synergy (tPAS)
2.2.2. Phage-Driven Antibiotic Sensitization
2.2.3. Evolutionary Trade-Offs in Phage–Antibiotic Synergy
2.3. Other Potential Synergistic Effects
2.3.1. Deep Dormant Cell Resuscitation
2.3.2. Purified Phage Proteins as Antimicrobial Adjuvants
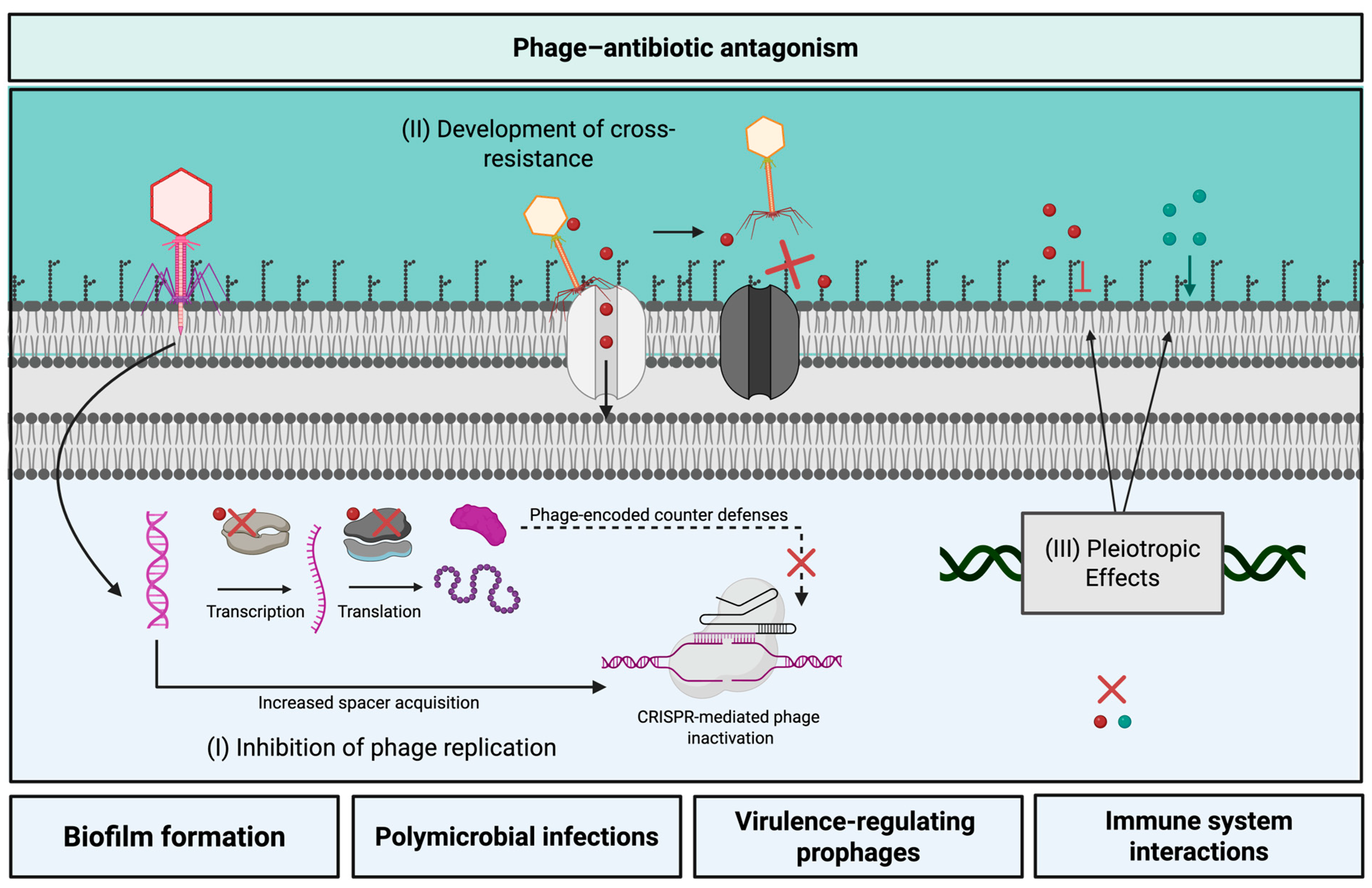
3. Phage–Antibiotic Antagonism
3.1. Possible Mechanisms of Phage–Antibiotic Antagonism
3.2. Challenges Toward Predicting Phage–Antibiotic Antagonism
3.2.1. Exceptions Are the Rule: Antibiotics
3.2.2. Antagonism Depends on the Specific Phage–Host System
3.2.3. In Vitro Differences Across Concentration, Time, and Condition
4. Development of Phage–Antibiotic Strategies
4.1. In Vitro Evaluation of Phage–Antibiotic Pairs
4.1.1. Agar-Based Methods
4.1.2. Broth-Based Methods
4.2. Timing of Application
4.3. Other Considerations
4.3.1. Endogenous Prophage Landscape
4.3.2. Interactions with Innate Immune System
4.3.3. Polymicrobial Infections
5. Conclusions
5.1. Summary of Insights
5.2. Research Gaps and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PAS | Phage–Antibiotic Synergy |
| AMR | Antimicrobial Resistance |
| LPS | Lipopolysaccharide |
| SOS | “Save-Our-Souls” |
References
- Naghavi, M.; Vollset, S.E.; Ikuta, K.S.; Swetschinski, L.R.; Gray, A.P.; Wool, E.E.; Aguilar, G.R.; Mestrovic, T.; Smith, G.; Han, C.; et al. Global burden of bacterial antimicrobial resistance 1990–2021: A systematic analysis with forecasts to 2050. Lancet 2024, 404, 1199–1226. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Bacterial Priority Pathogens List, 2024: Bacterial Pathogens of Public Health Importance to Guide Research, Development and Strategies to Prevent and Control Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- Jangra, M.; Travin, D.Y.; Aleksandrova, E.V.; Kaur, M.; Darwish, L.; Koteva, K.; Klepacki, D.; Wang, W.; Tiffany, M.; Sokaribo, A.; et al. A broad-spectrum lasso peptide antibiotic targeting the bacterial ribosome. Nature 2025, 640, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H. The antibiotic resistance crisis and the development of new antibiotics. Microb. Biotechnol. 2024, 17, e14510. [Google Scholar] [CrossRef]
- Butler, M.S.; Gigante, V.; Sati, H.; Paulin, S.; Al-Sulaiman, L.; Rex, J.H.; Fernandes, P.; Arias, C.A.; Paul, M.; Thwaites, G.E.; et al. Analysis of the Clinical Pipeline of Treatments for Drug-Resistant Bacterial Infections: Despite Progress, More Action Is Needed. Antimicrob. Agents Chemother. 2022, 66, e01991-21. [Google Scholar] [CrossRef]
- Darby, E.M.; Trampari, E.; Siasat, P.; Gaya, M.S.; Alav, I.; Webber, M.A.; Blair, J.M.A. Molecular mechanisms of antibiotic resistance revisited. Nat. Rev. Microbiol. 2023, 21, 280–295. [Google Scholar] [CrossRef]
- Prajapati, J.D.; Kleinekathöfer, U.; Winterhalter, M. How to Enter a Bacterium: Bacterial Porins and the Permeation of Antibiotics. Chem. Rev. 2021, 121, 5158–5192. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Bell, A. Antibiotic uptake into gram-negative bacteria. Eur. J. Clin. Microbiol. Infect. Dis. 1988, 7, 713–720. [Google Scholar] [CrossRef]
- Olesky, M.; Hobbs, M.; Nicholas, R.A. Identification and Analysis of Amino Acid Mutations in Porin IB That Mediate Intermediate-Level Resistance to Penicillin and Tetracycline in Neisseria gonorrhoeae. Antimicrob. Agents Chemother. 2002, 46, 2811–2820. [Google Scholar] [CrossRef] [PubMed]
- Masi, M.; Réfregiers, M.; Pos, K.M.; Pagès, J.-M. Mechanisms of envelope permeability and antibiotic influx and efflux in Gram-negative bacteria. Nat. Microbiol. 2017, 2, 17001. [Google Scholar] [CrossRef]
- Campos, M.A.; Vargas, M.A.; Regueiro, V.; Llompart, C.M.; Albertí, S.; Bengoechea, J.A. Capsule Polysaccharide Mediates Bacterial Resistance to Antimicrobial Peptides. Infect. Immun. 2004, 72, 7107–7114. [Google Scholar] [CrossRef]
- D’Angelo, F.; Rocha, E.P.C.; Rendueles, O. The Capsule Increases Susceptibility to Last-Resort Polymyxins, but Not to Other Antibiotics, in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2023, 67, e00127-23. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.N.; Bayer, A.S.; Tran, T.T.; Shamoo, Y.; Mileykovskaya, E.; Dowhan, W.; Guan, Z.; Arias, C.A. Daptomycin Resistance in Enterococci Is Associated with Distinct Alterations of Cell Membrane Phospholipid Content. PLoS ONE 2012, 7, e43958. [Google Scholar] [CrossRef]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef]
- Maeda, Y.; Kiba, A.; Ohnishi, K.; Hikichi, Y. Amino acid substitutions in GyrA of Burkholderia glumae are implicated in not only oxolinic acid resistance but also fitness on rice plants. Appl. Environ. Microbiol. 2007, 73, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef]
- Salmond, G.P.C.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef]
- Gerovac, M.; Chihara, K.; Wicke, L.; Böttcher, B.; Lavigne, R.; Vogel, J. Phage proteins target and co-opt host ribosomes immediately upon infection. Nat. Microbiol. 2024, 9, 787–800. [Google Scholar] [CrossRef]
- Nechaev, S.; Severinov, K. Bacteriophage-Induced Modifications of Host RNA Polymerase. Annu. Rev. Microbiol. 2003, 57, 301–322. [Google Scholar] [CrossRef]
- Drobysheva, A.V.; Panafidina, S.A.; Kolesnik, M.V.; Klimuk, E.I.; Minakhin, L.; Yakunina, M.V.; Borukhov, S.; Nilsson, E.; Holmfeldt, K.; Yutin, N.; et al. Structure and function of virion RNA polymerase of a crAss-like phage. Nature 2021, 589, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Yoshikawa, G.; Mihara, T.; Chatchawankanphanich, O.; Kawasaki, T.; Nakano, M.; Fujie, M.; Ogata, H.; Yamada, T. Replications of two closely related groups of jumbo phages show different level of dependence on host-encoded RNA polymerase. Front. Microbiol. 2017, 8, 1010. [Google Scholar] [CrossRef] [PubMed]
- Ceyssens, P.-J.; Minakhin, L.; Van den Bossche, A.; Yakunina, M.; Klimuk, E.; Blasdel, B.; De Smet, J.; Noben, J.-P.; Bläsi, U.; Severinov, K.; et al. Development of Giant Bacteriophage ϕKZ Is Independent of the Host Transcription Apparatus. J. Virol. 2014, 88, 10501–10510. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Mekalanos, J.J. Lysogenic conversion by a filamentous phage encoding Cholera toxin. Science 1996, 272, 1910–1914. [Google Scholar] [CrossRef]
- Shi, K.; Oakland, J.T.; Kurniawan, F.; Moeller, N.H.; Banerjee, S.; Aihara, H. Structural basis of superinfection exclusion by bacteriophage T4 Spackle. Commun. Biol. 2020, 3, 691. [Google Scholar] [CrossRef]
- Van den Berg, B.; Silale, A.; Baslé, A.; Brandner, A.F.; Mader, S.L.; Khalid, S. Structural basis for host recognition and superinfection exclusion by bacteriophage T5. Proc. Natl. Acad. Sci. USA 2022, 119, e2211672119. [Google Scholar] [CrossRef]
- Bucher, M.J.; Czyż, D.M. Phage against the machine: The SIE-ence of superinfection exclusion. Viruses 2024, 16, 1348. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef] [PubMed]
- Leavitt, J.C.; Woodbury, B.M.; Gilcrease, E.B.; Bridges, C.M.; Teschke, C.M.; Casjens, S.R. Bacteriophage P22 SieA-mediated superinfection exclusion. mBio 2024, 15, e02169-23. [Google Scholar] [CrossRef]
- Monteiro, R.; Pires, D.P.; Costa, A.R.; Azeredo, J. Phage therapy: Going temperate? Trends Microbiol. 2019, 27, 368–378. [Google Scholar] [CrossRef]
- Touchon, M.; Bernheim, A.; Rocha, E.P.C. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 2016, 10, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- Lauman, P.; Dennis, J.J. Synergistic Interactions among Burkholderia cepacia Complex-Targeting Phages Reveal a Novel Therapeutic Role for Lysogenization-Capable Phages. Microbiol. Spectr. 2023, 11, e04430-22. [Google Scholar] [CrossRef]
- Antine, S.P.; Johnson, A.G.; Mooney, S.E.; Leavitt, A.; Mayer, M.L.; Yirmiya, E.; Amitai, G.; Sorek, R.; Kranzusch, P.J. Structural basis of Gabija anti-phage defence and viral immune evasion. Nature 2024, 625, 360–365. [Google Scholar] [CrossRef]
- Georjon, H.; Bernheim, A. The highly diverse antiphage defence systems of bacteria. Nat. Rev. Microbiol. 2023, 21, 686–700. [Google Scholar] [CrossRef]
- Supina, B.S.I.; McCutcheon, J.G.; Peskett, S.R.; Stothard, P.; Dennis, J.J. A flagella-dependent Burkholderia jumbo phage controls rice seedling rot and steers Burkholderia glumae toward reduced virulence in rice seedlings. mBio 2025, 16, e02814-24. [Google Scholar] [CrossRef]
- Gordillo Altamirano, F.L.; Barr, J.J. Unlocking the next generation of phage therapy: The key is in the receptors. Curr. Opin. Biotechnol. 2021, 68, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, M.; Jee, S.-N.; Heu, S.; Ryu, S. Development of a bacteriophage cocktail against Pectobacterium carotovorum Subsp. carotovorum and its effects on Pectobacterium virulence. Appl. Environ. Microbiol. 2022, 88, e0076122. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shen, W.; Zhong, Q.; Chen, Q.; He, X.; Baker, J.L.; Xiong, K.; Jin, X.; Wang, J.; Hu, F.; et al. Development of a Bacteriophage Cocktail to Constrain the Emergence of Phage-Resistant Pseudomonas aeruginosa. Front. Microbiol. 2020, 11, 327. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Chen, Q.; Echterhof, A.; Pennetzdorfer, N.; McBride, R.C.; Banaei, N.; Burgener, E.B.; Milla, C.E.; Bollyky, P.L. A blueprint for broadly effective bacteriophage-antibiotic cocktails against bacterial infections. Nat. Commun. 2024, 15, 9987. [Google Scholar] [CrossRef]
- Zulk, J.J.; Clark, J.R.; Ottinger, S.; Ballard, M.B.; Mejia, M.E.; Mercado-Evans, V.; Heckmann, E.R.; Sanchez, B.C.; Trautner, B.W.; Maresso, A.W.; et al. Phage Resistance Accompanies Reduced Fitness of Uropathogenic Escherichia coli in the Urinary Environment. mSphere 2022, 7, e00345-22. [Google Scholar] [CrossRef]
- Gao, D.; Ji, H.; Wang, L.; Li, X.; Hu, D.; Zhao, J.; Wang, S.; Tao, P.; Li, X.; Qian, P. Fitness Trade-Offs in Phage Cocktail-Resistant Salmonella enterica Serovar Enteritidis Results in Increased Antibiotic Susceptibility and Reduced Virulence. Microbiol. Spectr. 2022, 10, e02914-22. [Google Scholar] [CrossRef] [PubMed]
- Gurney, J.; Pradier, L.; Griffin, J.S.; Gougat-Barbera, C.; Chan, B.K.; Turner, P.E.; Kaltz, O.; Hochberg, M.E. Phage steering of antibiotic-resistance evolution in the bacterial pathogen, Pseudomonas aeruginosa. Evol. Med. Public Health 2020, 2020, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Ruest, M.K.; Supina, B.S.I.; Dennis, J.J. Bacteriophage steering of Burkholderia cenocepacia toward reduced virulence and increased antibiotic sensitivity. J. Bacteriol. 2023, 205, e00196-23. [Google Scholar] [CrossRef] [PubMed]
- Segall, A.M.; Roach, D.R.; Strathdee, S.A. Stronger together? Perspectives on phage-antibiotic synergy in clinical applications of phage therapy. Curr. Opin. Microbiol. 2019, 51, 46–50. [Google Scholar] [CrossRef]
- Comeau, A.M.; Tétart, F.; Trojet, S.N.; Prère, M.-F.; Krisch, H.M. Phage-antibiotic synergy (PAS): β-lactam and quinolone antibiotics stimulate virulent phage growth. PLoS ONE 2007, 2, e799. [Google Scholar] [CrossRef]
- Nir-Paz, R.; Gelman, D.; Khouri, A.; Sisson, B.M.; Fackler, J.; Alkalay-Oren, S.; Khalifa, L.; Rimon, A.; Yerushalmy, O.; Bader, R.; et al. Successful Treatment of Antibiotic-resistant, Poly-microbial Bone Infection With Bacteriophages and Antibiotics Combination. Clin. Infect. Dis. 2019, 69, 2015–2018. [Google Scholar] [CrossRef]
- Critically Ill Patient with Multidrug-Resistant Acinetobacter Baumannii Respiratory Infection Successfully Treated with Intravenous and Nebulized Bacteriophage Therapy|Antimicrobial Agents and Chemotherapy. Available online: https://journals-asm-org.login.ezproxy.library.ualberta.ca/doi/10.1128/aac.00824-21 (accessed on 16 April 2025).
- Doern, C.D. When does 2 plus 2 equal 5? A review of antimicrobial synergy testing. J. Clin. Microbiol. 2014, 52, 4124–4128. [Google Scholar] [CrossRef]
- Kim, M.K.; Suh, G.A.; Cullen, G.D.; Rodriguez, S.P.; Dharmaraj, T.; Chang, T.H.W.; Li, Z.; Chen, Q.; Green, S.I.; Lavigne, R.; et al. Bacteriophage therapy for multidrug-resistant infections: Current technologies and therapeutic approaches. J. Clin. Investig. 2025, 135, e187996. [Google Scholar] [CrossRef]
- Knezevic, P.; Curcin, S.; Aleksic, V.; Petrusic, M.; Vlaski, L. Phage-antibiotic synergism: A possible approach to combatting Pseudomonas aeruginosa. Res. Microbiol. 2013, 164, 55–60. [Google Scholar] [CrossRef]
- Liu, C.; Hong, Q.; Chang, R.Y.K.; Kwok, P.C.L.; Chan, H.-K. Phage-antibiotic therapy as a promising strategy to combat multidrug-resistant infections and to enhance antimicrobial efficiency. Antibiot. Basel 2022, 11, 570. [Google Scholar] [CrossRef]
- Bulssico, J.; PapukashvilI, I.; Espinosa, L.; Gandon, S.; Ansaldi, M. Phage-antibiotic synergy: Cell filamentation is a key driver of successful phage predation. PLoS Pathog. 2023, 19, e1011602. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; McCutcheon, J.; Dennis, J. Aztreonam lysine increases the activity of phages E79 and phiKZ against Pseudomonas aeruginosa PA01. Microorganisms 2021, 9, 152. [Google Scholar] [CrossRef]
- Kim, M.; Jo, Y.; Hwang, Y.J.; Hong, H.W.; Hong, S.S.; Park, K.; Myung, H. Phage-Antibiotic Synergy via Delayed Lysis. Appl. Environ. Microbiol. 2018, 84, e02085-18. [Google Scholar] [CrossRef]
- Qin, K.; Shi, X.; Yang, K.; Xu, Q.; Wang, F.; Chen, S.; Xu, T.; Liu, J.; Wen, W.; Chen, R.; et al. Phage-antibiotic synergy suppresses resistance emergence of Klebsiella pneumoniae by altering the evolutionary fitness. mBio 2024, 15, e01393-24. [Google Scholar] [CrossRef] [PubMed]
- Al-Anany, A.M.; Fatima, R.; Nair, G.; Mayol, J.T.; Hynes, A.P. Temperate phage-antibiotic synergy across antibiotic classes reveals new mechanism for preventing lysogeny. mBio 2024, 15, e00504-24. [Google Scholar] [CrossRef]
- Al-Anany, A.M.; Fatima, R.; Hynes, A.P. Temperate phage-antibiotic synergy eradicates bacteria through depletion of lysogens. Cell Rep. 2021, 35, 109172. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Sistrom, M.; Wertz, J.E.; Kortright, K.E.; Narayan, D.; Turner, P.E. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 26717. [Google Scholar] [CrossRef]
- Gordillo Altamirano, F.L.; Kostoulias, X.; Subedi, D.; Korneev, D.; Peleg, A.Y.; Barr, J.J. Phage-antibiotic combination is a superior treatment against Acinetobacter baumannii in a preclinical study. eBioMedicine 2022, 80, 104045. [Google Scholar] [CrossRef]
- Parab, L.; Dherbey, J.R.; Rivera, N.; Schwarz, M.; Gallie, J.; Bertels, F. Chloramphenicol and gentamicin reduce the evolution of resistance to phage ΦX174 by suppressing a subset of E. coli LPS mutants. PLoS Biol. 2025, 23, e3002952. [Google Scholar] [CrossRef]
- Maffei, E.; Woischnig, A.-K.; Burkolter, M.R.; Heyer, Y.; Humolli, D.; Thürkauf, N.; Bock, T.; Schmidt, A.; Manfredi, P.; Egli, A.; et al. Phage Paride can kill dormant, antibiotic-tolerant cells of Pseudomonas aeruginosa by direct lytic replication. Nat. Commun. 2024, 15, 175. [Google Scholar] [CrossRef]
- Schuch, R.; Lee, H.M.; Schneider, B.C.; Sauve, K.L.; Law, C.; Khan, B.K.; Rotolo, J.A.; Horiuchi, Y.; Couto, D.E.; Raz, A.; et al. Combination therapy With lysin CF-301 and antibiotic is superior to antibiotic alone for treating methicillin-resistant Staphylococcus aureus–induced murine bacteremia. J. Infect. Dis. 2014, 209, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, M.; Zhang, P.; Xu, M.; Yuan, W.; Bian, L.; Liu, Y.; Xia, J.; Leung, S.S.Y. Phage-Derived Depolymerase as an Antibiotic Adjuvant Against Multidrug-Resistant Acinetobacter baumannii. Front. Microbiol. 2022, 13, 845500. [Google Scholar] [CrossRef] [PubMed]
- Justice, S.S.; Hunstad, D.A.; Cegelski, L.; Hultgren, S.J. Morphological plasticity as a bacterial survival strategy. Nat. Rev. Microbiol. 2008, 6, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.; Ali, M.A.; Lee, D.; Félix, M.-A.; Luallen, R.J. Bacterial filamentation as a mechanism for cell-to-cell spread within an animal host. Nat. Commun. 2022, 13, 693. [Google Scholar] [CrossRef]
- Mukherjee, A.; Cao, C.; Lutkenhaus, J. Inhibition of FtsZ polymerization by SulA, an inhibitor of septation in Escherichia coli. Proc. Natl. Acad. Sci. USA 1998, 95, 2885–2890. [Google Scholar] [CrossRef]
- Bos, J.; Zhang, Q.; Vyawahare, S.; Rogers, E.; Rosenberg, S.M.; Austin, R.H. Emergence of antibiotic resistance from multinucleated bacterial filaments. Proc. Natl. Acad. Sci. USA 2015, 112, 178–183. [Google Scholar] [CrossRef]
- Maiques, E.; Úbeda, C.; Campoy, S.; Salvador, N.; Lasa, Í.; Novick, R.P.; Barbé, J.; Penadés, J.R. β-Lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in Staphylococcus aureus. J. Bacteriol. 2006, 188, 2726–2729. [Google Scholar] [CrossRef]
- Miller, C.; Thomsen, L.E.; Gaggero, C.; Mosseri, R.; Ingmer, H.; Cohen, S.N. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science 2004, 305, 1629–1631. [Google Scholar] [CrossRef]
- Bush, N.G.; Diez-Santos, I.; Abbott, L.R.; Maxwell, A. Quinolones: Mechanism, Lethality and Their Contributions to Antibiotic Resistance. Molecules 2020, 25, 5662. [Google Scholar] [CrossRef]
- Halawa, E.M.; Fadel, M.; Al-Rabia, M.W.; Behairy, A.; Nouh, N.A.; Abdo, M.; Olga, R.; Fericean, L.; Atwa, A.M.; El-Nablaway, M.; et al. Antibiotic action and resistance: Updated review of mechanisms, spread, influencing factors, and alternative approaches for combating resistance. Front. Pharmacol. 2024, 14, 1305294. [Google Scholar] [CrossRef]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [PubMed]
- Sangurdekar, D.P.; Zhang, Z.; Khodursky, A.B. The association of DNA damage response and nucleotide level modulation with the antibacterial mechanism of the anti-folate drug Trimethoprim. BMC Genomics. 2011, 12, 583. [Google Scholar] [CrossRef]
- Kamal, F.; Dennis, J.J. Burkholderia cepacia complex Phage-Antibiotic Synergy (PAS): Antibiotics stimulate lytic phage activity. Appl. Environ. Microbiol. 2015, 81, 1132–1138. [Google Scholar] [CrossRef]
- Aframian, N.; Omer Bendori, S.; Kabel, S.; Guler, P.; Stokar-Avihail, A.; Manor, E.; Msaeed, K.; Lipsman, V.; Grinberg, I.; Mahagna, A.; et al. Dormant phages communicate via arbitrium to control exit from lysogeny. Nat. Microbiol. 2022, 7, 145–153. [Google Scholar] [CrossRef]
- Brady, A.; Felipe-Ruiz, A.; Gallego Del Sol, F.; Marina, A.; Quiles-Puchalt, N.; Penadés, J.R. Molecular basis of lysis-lysogeny decisions in Gram-positive phages. Annu. Rev. Microbiol. 2021, 75, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Fatima, R.; Hynes, A.P. Temperate phage-antibiotic synergy is widespread—Extending to Pseudomonas—But varies by phage, host strain, and antibiotic pairing. mBio 2024, 16, e02559-24. [Google Scholar] [CrossRef]
- Fu, Y.; Yin, M.; Cao, L.; Lu, Y.; Li, Y.; Zhang, L. Capsule mutations serve as a key strategy of phage resistance evolution of K54 hypervirulent Klebsiella pneumoniae. Commun. Biol. 2025, 8, 257. [Google Scholar] [CrossRef] [PubMed]
- Gordillo Altamirano, F.; Forsyth, J.H.; Patwa, R.; Kostoulias, X.; Trim, M.; Subedi, D.; Archer, S.K.; Morris, F.C.; Oliveira, C.; Kielty, L.; et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 2021, 6, 157–161. [Google Scholar] [CrossRef]
- Fujiki, J.; Nakamura, K.; Nakamura, T.; Iwano, H. Fitness trade-offs between phage and antibiotic sensitivity in phage-resistant variants: Molecular action and insights into clinical applications for phage therapy. Int. J. Mol. Sci. 2023, 24, 15628. [Google Scholar] [CrossRef]
- Fujiki, J.; Nakamura, K.; Ishiguro, Y.; Iwano, H. Using phage to drive selections toward restoring antibiotic sensitivity in Pseudomonas aeruginosa via chromosomal deletions. Front. Microbiol. 2024, 15, 1401234. [Google Scholar] [CrossRef]
- North, O.I.; Brown, E.D. Phage–antibiotic combinations: A promising approach to constrain resistance evolution in bacteria. Ann. N. Y. Acad. Sci. 2021, 1496, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Koderi Valappil, S.; Shetty, P.; Deim, Z.; Terhes, G.; Urbán, E.; Váczi, S.; Patai, R.; Polgár, T.; Pertics, B.Z.; Schneider, G.; et al. Survival comes at a cost: A coevolution of phage and Its host leads to phage resistance and antibiotic sensitivity of Pseudomonas aeruginosa multidrug resistant strains. Front. Microbiol. 2021, 12, 783722. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, A.R.; Fortier, A.; Roush, C.; Lessing, A.J.; Bender, R.G.; Barahman, R.; Grant, R.; Chan, B.K.; Turner, P.E. Pleiotropy complicates a trade-off between phage resistance and antibiotic resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 11207–11216. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Loh, B.; Gordillo Altamirano, F.; Yu, Y.; Hua, X.; Leptihn, S. Colistin-phage combinations decrease antibiotic resistance in Acinetobacter baumannii via changes in envelope architecture. Emerg. Microbes Infect. 2021, 10, 2205–2219. [Google Scholar] [CrossRef]
- Sabnis, A.; Hagart, K.L.; Klöckner, A.; Becce, M.; Evans, L.E.; Furniss, R.C.D.; Mavridou, D.A.; Murphy, R.; Stevens, M.M.; Davies, J.C.; et al. Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane. eLife 2021, 10, e65836. [Google Scholar] [CrossRef]
- Rhodes, K.A.; Schweizer, H.P. Antibiotic resistance in Burkholderia species. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 28, 82–90. [Google Scholar] [CrossRef]
- Ashworth, E.A.; Wright, R.C.T.; Shears, R.K.; Wong, J.K.L.; Hassan, A.; Hall, J.P.J.; Kadioglu, A.; Fothergill, J.L. Exploiting lung adaptation and phage steering to clear pan-resistant Pseudomonas aeruginosa infections in vivo. Nat. Commun. 2024, 15, 1547. [Google Scholar] [CrossRef]
- Feng, L.; Chen, H.; Qian, C.; Zhao, Y.; Wang, W.; Liu, Y.; Xu, M.; Cao, J.; Zhou, T.; Wu, Q. Resistance, mechanism, and fitness cost of specific bacteriophages for Pseudomonas aeruginosa. mSphere 2024, 9, e00553-23. [Google Scholar] [CrossRef]
- Niu, H.; Gu, J. and Zhang, Y. Bacterial persisters; molecular mechanisms and therapeutic development. Signal Transduct. Target. Ther. 2024, 9, 174. [Google Scholar] [CrossRef]
- Sanchez-Torres, V.; Kirigo, J.; Wood, T.K. Implications of lytic phage infections inducing persistence. Curr. Opin. Microbiol. 2024, 79, 102482. [Google Scholar] [CrossRef]
- Karthika, C.; Malligarjunan, N.; Jothi, R.; Kasthuri, T.; Alexpandi, R.; Ravi, A.V.; Pandian, S.K.; Gowrishankar, S. Two novel phages PSPa and APPa inhibit planktonic, sessile and persister populations of Pseudomonas aeruginosa, and mitigate its virulence in Zebrafish model. Sci. Rep. 2023, 13, 19033. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska, B.; Olszak, T.; Drulis-Kawa, Z. Applications of bacteriophages versus phage enzymes to combat and cure bacterial infections: An ambitious and also a realistic application? Appl. Microbiol. Biotechnol. 2018, 102, 2563–2581. [Google Scholar] [CrossRef]
- Watson, A.; Sauve, K.; Cassino, C.; Schuch, R. Exebacase demonstrates in vitro synergy with a broad range of antibiotics against both methicillin-resistant and methicillin-susceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 2020, 64, e01885-19. [Google Scholar] [CrossRef]
- Asempa, T.E.; Abdelraouf, K.; Carabeo, T.; Schuch, R.; Nicolau, D.P. Synergistic activity of exebacase (CF-301) in addition to daptomycin against Staphylococcus aureus in a neutropenic murine thigh infection model. Antimicrob. Agents Chemother. 2020, 64, e02176-19. [Google Scholar] [CrossRef]
- Fowler, V.-G.; Das, A.F.; Lipka-Diamond, J.; Schuch, R.; Pomerantz, R.; Jáuregui-Peredo, L.; Bressler, A.; Evans, D.; Moran, G.J.; Rupp, M.E.; et al. Exebacase for patients with Staphylococcus aureus bloodstream infection and endocarditis. J. Clin. Investig. 2020, 130, 3750–3760. [Google Scholar] [CrossRef]
- Fowler, V.G.; Das, A.F.; Lipka-Diamond, J.; Ambler, J.E.; Schuch, R.; Pomerantz, R.; Cassino, C.; Jáuregui-Peredo, L.; Moran, G.J.; Rupp, M.E.; et al. Exebacase in addition to standard-of-care antibiotics for Staphylococcus aureus bloodstream infections and right-sided infective endocarditis: A Phase 3, superiority-design, placebo-controlled, randomized clinical trial (DISRUPT). Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2024, 78, 1473–1481. [Google Scholar] [CrossRef]
- Nair, S.; Poonacha, N.; Desai, S.; Hiremath, D.; Tuppad, D.; Mohan, T.; Chikkamadaiah, R.; Durgaiah, M.; Kumar, S.; Channabasappa, S.; et al. Restoration of sensitivity of a diverse set of drug-resistant Staphylococcus clinical strains by bactericidal protein P128. J. Med. Microbiol. 2018, 67, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Frontiers|Enhanced Antibacterial Activity of Acinetobacter Baumannii Bacteriophage ØABP-01 Endolysin (LysABP-01) in Combination with Colistin. Available online: https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2016.01402/full (accessed on 5 April 2025).
- Abdelkader, K.; Gutiérrez, D.; Grimon, D.; Ruas-Madiedo, P.; Lood, C.; Lavigne, R.; Safaan, A.; Khairalla, A.S.; Gaber, Y.; Dishisha, T.; et al. Lysin LysMK34 of Acinetobacter baumannii Bacteriophage PMK34 Has a Turgor Pressure-Dependent Intrinsic Antibacterial Activity and Reverts Colistin Resistance. Appl. Environ. Microbiol. 2020, 86, e01311-20. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.-W.; Kim, Y.D.; Jang, J.; Kim, M.S.; Song, M.; Myung, H. Combination effect of engineered endolysin EC340 with antibiotics. Front. Microbiol. 2022, 13, 821936. [Google Scholar] [CrossRef]
- Abdelkader, K.; Gutiérrez, D.; Tamés-Caunedo, H.; Ruas-Madiedo, P.; Safaan, A.; Khairalla, A.S.; Gaber, Y.; Dishisha, T.; Briers, Y. Engineering a Lysin with Intrinsic Antibacterial Activity (LysMK34) by Cecropin A Fusion Enhances Its Antibacterial Properties against Acinetobacter baumannii. Appl. Environ. Microbiol. 2022, 88, e01515-21. [Google Scholar] [CrossRef]
- Gorodnichev, R.B.; Krivulia, A.O.; Kornienko, M.A.; Abdraimova, N.K.; Malakhova, M.V.; Zaychikova, M.V.; Bespiatykh, D.A.; Manuvera, V.A.; Shitikov, E.A. Phage-antibiotic combinations against Klebsiella pneumoniae: Impact of methodological approaches on effect evaluation. Front. Microbiol. 2025, 16, 1530819. [Google Scholar] [CrossRef] [PubMed]
- Kever, L.; Hardy, A.; Luthe, T.; Hünnefeld, M.; Gätgens, C.; Milke, L.; Wiechert, J.; Wittmann, J.; Moraru, C.; Marienhagen, J.; et al. Aminoglycoside antibiotics inhibit phage infection by blocking an early step of the infection cycle. mBio 2022, 13, e00783-22. [Google Scholar] [CrossRef]
- Kunz Coyne, A.J.; Eshaya, M.; Bleick, C.; Vader, S.; Biswas, B.; Wilson, M.; Deschenes, M.V.; Alexander, J.; Lehman, S.M.; Rybak, M.J. Exploring synergistic and antagonistic interactions in phage-antibiotic combinations against ESKAPE pathogens. Microbiol. Spectr. 2024, 12, e00427-24. [Google Scholar] [CrossRef] [PubMed]
- Zuo, P.; Yu, P.; Alvarez, P.J.J. Aminoglycosides antagonize bacteriophage proliferation, attenuating phage suppression of bacterial growth, biofilm formation, and antibiotic resistance. Appl. Environ. Microbiol. 2021, 87, e00468-21. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Phage-antibiotic combination treatments: Antagonistic impacts of antibiotics on the pharmacodynamics of phage therapy? Antibiotics 2019, 8, 182. [Google Scholar] [CrossRef]
- Dimitriu, T.; Kurilovich, E.; Łapińska, U.; Severinov, K.; Pagliara, S.; Szczelkun, M.D.; Westra, E.R. Bacteriostatic antibiotics promote CRISPR-Cas adaptive immunity by enabling increased spacer acquisition. Cell Host Microbe 2022, 30, 31–40.e5. [Google Scholar] [CrossRef]
- Pons, B.J.; Dimitriu, T.; Westra, E.R.; van Houte, S. Antibiotics that affect translation can antagonize phage infectivity by interfering with the deployment of counter-defenses. Proc. Natl. Acad. Sci. USA 2023, 120, e2216084120. [Google Scholar] [CrossRef]
- Kortright, K.E.; Doss-Gollin, S.; Chan, B.K.; Turner, P.E. Evolution of Bacterial Cross-Resistance to Lytic Phages and Albicidin Antibiotic. Front. Microbiol. 2021, 12, 658374. [Google Scholar] [CrossRef]
- Jiang, Z.; Wei, J.; Liang, Y.; Peng, N.; Li, Y. Aminoglycoside antibiotics inhibit mycobacteriophage infection. Antibiotics 2020, 9, 714. [Google Scholar] [CrossRef]
- Baird, J.P.; Bourguignon, G.J.; Sternglanz, R. Effect of nalidixic acid on the growth of deoxyribonucleic acid bacteriophages. J. Virol. 1972, 9, 17–21. [Google Scholar] [CrossRef]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef] [PubMed]
- Hosseinidoust, Z.; Tufenkji, N.; van de Ven, T.G.M. Formation of biofilms under phage predation: Considerations concerning a biofilm increase. Biofouling 2013, 29, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; González, S.; Campelo, A.B.; Martínez, B.; Rodríguez, A.; García, P. Low-level predation by lytic phage phiIPLA-RODI promotes biofilm formation and triggers the stringent response in Staphylococcus aureus. Sci. Rep. 2017, 7, 40965. [Google Scholar] [CrossRef]
- Akturk, E.; Melo, L.D.R.; Oliveira, H.; Crabbé, A.; Coenye, T.; Azeredo, J. Combining phages and antibiotic to enhance antibiofilm efficacy against an in vitro dual species wound biofilm. Biofilm 2023, 6, 100147. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Li, L.; Han, K.; Wang, L.; Cao, Y.; Zhou, Y.; Chen, H.; Wang, X. The antagonistic interactions between a polyvalent phage SaP7 and β-lactam antibiotics on combined therapies. Vet. Microbiol. 2022, 266, 109332. [Google Scholar] [CrossRef]
- Danis-Wlodarczyk, K.M.; Cai, A.; Chen, A.; Gittrich, M.R.; Sullivan, M.B.; Wozniak, D.J.; Abedon, S.T. Friends or Foes? Rapid Determination of Dissimilar Colistin and Ciprofloxacin Antagonism of Pseudomonas aeruginosa Phages. Pharmaceuticals 2021, 14, 1162. [Google Scholar] [CrossRef]
- Orozco-Ochoa, A.K.; González-Gómez, J.P.; Quiñones, B.; Castro-del Campo, N.; Valdez-Torres, J.B.; Chaidez-Quiroz, C. Bacteriophage Indie resensitizes multidrug-resistant Acinetobacter baumannii to antibiotics in vitro. Sci. Rep. 2025, 15, 11578. [Google Scholar] [CrossRef]
- Bhunchoth, A.; Blanc-Mathieu, R.; Mihara, T.; Nishimura, Y.; Askora, A.; Phironrit, N.; Leksomboon, C.; Chatchawankanphanich, O.; Kawasaki, T.; Nakano, M.; et al. Two asian jumbo phages, ϕRSL2 and ϕRSF1, infect Ralstonia solanacearum and show common features of ϕKZ-related phages. Virology 2016, 494, 56–66. [Google Scholar] [CrossRef]
- Gu Liu, C.; Green, S.I.; Min, L.; Clark, J.R.; Salazar, K.C.; Terwilliger, A.L.; Kaplan, H.B.; Trautner, B.W.; Ramig, R.F.; Maresso, A.W. Phage-antibiotic synergy Is driven by a unique combination of antibacterial mechanism of action and stoichiometry. mBio 2020, 11, 10–1128. [Google Scholar] [CrossRef]
- Torres-Barceló, C.; Gurney, J.; Gougat-Barberá, C.; Vasse, M.; Hochberg, M.E. Transient negative effects of antibiotics on phages do not jeopardise the advantages of combination therapies. FEMS Microbiol. Ecol. 2018, 94, fiy107. [Google Scholar] [CrossRef]
- Nicholls, P.; Clark, J.R.; Gu Liu, C.; Terwilliger, A.; Maresso, A.W. Class-driven synergy and antagonism between a Pseudomonas phage and antibiotics. Infect. Immun. 2023, 91, e00065-23. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, I.; Vukovic, D.; Gavric, D.; Cvetanovic, J.; Aleksic Sabo, V.; Gostimirovic, S.; Narancic, J.; Knezevic, P. An optimized checkerboard method for phage-antibiotic synergy detection. Viruses 2022, 14, 1542. [Google Scholar] [CrossRef] [PubMed]
- Khong, E.; Oh, J.J.; Jimenez, J.M.; Liu, R.; Dunham, S.; Monsibais, A.; Rhoads, A.; Ghatbale, P.; Garcia, A.; Cobián Güemes, A.G.; et al. A simple solid media assay for detection of synergy between bacteriophages and antibiotics. Microbiol. Spectr. 2024, 12, e03221-23. [Google Scholar] [CrossRef]
- Stachurska, X.; Roszak, M.; Jabłońska, J.; Mizielińska, M.; Nawrotek, P. Double-Layer Agar (DLA) Modifications for the First Step of the Phage-Antibiotic Synergy (PAS) Identification. Antibiotics 2021, 10, 1306. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, M. Jumbo Bacteriophages: An Overview. Front. Microbiol. 2017, 8, 403. [Google Scholar] [CrossRef]
- Loganathan, A.; Bozdogan, B.; Manohar, P.; Nachimuthu, R. Phage-antibiotic combinations in various treatment modalities to manage MRSA infections. Front. Pharmacol. 2024, 15, 1356179. [Google Scholar] [CrossRef]
- Mouton, J.W.; Vinks, A.A. Pharmacokinetic/Pharmacodynamic Modelling of Antibacterials In Vitro and In Vivo Using Bacterial Growth and Kill Kinetics. Clin. Pharmacokinet. 2005, 44, 201–210. [Google Scholar] [CrossRef]
- Brown, D.C.; Turner, R.J. Assessing microbial monitoring methods for challenging environmental strains and cultures. Microbiol. Res. 2022, 13, 235–257. [Google Scholar] [CrossRef]
- Grygorcewicz, B.; Roszak, M.; Rakoczy, R.; Augustyniak, A.; Konopacki, M.; Jabłońska, J.; Serwin, N.; Cecerska-Heryć, E.; Kordas, M.; Galant, K.; et al. PhageScore-based analysis of Acinetobacter baumannii infecting phages antibiotic interaction in liquid medium. Arch. Microbiol. 2022, 204, 421. [Google Scholar] [CrossRef]
- Chaudhry, W.N.; Concepción-Acevedo, J.; Park, T.; Andleeb, S.; Bull, J.J.; Levin, B.R. Synergy and order effects of antibiotics and phages in killing Pseudomonas aeruginosa biofilms. PLoS ONE 2017, 12, e0168615. [Google Scholar] [CrossRef]
- Kumaran, D.; Taha, M.; Yi, Q.; Ramirez-Arcos, S.; Diallo, J.-S.; Carli, A.; Abdelbary, H. Does Treatment Order Matter? Investigating the Ability of Bacteriophage to Augment Antibiotic Activity against Staphylococcus aureus Biofilms. Front. Microbiol. 2018, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Torres-Barceló, C.; Arias-Sánchez, F.I.; Vasse, M.; Ramsayer, J.; Kaltz, O.; Hochberg, M.E. A Window of Opportunity to Control the Bacterial Pathogen Pseudomonas aeruginosa Combining Antibiotics and Phages. PLoS ONE 2014, 9, e106628. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Zhang, P.; To, K.K.W.; Liu, Y.; Bai, C.; Leung, S.S.Y. Sequential treatment effects on phage–antibiotic synergistic application against multi-drug-resistant Acinetobacter baumannii. Int. J. Antimicrob. Agents 2023, 62, 106951. [Google Scholar] [CrossRef]
- Wright, R.C.T.; Friman, V.-P.; Smith, M.C.M.; Brockhurst, M.A. Resistance Evolution against Phage Combinations Depends on the Timing and Order of Exposure. mBio 2019, 10, e01652-19. [Google Scholar] [CrossRef]
- Sutcliffe, S.G.; Shamash, M.; Hynes, A.P.; Maurice, C.F. Common oral medications lead to prophage induction in bacterial isolates from the human gut. Viruses 2021, 13, 455. [Google Scholar] [CrossRef]
- Goerke, C.; Köller, J.; Wolz, C. Ciprofloxacin and trimethoprim cause phage induction and virulence modulation in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Rohmer, C.; Wolz, C. The Role of hlb-converting bacteriophages in Staphylococcus aureus host adaption. Microb. Physiol. 2021, 31, 109–122. [Google Scholar] [CrossRef]
- Tran, P.M.; Feiss, M.; Kinney, K.J.; Salgado-Pabón, W. ϕSa3mw prophage as a molecular regulatory switch of Staphylococcus aureus β-toxin production. J. Bacteriol. 2019, 201, e00766-18. [Google Scholar] [CrossRef]
- Rabinovich, L.; Sigal, N.; Borovok, I.; Nir-Paz, R.; Herskovits, A.A. Prophage Excision Activates Listeria Competence Genes that Promote Phagosomal Escape and Virulence. Cell 2012, 150, 792–802. [Google Scholar] [CrossRef]
- Champagne-Jorgensen, K.; Luong, T.; Darby, T.; Roach, D.R. Immunogenicity of bacteriophages. Trends Microbiol. 2023, 31, 1058–1071. [Google Scholar] [CrossRef]
- Berkson, J.D.; Wate, C.E.; Allen, G.B.; Schubert, A.M.; Dunbar, K.E.; Coryell, M.P.; Sava, R.L.; Gao, Y.; Hastie, J.L.; Smith, E.M.; et al. Phage-specific immunity impairs efficacy of bacteriophage targeting Vancomycin Resistant Enterococcus in a murine model. Nat. Commun. 2024, 15, 2993. [Google Scholar] [CrossRef] [PubMed]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363, eaat9691. [Google Scholar] [CrossRef]
- Górski, A.; Międzybrodzki, R.; Jończyk-Matysiak, E.; Kniotek, M.; Letkiewicz, S. Therapeutic Phages as Modulators of the Immune Response: Practical Implications. Clin. Infect. Dis. 2023, 77, S433–S439. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the Host Immune System and Bacteriophage Is Essential for Successful Phage Therapy against an Acute Respiratory Pathogen. Cell Host Microbe 2017, 22, 38–47.e4. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gonzalez, R.A.; Leung, C.Y.; Chan, B.K.; Turner, P.E.; Weitz, J.S. Quantitative models of phage-antibiotic combination therapy. mSystems 2020, 5, 10–1128. [Google Scholar] [CrossRef]
- Indiani, C.; Sauve, K.; Raz, A.; Abdelhady, W.; Xiong, Y.Q.; Cassino, C.; Bayer, A.S.; Schuch, R. The antistaphylococcal lysin, CF-301, activates key host factors in human blood to potentiate methicillin-resistant Staphylococcus aureus bacteriolysis. Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef]
- Black, C.; Al Mahmud, H.; Howle, V.; Wilson, S.; Smith, A.C.; Wakeman, C.A. Development of a polymicrobial checkerboard assay as a tool for determining combinatorial antibiotic effectiveness in polymicrobial communities. Antibiotics 2023, 12, 1207. [Google Scholar] [CrossRef]
- Van Nieuwenhuyse, B.; Galant, C.; Brichard, B.; Docquier, P.-L.; Djebara, S.; Pirnay, J.-P.; Van der Linden, D.; Merabishvili, M.; Chatzis, O. A case of In situ phage therapy against Staphylococcus aureus in a bone allograft polymicrobial biofilm infection: Outcomes and phage-antibiotic interactions. Viruses 2021, 13, 1898. [Google Scholar] [CrossRef]
- Akturk, E.; Oliveira, H.; Santos, S.B.; Costa, S.; Kuyumcu, S.; Melo, L.D.R.; Azeredo, J. Synergistic Action of Phage and Antibiotics: Parameters to Enhance the Killing Efficacy Against Mono and Dual-Species Biofilms. Antibiotics 2019, 8, 103. [Google Scholar] [CrossRef]
- Alseth, E.O.; Pursey, E.; Luján, A.M.; McLeod, I.; Rollie, C.; Westra, E.R. Bacterial biodiversity drives the evolution of CRISPR-based phage resistance. Nature 2019, 574, 549–552. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Supina, B.S.I.; Dennis, J.J. The Current Landscape of Phage–Antibiotic Synergistic (PAS) Interactions. Antibiotics 2025, 14, 545. https://doi.org/10.3390/antibiotics14060545
Supina BSI, Dennis JJ. The Current Landscape of Phage–Antibiotic Synergistic (PAS) Interactions. Antibiotics. 2025; 14(6):545. https://doi.org/10.3390/antibiotics14060545
Chicago/Turabian StyleSupina, Brittany S. I., and Jonathan J. Dennis. 2025. "The Current Landscape of Phage–Antibiotic Synergistic (PAS) Interactions" Antibiotics 14, no. 6: 545. https://doi.org/10.3390/antibiotics14060545
APA StyleSupina, B. S. I., & Dennis, J. J. (2025). The Current Landscape of Phage–Antibiotic Synergistic (PAS) Interactions. Antibiotics, 14(6), 545. https://doi.org/10.3390/antibiotics14060545