
Metagenomics as a Transformative Tool for Antibiotic Resistance Surveillance: Highlighting the Impact of Mobile Genetic Elements with a Focus on the Complex Role of Phages


Abstract
1. Introduction
2. Traditional Tools Used for AMR Surveillance
3. Metagenomics as a Transformative Tool for AMR Surveillance
Addressing Limitations and Challenges in Applying Metagenomics in AMR Surveillance
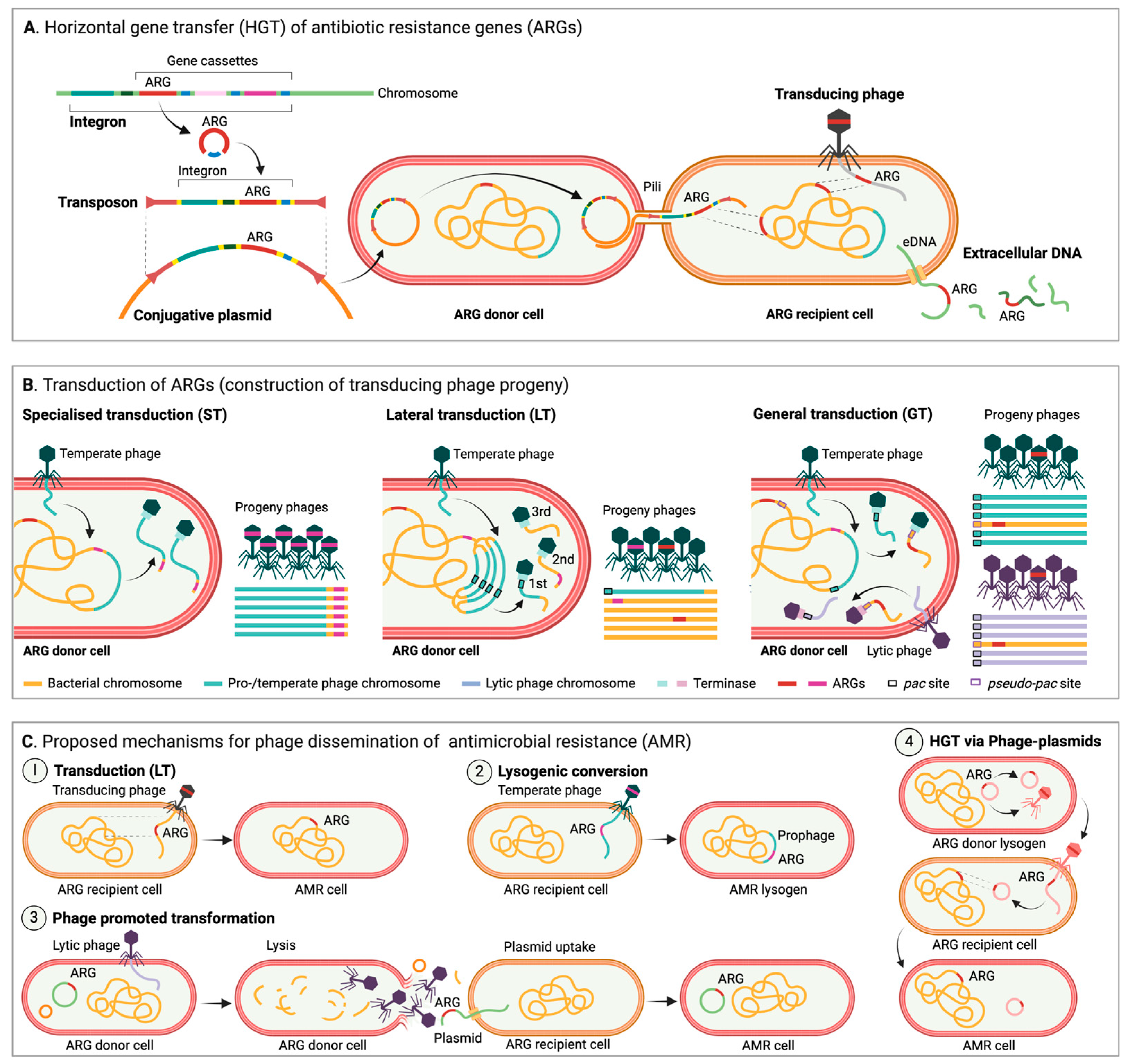
4. Overview of MGEs and Their Impact on Occurrence and Spread of AMR
The Role of Phages, an Underestimated Driver of AMR Transmission?
5. Integration of MGE-Based Metagenomic Data for Improved AMR Surveillance
6. Phage Therapy, a Renewed Approach to Treat (AMR) Bacterial Infections?
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Emran, T.B.; Dhama, K.; Ripon, M.K.H.; Gajdacs, M.; Sahibzada, M.U.K.; et al. Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. J. Infect. Public Health 2021, 14, 1750–1766. [Google Scholar] [CrossRef] [PubMed]
- MacLean, R.C.; San Millan, A. The evolution of antibiotic resistance. Science 2019, 365, 1082–1083. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Nathan, C.; Cars, O. Antibiotic resistance--problems, progress, and prospects. N. Engl. J. Med. 2014, 371, 1761–1763. [Google Scholar] [CrossRef]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef]
- Muteeb, G.; Rehman, M.T.; Shahwan, M.; Aatif, M. Origin of Antibiotics and Antibiotic Resistance, and Their Impacts on Drug Development: A Narrative Review. Pharmaceuticals 2023, 16, 1615. [Google Scholar] [CrossRef]
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- O’Neill, J. Review on Antimicrobial Resistance: Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Wellcome Trust: London, UK, 2016. [Google Scholar]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef]
- Witte, W.; Klare, I.; Werner, G. Selective pressure by antibiotics as feed additives. Infection 1999, 27 (Suppl. S2), S35–S38. [Google Scholar] [CrossRef]
- Frost, L.S.; Leplae, R.; Summers, A.O.; Toussaint, A. Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 2005, 3, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Haudiquet, M.; de Sousa, J.M.; Touchon, M.; Rocha, E.P.C. Selfish, promiscuous and sometimes useful: How mobile genetic elements drive horizontal gene transfer in microbial populations. Philos. Trans. R. Soc. B Biol. Sci. 2022, 377, 20210234. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, M.; Shintani, M. Microbial evolution through horizontal gene transfer by mobile genetic elements. Microb. Biotechnol. 2024, 17, e14408. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Spellberg, B.; Gilbert, D.N. The future of antibiotics and resistance: A tribute to a career of leadership by John Bartlett. Clin. Infect. Dis. 2014, 59 (Suppl. S2), S71–S75. [Google Scholar] [CrossRef]
- Walsh, T.R.; Gales, A.C.; Laxminarayan, R.; Dodd, P.C. Antimicrobial Resistance: Addressing a Global Threat to Humanity. PLoS Med. 2023, 20, e1004264. [Google Scholar] [CrossRef]
- Fuhrmeister, A.S.; Jones, R.N. The Importance of Antimicrobial Resistance Monitoring Worldwide and the Origins of SENTRY Antimicrobial Surveillance Program. Open Forum Infect. Dis. 2019, 6, S1–S4. [Google Scholar] [CrossRef]
- WHO. Global Antimicrobial Resistance Surveillance System. Manual for Early Implementation. Available online: https://iris.who.int/bitstream/handle/10665/188783/9789241549400_eng.pdf (accessed on 22 December 2024).
- Lappan, R.; Chown, S.L.; French, M.; Perlaza-Jimenez, L.; Macesic, N.; Davis, M.; Brown, R.; Cheng, A.; Clasen, T.; Conlan, L.; et al. Towards integrated cross-sectoral surveillance of pathogens and antimicrobial resistance: Needs, approaches, and considerations for linking surveillance to action. Environ. Int. 2024, 192, 109046. [Google Scholar] [CrossRef]
- Miteu, G.D.; Achinebiri, P.; Raghunathan, N.; Sankaran, S. Closing potential drivers of antimicrobial resistance: Last-resort antimicrobials with the potential of being misused, the way forward—A short communication. Ann. Med. Surg. 2023, 85, 3226–3231. [Google Scholar] [CrossRef]
- Zanichelli, V.; Sharland, M.; Cappello, B.; Moja, L.; Getahun, H.; Pessoa-Silva, C.; Sati, H.; van Weezenbeek, C.; Balkhy, H.; Simão, M.; et al. The WHO AWaRe (Access, Watch, Reserve) antibiotic book and prevention of antimicrobial resistance. Bull. World Health Organ. 2023, 101, 290–296. [Google Scholar] [CrossRef]
- Stroud, C.; Kaplan, B.; Logan, J.E.; Gray, G.C. One Health training, research, and outreach in North America. Infect. Ecol. Epidemiol. 2016, 6, 33680. [Google Scholar] [CrossRef] [PubMed]
- Sagar, P.; Aseem, A.; Banjara, S.K.; Veleri, S. The role of food chain in antimicrobial resistance spread and One Health approach to reduce risks. Int. J. Food Microbiol. 2023, 391–393, 110148. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Antimicrobial Resistance Surveillance System (GLASS) Report Early Implementation 2017–2018. Available online: https://www.who.int/publications/i/item/9789241515061 (accessed on 22 December 2024).
- WHO. Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report 2022. Available online: https://www.who.int/publications/i/item/9789240062702 (accessed on 22 December 2024).
- Franco-Duarte, R.; Cernakova, L.; Kadam, S.; Kaushik, K.S.; Salehi, B.; Bevilacqua, A.; Corbo, M.R.; Antolak, H.; Dybka-Stepien, K.; Leszczewicz, M.; et al. Advances in Chemical and Biological Methods to Identify Microorganisms-From Past to Present. Microorganisms 2019, 7, 130. [Google Scholar] [CrossRef]
- Gajic, I.; Kabic, J.; Kekic, D.; Jovicevic, M.; Milenkovic, M.; Mitic Culafic, D.; Trudic, A.; Ranin, L.; Opavski, N. Antimicrobial Susceptibility Testing: A Comprehensive Review of Currently Used Methods. Antibiotics 2022, 11, 427. [Google Scholar] [CrossRef]
- Biemer, J.J. Antimicrobial susceptibility testing by the Kirby-Bauer disc diffusion method. Ann. Clin. Lab. Sci. 1973, 3, 135–140. [Google Scholar]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Idelevich, E.A.; Becker, K. How to accelerate antimicrobial susceptibility testing. Clin. Microbiol. Infect. 2019, 25, 1347–1355. [Google Scholar] [CrossRef]
- Khan, Z.A.; Siddiqui, M.F.; Park, S. Current and Emerging Methods of Antibiotic Susceptibility Testing. Diagnostics 2019, 9, 49. [Google Scholar] [CrossRef]
- Zagajewski, A.; Turner, P.; Feehily, C.; El Sayyed, H.; Andersson, M.; Barrett, L.; Oakley, S.; Stracy, M.; Crook, D.; Nellaker, C.; et al. Deep learning and single-cell phenotyping for rapid antimicrobial susceptibility detection in Escherichia coli. Commun. Biol. 2023, 6, 1164. [Google Scholar] [CrossRef]
- Doern, G.V.; Brecher, S.M. The Clinical Predictive Value (or Lack Thereof) of the Results of In Vitro Antimicrobial Susceptibility Tests. J. Clin. Microbiol. 2011, 49, S11–S14. [Google Scholar] [CrossRef]
- Karp, B.E.; Tate, H.; Plumblee, J.R.; Dessai, U.; Whichard, J.M.; Thacker, E.L.; Hale, K.R.; Wilson, W.; Friedman, C.R.; Griffin, P.M.; et al. National Antimicrobial Resistance Monitoring System: Two Decades of Advancing Public Health Through Integrated Surveillance of Antimicrobial Resistance. Foodborne Pathog. Dis. 2017, 14, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Gagliotti, C.; Balode, A.; Baquero, F.; Degener, J.; Grundmann, H.; Gur, D.; Jarlier, V.; Kahlmeter, G.; Monen, J.; Monnet, D.L.; et al. Escherichia coli and Staphylococcus aureus: Bad news and good news from the European Antimicrobial Resistance Surveillance Network (EARS-Net, formerly EARSS), 2002 to 2009. Eurosurveillance 2011, 16, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Ataker, F.; Hedin, G.; Dornbusch, K. Molecular epidemiology of extended-spectrum beta-lactamases among Escherichia coli isolates collected in a Swedish hospital and its associated health care facilities from 2001 to 2006. J. Clin. Microbiol. 2008, 46, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Van der Zee, A.; Roorda, L.; Bosman, G.; Fluit, A.C.; Hermans, M.; Smits, P.H.; van der Zanden, A.G.; Te Witt, R.; Bruijnesteijn van Coppenraet, L.E.; Cohen Stuart, J.; et al. Multi-centre evaluation of real-time multiplex PCR for detection of carbapenemase genes OXA-48, VIM, IMP, NDM and KPC. BMC Infect. Dis. 2014, 14, 27. [Google Scholar] [CrossRef]
- Strommenger, B.; Kettlitz, C.; Werner, G.; Witte, W. Multiplex PCR assay for simultaneous detection of nine clinically relevant antibiotic resistance genes in Staphylococcus aureus. J. Clin. Microbiol. 2003, 41, 4089–4094. [Google Scholar] [CrossRef]
- Nair, D.; Shashindran, N.; Kumar, A.; Vinodh, V.; Biswas, L.; Biswas, R. Comparison of Phenotypic MRSA Detection Methods with PCR for mecA Gene in the Background of Emergence of Oxacillin-Susceptible MRSA. Microb. Drug Resist. 2021, 27, 1190–1194. [Google Scholar] [CrossRef]
- Yamin, D.; Uskokovic, V.; Wakil, A.M.; Goni, M.D.; Shamsuddin, S.H.; Mustafa, F.H.; Alfouzan, W.A.; Alissa, M.; Alshengeti, A.; Almaghrabi, R.H.; et al. Current and Future Technologies for the Detection of Antibiotic-Resistant Bacteria. Diagnostics 2023, 13, 3246. [Google Scholar] [CrossRef]
- Call, D.R.; Bakko, M.K.; Krug, M.J.; Roberts, M.C. Identifying antimicrobial resistance genes with DNA microarrays. Antimicrob. Agents Chemother. 2003, 47, 3290–3295. [Google Scholar] [CrossRef]
- Jaksik, R.; Iwanaszko, M.; Rzeszowska-Wolny, J.; Kimmel, M. Microarray experiments and factors which affect their reliability. Biol. Direct 2015, 10, 46. [Google Scholar] [CrossRef]
- Saleem, Z.; Hassali, M.A.; Godman, B.; Versporten, A.; Hashmi, F.K.; Saeed, H.; Saleem, F.; Salman, M.; Rehman, I.U.; Khan, T.M. Point prevalence surveys of antimicrobial use: A systematic review and the implications. Expert Rev. Anti-Infect. Ther. 2020, 18, 897–910. [Google Scholar] [CrossRef]
- Pauwels, I.; Versporten, A.; Vermeulen, H.; Vlieghe, E.; Goossens, H. Assessing the impact of the Global Point Prevalence Survey of Antimicrobial Consumption and Resistance (Global-PPS) on hospital antimicrobial stewardship programmes: Results of a worldwide survey. Antimicrob. Resist. Infect. Control 2021, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Iskandar, K.; Molinier, L.; Hallit, S.; Sartelli, M.; Hardcastle, T.C.; Haque, M.; Lugova, H.; Dhingra, S.; Sharma, P.; Islam, S.; et al. Surveillance of antimicrobial resistance in low- and middle-income countries: A scattered picture. Antimicrob. Resist. Infect. Control 2021, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, H. Targeted metagenomics: A high-resolution metagenomics approach for specific gene clusters in complex microbial communities. Environ. Microbiol. 2012, 14, 13–22. [Google Scholar] [CrossRef]
- Cowan, D.A.; Ramond, J.B.; Makhalanyane, T.P.; De Maayer, P. Metagenomics of extreme environments. Curr. Opin. Microbiol. 2015, 25, 97–102. [Google Scholar] [CrossRef]
- Munk, P.; Andersen, V.D.; de Knegt, L.; Jensen, M.S.; Knudsen, B.E.; Lukjancenko, O.; Mordhorst, H.; Clasen, J.; Agerso, Y.; Folkesson, A.; et al. A sampling and metagenomic sequencing-based methodology for monitoring antimicrobial resistance in swine herds. J. Antimicrob. Chemother. 2017, 72, 385–392. [Google Scholar] [CrossRef]
- Morgan, X.C.; Huttenhower, C. Chapter 12: Human microbiome analysis. PLoS Comput. Biol. 2012, 8, e1002808. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef]
- Manoharan, R.K.; Srinivasan, S.; Shanmugam, G.; Ahn, Y.H. Shotgun metagenomic analysis reveals the prevalence of antibiotic resistance genes and mobile genetic elements in full scale hospital wastewater treatment plants. J. Environ. Manag. 2021, 296, 113270. [Google Scholar] [CrossRef]
- Purushothaman, S.; Meola, M.; Roloff, T.; Rooney, A.M.; Egli, A. Evaluation of DNA extraction kits for long-read shotgun metagenomics using Oxford Nanopore sequencing for rapid taxonomic and antimicrobial resistance detection. Sci. Rep. 2024, 14, 29531. [Google Scholar] [CrossRef] [PubMed]
- Olsen, G.J.; Lane, D.J.; Giovannoni, S.J.; Pace, N.R.; Stahl, D.A. Microbial ecology and evolution: A ribosomal RNA approach. Annu. Rev. Microbiol. 1986, 40, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Hamady, M.; Knight, R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res. 2009, 19, 1141–1152. [Google Scholar] [CrossRef]
- Matchado, M.S.; Ruhlemann, M.; Reitmeier, S.; Kacprowski, T.; Frost, F.; Haller, D.; Baumbach, J.; List, M. On the limits of 16S rRNA gene-based metagenome prediction and functional profiling. Microb. Genom. 2024, 10, 1203. [Google Scholar] [CrossRef]
- Bars-Cortina, D.; Ramon, E.; Rius-Sansalvador, B.; Guino, E.; Garcia-Serrano, A.; Mach, N.; Khannous-Lleiffe, O.; Saus, E.; Gabaldon, T.; Ibanez-Sanz, G.; et al. Comparison between 16S rRNA and shotgun sequencing in colorectal cancer, advanced colorectal lesions, and healthy human gut microbiota. BMC Genom. 2024, 25, 730. [Google Scholar] [CrossRef]
- Yee, R.; Simner, P.J. Next-Generation Sequencing Approaches to Predicting Antimicrobial Susceptibility Testing Results. Clin. Lab. Med. 2022, 42, 557–572. [Google Scholar] [CrossRef]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; AbuOun, M.; Stubberfield, E.; Hoosdally, S.J.; et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb. Genom. 2019, 5, e000294. [Google Scholar] [CrossRef]
- Boolchandani, M.; D’Souza, A.W.; Dantas, G. Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 2019, 20, 356–370. [Google Scholar] [CrossRef]
- Peona, V.; Blom, M.P.K.; Xu, L.; Burri, R.; Sullivan, S.; Bunikis, I.; Liachko, I.; Haryoko, T.; Jonsson, K.A.; Zhou, Q.; et al. Identifying the causes and consequences of assembly gaps using a multiplatform genome assembly of a bird-of-paradise. Mol. Ecol. Resour. 2021, 21, 263–286. [Google Scholar] [CrossRef]
- Maguire, F.; Jia, B.; Gray, K.L.; Lau, W.Y.V.; Beiko, R.G.; Brinkman, F.S.L. Metagenome-assembled genome binning methods with short reads disproportionately fail for plasmids and genomic Islands. Microb. Genom. 2020, 6, mgen000436. [Google Scholar] [CrossRef]
- Espinosa, E.; Bautista, R.; Larrosa, R.; Plata, O. Advancements in long-read genome sequencing technologies and algorithms. Genomics 2024, 116, 110842. [Google Scholar] [CrossRef] [PubMed]
- Shafin, K.; Pesout, T.; Lorig-Roach, R.; Haukness, M.; Olsen, H.E.; Bosworth, C.; Armstrong, J.; Tigyi, K.; Maurer, N.; Koren, S.; et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 2020, 38, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Sauerborn, E.; Corredor, N.C.; Reska, T.; Perlas, A.; Vargas da Fonseca Atum, S.; Goldman, N.; Wantia, N.; Prazeres da Costa, C.; Foster-Nyarko, E.; Urban, L. Detection of hidden antibiotic resistance through real-time genomics. Nat. Commun. 2024, 15, 5494. [Google Scholar] [CrossRef]
- Taxt, A.M.; Avershina, E.; Frye, S.A.; Naseer, U.; Ahmad, R. Rapid identification of pathogens, antibiotic resistance genes and plasmids in blood cultures by nanopore sequencing. Sci. Rep. 2020, 10, 7622. [Google Scholar] [CrossRef]
- Leggett, R.M.; Alcon-Giner, C.; Heavens, D.; Caim, S.; Brook, T.C.; Kujawska, M.; Martin, S.; Peel, N.; Acford-Palmer, H.; Hoyles, L.; et al. Rapid MinION profiling of preterm microbiota and antimicrobial-resistant pathogens. Nat. Microbiol. 2020, 5, 430–442. [Google Scholar] [CrossRef]
- Athanasopoulou, K.; Boti, M.A.; Adamopoulos, P.G.; Skourou, P.C.; Scorilas, A. Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics. Life 2021, 12, 30. [Google Scholar] [CrossRef]
- Berbers, B.; Vanneste, K.; Roosens, N.; Marchal, K.; Ceyssens, P.J.; De Keersmaecker, S.C.J. Using a combination of short- and long-read sequencing to investigate the diversity in plasmid- and chromosomally encoded extended-spectrum beta-lactamases (ESBLs) in clinical Shigella and Salmonella isolates in Belgium. Microb. Genom. 2023, 9, 925. [Google Scholar] [CrossRef]
- Chen, Y.H.; Chiang, P.W.; Rogozin, D.Y.; Degermendzhy, A.G.; Chiu, H.H.; Tang, S.L. Salvaging high-quality genomes of microbial species from a meromictic lake using a hybrid sequencing approach. Commun. Biol. 2021, 4, 996. [Google Scholar] [CrossRef]
- Steen, A.D.; Crits-Christoph, A.; Carini, P.; DeAngelis, K.M.; Fierer, N.; Lloyd, K.G.; Cameron Thrash, J. High proportions of bacteria and archaea across most biomes remain uncultured. ISME J. 2019, 13, 3126–3130. [Google Scholar] [CrossRef]
- Garza, D.R.; Dutilh, B.E. From cultured to uncultured genome sequences: Metagenomics and modeling microbial ecosystems. Cell. Mol. Life Sci. 2015, 72, 4287–4308. [Google Scholar] [CrossRef]
- Pillay, S.; Calderon-Franco, D.; Urhan, A.; Abeel, T. Metagenomic-based surveillance systems for antibiotic resistance in non-clinical settings. Front. Microbiol. 2022, 13, 1066995. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yin, Y. Critical assessment of pan-genomic analysis of metagenome-assembled genomes. Brief. Bioinform. 2022, 23, bbac413. [Google Scholar] [CrossRef]
- NARMS. Research to Advance Antimicrobial Resistance Monitoring. Available online: https://www.fda.gov/animal-veterinary/national-antimicrobial-resistance-monitoring-system/research-advance-antimicrobial-resistance-monitoring (accessed on 16 February 2025).
- GLASS. Whole-Genome Sequencing for Surveillance of Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2020; Available online: https://www.who.int/docs/default-source/antimicrobial-resistance/glass_wgs_report_v8_web.pdf?sfvrsn=9ef1b4a5_1 (accessed on 16 February 2025).
- Hendriksen, R.S.; Bortolaia, V.; Tate, H.; Tyson, G.H.; Aarestrup, F.M.; McDermott, P.F. Using Genomics to Track Global Antimicrobial Resistance. Front. Public Health 2019, 7, 242. [Google Scholar] [CrossRef]
- Martinez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2015, 13, 116–123. [Google Scholar] [CrossRef]
- Liao, H.; Liu, C.; Zhou, S.; Liu, C.; Eldridge, D.J.; Ai, C.; Wilhelm, S.W.; Singh, B.K.; Liang, X.; Radosevich, M.; et al. Prophage-encoded antibiotic resistance genes are enriched in human-impacted environments. Nat. Commun. 2024, 15, 8315. [Google Scholar] [CrossRef]
- Mahfouz, N.; Ferreira, I.; Beisken, S.; von Haeseler, A.; Posch, A.E. Large-scale assessment of antimicrobial resistance marker databases for genetic phenotype prediction: A systematic review. J. Antimicrob. Chemother. 2020, 75, 3099–3108. [Google Scholar] [CrossRef]
- Ellington, M.J.; Ekelund, O.; Aarestrup, F.M.; Canton, R.; Doumith, M.; Giske, C.; Grundman, H.; Hasman, H.; Holden, M.T.G.; Hopkins, K.L.; et al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: Report from the EUCAST Subcommittee. Clin. Microbiol. Infect. 2017, 23, 2–22. [Google Scholar] [CrossRef]
- Nielsen, T.K.; Browne, P.D.; Hansen, L.H. Antibiotic resistance genes are differentially mobilized according to resistance mechanism. Gigascience 2022, 11, giac072. [Google Scholar] [CrossRef]
- Castanheira, M.; Simner, P.J.; Bradford, P.A. Extended-spectrum beta-lactamases: An update on their characteristics, epidemiology and detection. JAC Antimicrob. Resist. 2021, 3, dlab092. [Google Scholar] [CrossRef]
- Zhang, A.N.; Gaston, J.M.; Dai, C.L.; Zhao, S.; Poyet, M.; Groussin, M.; Yin, X.; Li, L.G.; van Loosdrecht, M.C.M.; Topp, E.; et al. An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commun. 2021, 12, 4765. [Google Scholar] [CrossRef] [PubMed]
- Yagimoto, K.; Hosoda, S.; Sato, M.; Hamada, M. Prediction of antibiotic resistance mechanisms using a protein language model. Bioinformatics 2024, 40, btae550. [Google Scholar] [CrossRef] [PubMed]
- Harandi, H.; Shafaati, M.; Salehi, M.; Roozbahani, M.M.; Mohammadi, K.; Akbarpour, S.; Rahimnia, R.; Hassanpour, G.; Rahmani, Y.; Seifi, A. Artificial intelligence-driven approaches in antibiotic stewardship programs and optimizing prescription practices: A systematic review. Artif. Intell. Med. 2025, 162, 103089. [Google Scholar] [CrossRef]
- Bilal, H.; Khan, M.N.; Khan, S.; Shafiq, M.; Fang, W.; Khan, R.U.; Rahman, M.U.; Li, X.; Lv, Q.L.; Xu, B. The role of artificial intelligence and machine learning in predicting and combating antimicrobial resistance. Comput. Struct. Biotechnol. J. 2025, 27, 423–439. [Google Scholar] [CrossRef]
- Maeda, T.; Furusawa, C. Laboratory Evolution of Antimicrobial Resistance in Bacteria to Develop Rational Treatment Strategies. Antibiotics 2024, 13, 94. [Google Scholar] [CrossRef]
- Van Dijk, B.; Buffard, P.; Farr, A.D.; Giersdorf, F.; Meijer, J.; Dutilh, B.E.; Rainey, P.B. Identifying and tracking mobile elements in evolving compost communities yields insights into the nanobiome. ISME Commun. 2023, 3, 90. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef]
- Hall, R.M.; Collis, C.M. Mobile gene cassettes and integrons: Capture and spread of genes by site-specific recombination. Mol. Microbiol. 1995, 15, 593–600. [Google Scholar] [CrossRef]
- Mazel, D. Integrons: Agents of bacterial evolution. Nat. Rev. Microbiol. 2006, 4, 608–620. [Google Scholar] [CrossRef]
- Schmitz, M.; Querques, I. DNA on the move: Mechanisms, functions and applications of transposable elements. FEBS Open Bio 2024, 14, 13–22. [Google Scholar] [CrossRef]
- Munoz-Lopez, M.; Garcia-Perez, J.L. DNA transposons: Nature and applications in genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- del Solar, G.; Giraldo, R.; Ruiz-Echevarria, M.J.; Espinosa, M.; Diaz-Orejas, R. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 1998, 62, 434–464. [Google Scholar] [CrossRef]
- Harrison, E.; Brockhurst, M.A. Plasmid-mediated horizontal gene transfer is a coevolutionary process. Trends Microbiol. 2012, 20, 262–267. [Google Scholar] [CrossRef]
- Halary, S.; Leigh, J.W.; Cheaib, B.; Lopez, P.; Bapteste, E. Network analyses structure genetic diversity in independent genetic worlds. Proc. Natl. Acad. Sci. USA 2010, 107, 127–132. [Google Scholar] [CrossRef]
- Sprague, G.F., Jr. Genetic exchange between kingdoms. Curr. Opin. Genet. Dev. 1991, 1, 530–533. [Google Scholar] [CrossRef]
- Rozwandowicz, M.; Brouwer, M.S.M.; Fischer, J.; Wagenaar, J.A.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018, 73, 1121–1137. [Google Scholar] [CrossRef]
- Young, R. Bacteriophage lysis: Mechanism and regulation. Microbiol. Rev. 1992, 56, 430–481. [Google Scholar] [CrossRef]
- Feiner, R.; Argov, T.; Rabinovich, L.; Sigal, N.; Borovok, I.; Herskovits, A.A. A new perspective on lysogeny: Prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 2015, 13, 641–650. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, T.; Yu, M.; Chen, Y.L.; Jin, M. The Life Cycle Transitions of Temperate Phages: Regulating Factors and Potential Ecological Implications. Viruses 2022, 14, 1904. [Google Scholar] [CrossRef]
- Liu, F.; Luo, Y.; Xu, T.; Lin, H.; Qiu, Y.; Li, B. Current examining methods and mathematical models of horizontal transfer of antibiotic resistance genes in the environment. Front. Microbiol. 2024, 15, 1371388. [Google Scholar] [CrossRef]
- Wachino, J.I.; Jin, W.; Kimura, K.; Arakawa, Y. Intercellular Transfer of Chromosomal Antimicrobial Resistance Genes Between Acinetobacter baumannii Strains Mediated by Prophages. Antimicrob. Agents Chemother. 2019, 63, e00334-19. [Google Scholar] [CrossRef] [PubMed]
- Stanczak-Mrozek, K.I.; Laing, K.G.; Lindsay, J.A. Resistance gene transfer: Induction of transducing phage by sub-inhibitory concentrations of antimicrobials is not correlated to induction of lytic phage. J. Antimicrob. Chemother. 2017, 72, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Bearson, B.L.; Allen, H.K.; Brunelle, B.W.; Lee, I.S.; Casjens, S.R.; Stanton, T.B. The agricultural antibiotic carbadox induces phage-mediated gene transfer in Salmonella. Front. Microbiol. 2014, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Bearson, B.L.; Brunelle, B.W. Fluoroquinolone induction of phage-mediated gene transfer in multidrug-resistant Salmonella. Int. J. Antimicrob. Agents 2015, 46, 201–204. [Google Scholar] [CrossRef]
- Fillol-Salom, A.; Alsaadi, A.; Sousa, J.A.M.; Zhong, L.; Foster, K.R.; Rocha, E.P.C.; Penades, J.R.; Ingmer, H.; Haaber, J. Bacteriophages benefit from generalized transduction. PLoS Pathog. 2019, 15, e1007888. [Google Scholar] [CrossRef]
- Leclerc, Q.J.; Wildfire, J.; Gupta, A.; Lindsay, J.A.; Knight, G.M. Growth-Dependent Predation and Generalized Transduction of Antimicrobial Resistance by Bacteriophage. mSystems 2022, 7, e00135-22. [Google Scholar] [CrossRef]
- Maslanova, I.; Doskar, J.; Varga, M.; Kuntova, L.; Muzik, J.; Maluskova, D.; Ruzickova, V.; Pantucek, R. Bacteriophages of Staphylococcus aureus efficiently package various bacterial genes and mobile genetic elements including SCCmec with different frequencies. Environ. Microbiol. Rep. 2013, 5, 66–73. [Google Scholar] [CrossRef]
- Wolput, S.; Lood, C.; Fillol-Salom, A.; Casters, Y.; Albasiony, A.; Cenens, W.; Vanoirbeek, K.; Kerremans, A.; Lavigne, R.; Penades, J.R.; et al. Phage-host co-evolution has led to distinct generalized transduction strategies. Nucleic Acids Res. 2024, 52, 7780–7791. [Google Scholar] [CrossRef]
- Liu, J.; Liu, P.; Feng, F.; Zhang, J.; Li, F.; Wang, M.; Sun, Y. Evaluation of Potential ARG Packaging by Two Environmental T7-Like Phage during Phage-Host Interaction. Viruses 2020, 12, 1060. [Google Scholar] [CrossRef]
- Gottesman, M.E.; Weisberg, R.A. Little lambda, who made thee? Microbiol. Mol. Biol. Rev. 2004, 68, 796–813. [Google Scholar] [CrossRef]
- Kant, P.; Petersen, B.; Sicheritz-Pontén, T.; Kondabagil, K. The low abundance of antimicrobial resistance genes (ARGs) in bacteriophages and their transfer bottlenecks limit the ability of phages to contribute to the spread of ARGs. bioRxiv 2024. [Google Scholar] [CrossRef]
- Bowring, J.Z.; Su, Y.; Alsaadi, A.; Svenningsen, S.L.; Parkhill, J.; Ingmer, H. Screening for Highly Transduced Genes in Staphylococcus aureus Revealed Both Lateral and Specialized Transduction. Microbiol. Spectr. 2022, 10, e02423-21. [Google Scholar] [CrossRef]
- Michaelis, C.; Grohmann, E. Horizontal Gene Transfer of Antibiotic Resistance Genes in Biofilms. Antibiotics 2023, 12, 328. [Google Scholar] [CrossRef]
- Humphrey, S.; Fillol-Salom, A.; Quiles-Puchalt, N.; Ibarra-Chavez, R.; Haag, A.F.; Chen, J.; Penades, J.R. Bacterial chromosomal mobility via lateral transduction exceeds that of classical mobile genetic elements. Nat. Commun. 2021, 12, 6509. [Google Scholar] [CrossRef]
- Chen, J.; Quiles-Puchalt, N.; Chiang, Y.N.; Bacigalupe, R.; Fillol-Salom, A.; Chee, M.S.J.; Fitzgerald, J.R.; Penades, J.R. Genome hypermobility by lateral transduction. Science 2018, 362, 207–212. [Google Scholar] [CrossRef]
- Fisarova, L.; Botka, T.; Du, X.; Maslanova, I.; Bardy, P.; Pantucek, R.; Benesik, M.; Roudnicky, P.; Winstel, V.; Larsen, J.; et al. Staphylococcus epidermidis Phages Transduce Antimicrobial Resistance Plasmids and Mobilize Chromosomal Islands. mSphere 2021, 6, e00223-21. [Google Scholar] [CrossRef]
- Mazaheri Nezhad Fard, R.; Barton, M.D.; Heuzenroeder, M.W. Bacteriophage-mediated transduction of antibiotic resistance in enterococci. Lett. Appl. Microbiol. 2011, 52, 559–564. [Google Scholar] [CrossRef]
- Georjon, H.; Bernheim, A. The highly diverse antiphage defence systems of bacteria. Nat. Rev. Microbiol. 2023, 21, 686–700. [Google Scholar] [CrossRef]
- Borodovich, T.; Shkoporov, A.N.; Ross, R.P.; Hill, C. Phage-mediated horizontal gene transfer and its implications for the human gut microbiome. Gastroenterol. Rep. 2022, 10, goac012. [Google Scholar] [CrossRef]
- Enault, F.; Briet, A.; Bouteille, L.; Roux, S.; Sullivan, M.B.; Petit, M.A. Phages rarely encode antibiotic resistance genes: A cautionary tale for virome analyses. ISME J. 2017, 11, 237–247. [Google Scholar] [CrossRef]
- Calero-Caceres, W.; Ye, M.; Balcazar, J.L. Bacteriophages as Environmental Reservoirs of Antibiotic Resistance. Trends Microbiol. 2019, 27, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, E.; Bonnin, R.A.; Rocha, E.P.C. Phage-Plasmids Spread Antibiotic Resistance Genes Through Infection and Lysogenic Conversion. mBio 2022, 13, e01851-22. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, E.; Rocha, E.P.C. Phage-plasmids promote recombination and emergence of phages and plasmids. Nat. Commun. 2024, 15, 1545. [Google Scholar] [CrossRef]
- Keen, E.C.; Bliskovsky, V.V.; Malagon, F.; Baker, J.D.; Prince, J.S.; Klaus, J.S.; Adhya, S.L. Novel “Superspreader” Bacteriophages Promote Horizontal Gene Transfer by Transformation. mBio 2017, 8, e02115-16. [Google Scholar] [CrossRef]
- Che, Y.; Yang, Y.; Xu, X.; Brinda, K.; Polz, M.F.; Hanage, W.P.; Zhang, T. Conjugative plasmids interact with insertion sequences to shape the horizontal transfer of antimicrobial resistance genes. Proc. Natl. Acad. Sci. USA 2021, 118, e2008731118. [Google Scholar] [CrossRef]
- Shintani, M.; Vestergaard, G.; Milakovic, M.; Kublik, S.; Smalla, K.; Schloter, M.; Udikovic-Kolic, N. Integrons, transposons and IS elements promote diversification of multidrug resistance plasmids and adaptation of their hosts to antibiotic pollutants from pharmaceutical companies. Environ. Microbiol. 2023, 25, 3035–3051. [Google Scholar] [CrossRef]
- Wang, Y.; Dagan, T. The evolution of antibiotic resistance islands occurs within the framework of plasmid lineages. Nat. Commun. 2024, 15, 4555. [Google Scholar] [CrossRef]
- Davis, B.C.; Brown, C.; Gupta, S.; Calarco, J.; Liguori, K.; Milligan, E.; Harwood, V.J.; Pruden, A.; Keenum, I. Recommendations for the use of metagenomics for routine monitoring of antibiotic resistance in wastewater and impacted aquatic environments. Crit. Rev. Environ. Sci. Technol. 2023, 53, 1731–1756. [Google Scholar] [CrossRef]
- Camargo, A.P.; Roux, S.; Schulz, F.; Babinski, M.; Xu, Y.; Hu, B.; Chain, P.S.G.; Nayfach, S.; Kyrpides, N.C. Identification of mobile genetic elements with geNomad. Nat. Biotechnol. 2024, 42, 1303–1312. [Google Scholar] [CrossRef]
- Parnanen, K.; Karkman, A.; Hultman, J.; Lyra, C.; Bengtsson-Palme, J.; Larsson, D.G.J.; Rautava, S.; Isolauri, E.; Salminen, S.; Kumar, H.; et al. Maternal gut and breast milk microbiota affect infant gut antibiotic resistome and mobile genetic elements. Nat. Commun. 2018, 9, 3891. [Google Scholar] [CrossRef] [PubMed]
- Durrant, M.G.; Li, M.M.; Siranosian, B.A.; Montgomery, S.B.; Bhatt, A.S. A Bioinformatic Analysis of Integrative Mobile Genetic Elements Highlights Their Role in Bacterial Adaptation. Cell Host Microbe 2020, 28, 767. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 2020, 76, 101–109. [Google Scholar] [CrossRef]
- Li, X.B.; Xie, Y.Z.; Liu, M.; Tai, C.; Sung, J.Y.; Deng, Z.X.; Ou, H.Y. oriTfinder: A web-based tool for the identification of origin of transfers in DNA sequences of bacterial mobile genetic elements. Nucleic Acids Res. 2018, 46, W229–W234. [Google Scholar] [CrossRef]
- Liu, M.; Li, X.; Xie, Y.; Bi, D.; Sun, J.; Li, J.; Tai, C.; Deng, Z.; Ou, H.Y. ICEberg 2.0: An updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 2019, 47, D660–D665. [Google Scholar] [CrossRef]
- Neron, B.; Littner, E.; Haudiquet, M.; Perrin, A.; Cury, J.; Rocha, E.P.C. IntegronFinder 2.0: Identification and Analysis of Integrons Across Bacteria, with a Focus on Antibiotic Resistance in Klebsiella. Microorganisms 2022, 10, 700. [Google Scholar] [CrossRef]
- Xie, Z.; Tang, H. ISEScan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef]
- Robertson, J.; Nash, J.H.E. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef]
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: Predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef]
- Pellow, D.; Mizrahi, I.; Shamir, R. PlasClass improves plasmid sequence classification. PLoS Comput. Biol. 2020, 16, e1007781. [Google Scholar] [CrossRef]
- Carattoli, A.; Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods Mol. Biol. 2020, 2075, 285–294. [Google Scholar] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Han, S.; Saha, S.; Oler, E.; Peters, H.; Grant, J.R.; Stothard, P.; Gautam, V. PHASTEST: Faster than PHASTER, better than PHAST. Nucleic Acids Res. 2023, 51, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Zhou, Z.C.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Guo, J.R.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef]
- Hua, X.T.; Liang, Q.; Deng, M.; He, J.T.; Wang, M.X.; Hong, W.J.; Wu, J.; Lu, B.; Leptihn, S.; Yu, Y.S.; et al. BacAnt: A Combination Annotation Server for Bacterial DNA Sequences to Identify Antibiotic Resistance Genes, Integrons, and Transposable Elements. Front. Microbiol. 2021, 12, 649969. [Google Scholar] [CrossRef]
- Carr, V.R.; Shkoporov, A.; Hill, C.; Mullany, P.; Moyes, D.L. Probing the Mobilome: Discoveries in the Dynamic Microbiome. Trends Microbiol. 2021, 29, 158–170. [Google Scholar] [CrossRef]
- Ferdinand, A.S.; Kelaher, M.; Lane, C.R.; da Silva, A.G.; Sherry, N.L.; Ballard, S.A.; Andersson, P.; Hoang, T.; Denholm, J.T.; Easton, M.; et al. An implementation science approach to evaluating pathogen whole genome sequencing in public health. Genome Med. 2021, 13, 121. [Google Scholar] [CrossRef]
- Mustafa, A. Whole Genome Sequencing: Applications in Clinical Bacteriology. Med. Princ. Pract. 2024, 33, 185–197. [Google Scholar] [CrossRef]
- Sczyrba, A.; Hofmann, P.; Belmann, P.; Koslicki, D.; Janssen, S.; Dröge, J.; Gregor, I.; Majda, S.; Fiedler, J.; Dahms, E.; et al. Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software. Nat. Methods 2017, 14, 1063–1071. [Google Scholar] [CrossRef]
- Brito, I.L. Examining horizontal gene transfer in microbial communities. Nat. Rev. Microbiol. 2021, 19, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Roy, G.; Prifti, E.; Belda, E.; Zucker, J.D. Deep learning methods in metagenomics: A review. Microb. Genom. 2024, 10, 1231. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.X.; Meyer, F.; Deng, Z.L.; Asgari, E.; Kuo, T.H.; Muench, P.C.; Mchardy, A.C. Assessing computational predictions of antimicrobial resistance phenotypes from microbial genomes. Brief. Bioinform. 2024, 25, bbae206. [Google Scholar] [CrossRef] [PubMed]
- Pesesky, M.W.; Hussain, T.; Wallace, M.; Patel, S.; Andleeb, S.; Burnham, C.A.D.; Dantas, G. Evaluation of Machine Learning and Rules-Based Approaches for Predicting Antimicrobial Resistance Profiles in Gram-negative Bacilli from Whole Genome Sequence Data. Front. Microbiol. 2016, 7, 1887. [Google Scholar] [CrossRef]
- Lüftinger, L.; Májek, P.; Rattei, T.; Beisken, S. Metagenomic Antimicrobial Susceptibility Testing from Simulated Native Patient Samples. Antibiotics 2023, 12, 366. [Google Scholar] [CrossRef]
- Forde, B.M.; De Oliveira, D.M.P.; Falconer, C.; Graves, B.; Harris, P.N.A. Strengths and caveats of identifying resistance genes from whole genome sequencing data. Expert Rev. Anti-Infect. Ther. 2022, 20, 533–547. [Google Scholar] [CrossRef]
- Fenza, G.; Gallo, M.; Loia, V.; Orciuoli, F.; Herrera-Viedma, E. Data set quality in Machine Learning: Consistency measure based on Group Decision Making. Appl. Soft Comput. 2021, 106, 107366. [Google Scholar] [CrossRef]
- Saak, C.C.; Dinh, C.B.; Dutton, R.J. Experimental approaches to tracking mobile genetic elements in microbial communities. FEMS Microbiol. Rev. 2020, 44, 606–630. [Google Scholar] [CrossRef]
- Forster, S.C.; Liu, J.Y.; Kumar, N.; Gulliver, E.L.; Gould, J.A.; Escobar-Zepeda, A.; Mkandawire, T.; Pike, L.J.; Shao, Y.; Stares, M.D.; et al. Strain-level characterization of broad host range mobile genetic elements transferring antibiotic resistance from the human microbiome. Nat. Commun. 2022, 13, 1445. [Google Scholar] [CrossRef]
- Grodner, B.; Shi, H.; Farchione, O.; Vill, A.C.; Ntekas, I.; Diebold, P.J.; Wu, D.T.; Chen, C.Y.; Kim, D.M.; Zipfel, W.R.; et al. Spatial mapping of mobile genetic elements and their bacterial hosts in complex microbiomes. Nat. Microbiol. 2024, 9, 2262–2277. [Google Scholar] [CrossRef]
- Aytan-Aktug, D.; Clausen, P.; Szarvas, J.; Munk, P.; Otani, S.; Nguyen, M.; Davis, J.J.; Lund, O.; Aarestrup, F.M. PlasmidHostFinder: Prediction of Plasmid Hosts Using Random Forest. mSystems 2022, 7, e01180-21. [Google Scholar] [CrossRef] [PubMed]
- Beaulaurier, J.; Zhu, S.; Deikus, G.; Mogno, I.; Zhang, X.S.; Davis-Richardson, A.; Canepa, R.; Triplett, E.W.; Faith, J.J.; Sebra, R.; et al. Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat. Biotechnol. 2018, 36, 61–69. [Google Scholar] [CrossRef] [PubMed]
- De Melo, A.G.; Morency, C.; Moineau, S. Virulence-associated factors as targets for phage infection. Curr. Opin Microbiol. 2024, 79, 102471. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, J.; Nakamura, K.; Nakamura, T.; Iwano, H. Fitness Trade-Offs between Phage and Antibiotic Sensitivity in Phage-Resistant Variants: Molecular Action and Insights into Clinical Applications for Phage Therapy. Int. J. Mol. Sci. 2023, 24, 15628. [Google Scholar] [CrossRef]
- Strathdee, S.A.; Hatfull, G.F.; Mutalik, V.K.; Schooley, R.T. Phage therapy: From biological mechanisms to future directions. Cell 2023, 186, 17–31. [Google Scholar] [CrossRef]
- Pirnay, J.P.; Djebara, S.; Steurs, G.; Griselain, J.; Cochez, C.; De Soir, S.; Glonti, T.; Spiessens, A.; Vanden Berghe, E.; Green, S.; et al. Personalized bacteriophage therapy outcomes for 100 consecutive cases: A multicentre, multinational, retrospective observational study. Nat. Microbiol. 2024, 9, 1434–1453. [Google Scholar] [CrossRef]
- Skurnik, M.; Alkalay-Oren, S.; Boon, M.; Clokie, M.; Sicheritz-Pontén, T.; Dąbrowska, K.; Hatfull, G.F.; Hazan, R.; Jalasvuori, M.; Kiljunen, S.; et al. Phage therapy. Nat. Rev. Methods Primers 2025, 5, 9. [Google Scholar] [CrossRef]
- Turner, P.E.; Azeredo, J.; Buurman, E.T.; Green, S.; Haaber, J.K.; Haggstrom, D.; Carvalho, K.K.D.; Kirchhelle, C.; Moreno, M.G.; Pirnay, J.P.; et al. Addressing the Research and Development Gaps in Modern Phage Therapy. Phage-Ther. Appl. Res. 2024, 5, 30–39. [Google Scholar] [CrossRef]
- Karn, S.L.; Gangwar, M.; Kumar, R.; Bhartiya, S.K.; Nath, G. Phage therapy: A revolutionary shift in the management of bacterial infections, pioneering new horizons in clinical practice, and reimagining the arsenal against microbial pathogens. Front. Med. 2023, 10, 1209782. [Google Scholar] [CrossRef]
- Faltus, T. The Medicinal Phage-Regulatory Roadmap for Phage Therapy under EU Pharmaceutical Legislation. Viruses 2024, 16, 443. [Google Scholar] [CrossRef]
- Bosco, K.; Lynch, S.; Sandaradura, I.; Khatami, A. Therapeutic Phage Monitoring: A Review. Clin. Infect. Dis. 2023, 77, S384–S394. [Google Scholar] [CrossRef]

{kind=link}
| Name | Type | Description | Strengths | Limitations |
|---|---|---|---|---|
| MobileGeneticElementDatabase [137] | Varied MGEs | Non-redundant fasta format database with annotation files. | Cross-reference with other databases; links MGEs to broader functional contexts. Includes metadata, e.g., host and environment. | May not always capture the most recently discovered or rare MGEs. Updates and curation frequency vary. |
| MGEfinder [138] | iMGEs | Identifies iMGEs (primarily IS and transposable elements) and their insertion sites using short reads. | Precise annotations of MGE-host interactions (insight into HGT events). | Highly fragmented assemblies may reduce accuracy. Relies on known sequence features, might miss novel elements. |
| MobileElementFinder [139] | iMGEs and conjugative Tns | Webserver; detects iMGEs (MITEs, ISs, ComTns, PCTs, Tns and conjugative Tns (ICEs, IMEs and CIMEs)), annotates in relation to AMR. | User-friendly and highly specific detection of iMGEs in bacterial genomes based on curated databases and sequence motifs. Links MGEs to ARGs. | May not perform as well on high complexity metagenomic datasets. Relies on known databases; limited detection of novel iMGEs. Requires high-quality assemblies. |
| OriTfinder [140] | ICEs, conjugative plasmids, AMR plasmids. | Identifies and annotates oriT regions in bacterial DNA sequences of conjugative plasmids and ICEs. | Insight into HGT/mobility of plasmids and their role in spread ARGs. Integrates with multiple databases to predict plasmid transfer genes. | Focused on oriT and relaxase genes. No comprehensive overview of MGEs. Limited to known oriT and relaxase sequences, potentially missing novel elements. |
| geNomad [136] | MGEs, specifically plasmids, virus and prophages. | Integrates ML approaches to identify and classify MGEs in meta-/genomic datasets, based on nucleotide composition. | Advanced algorithms and ML enable reliable classification and accurate detection of varied MGEs, incl. atypical features. Prediction of bacterial host(s). Suitable for large metagenomic datasets. | Accuracy depends on training-dataset quality; novel/rare MGEs might be missed. Requires significant computational power for large-scale analyses. Host range prediction is probabilistic and might not be accurate for MGEs with unknown associations. |
| ICEBERG 2.0 [141] | ICEs | Specialized database for identification, classification and annotation of ICEs in bacterial genomes. | Tools to identify ICEs based on integrase genes, attachment sites, and conjugative machinery. Facilitates comparative analysis by linking ICEs to host genomes. | Limited to identifying known ICE features; novel ICEs may not be detected. Requires high-quality genome assemblies for accurate identification. |
| IntegronFinder 2.0 [142] | Integrons | Identifies and annotates integrons and their associated gene cassettes in bacterial meta-/genomes. | Supports the detection of atypical integrons. Can process large-scale datasets, including draft genomes and metagenomes. Visualization of integron structures. | Relies on sequence quality; low quality may result in incomplete detection. Reduced speed at large datasets. Focused on integrons and their components. |
| ISEScan [143] | IS elements | Based on profile hidden Markov models constructed from manually curated IS elements. Identification of ISs in bacterial meta-/genomes. | Capable of identifying a wide range of IS families. Provides detailed annotations of IS elements, including their functional domains. | Requires well-assembled sequences for optimal performance. Limited to ISs; does not detect other MGEs. |
| MOB-suite [144] | Plasmids | Characterization, clustering and replicon typing of plasmids from WGS draft assemblies. | Prediction of plasmid replication host range and mobility potential. Incorporates up-to-date plasmid sequence data. Links ARGs to specific plasmids, relevant in the context of AMR dissemination. | Performance is limited by the reference database, with reduced accuracy for poorly characterized or novel plasmids. Computationally demanding for large datasets, requires some bioinformatics expertise. |
| PlasFlow [145] | Plasmids | Neural network-based tool for predicting and classifying plasmid sequences in metagenomic contigs from environmental samples. | High accuracy in distinguishing plasmid-derived sequences from chromosomal DNA. Suitable for large-scale metagenomic datasets. | Supervised ML approach, limited by training test; may not classify novel sequences well. Similar backbone elements and short sequences might complicate plasmid classifications. |
| PlasClass [146] | Plasmids | Algorithm (ML) that classifies contigs in meta-/genomic assemblies as plasmid or chromosomal DNA. | Useful for metagenomic datasets with short, fragmented or incomplete sequences. Can identify plasmid sequences even without known replicons. | Accuracy may vary with highly fragmented assemblies. Novel plasmid types might not be well-represented in training datasets. |
| PlasmidFinder [147] | Plasmids | Web-based; detects and identifies plasmid replicons in bacterial genomes. | Highly accurate identification of known plasmid replicons including Inc group assignment. Immediate plasmid classification. For WGS, accepts raw sequence data. | Relies on known plasmid replicon sequences; does not detect novel plasmid types. Does not provide detailed information on plasmid structure or the specific genes carried on the plasmid. |
| PHASTER [148] | Prophages | Identifies and annotates prophage sequences within bacterial genomes and plasmids. | User-friendly web interface. Detailed visual output of prophage regions. Continuously updated database improves accuracy. Suitable for complete and draft bacterial genomes (meta-/genomic). | Performance declines with highly fragmented genomes/incomplete assemblies. Longer processing times for large datasets (faster version; PHASTEST [149] for metagenomic/fragmented datasets, cursory annotation). |
| VIBRANT [150] | Phages, prophages | ML; identification and functional annotation of phages in metagenome data, especially prophages. | Accurate prophage detection, activity assessment and infection mechanism. Interactive outputs, including detailed annotations and visualization. Can identify a wide range of phages, including novel ones. | Computationally demanding for large datasets. Needs command-line expertise for local installation. |
| VirSorter2 [151] | Phages, prophages | DNA and RNA virus identification and multi-classifier tool, differentiates between prophages and free viruses in meta-/genomic datasets. | Detects diverse viral sequences (complete and partial), including novel viruses and prophages, using ML and curated regularly updated databases. Can process large-scale metagenomic datasets. | May classify some host-derived sequences as viral, leading to false positives. Accuracy depends on the completeness of viral reference databases and may miss highly divergent viruses. Computationally demanding for large datasets. |
| BacAnt [152] | ARGs, integrons, transposable elements | Web-based tool tailored for predicting ARGs, integrons and transposable elements in genome sequences. | Provides detailed functional annotations of ARGs, integrons and transposable elements in a single step. Generates Genbank files for comparative genomic analysis. | Limited database coverage for rare or emerging genes; novel elements may be missed. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olsen, N.S.; Riber, L. Metagenomics as a Transformative Tool for Antibiotic Resistance Surveillance: Highlighting the Impact of Mobile Genetic Elements with a Focus on the Complex Role of Phages. Antibiotics 2025, 14, 296. https://doi.org/10.3390/antibiotics14030296
Olsen NS, Riber L. Metagenomics as a Transformative Tool for Antibiotic Resistance Surveillance: Highlighting the Impact of Mobile Genetic Elements with a Focus on the Complex Role of Phages. Antibiotics. 2025; 14(3):296. https://doi.org/10.3390/antibiotics14030296
Chicago/Turabian StyleOlsen, Nikoline S., and Leise Riber. 2025. "Metagenomics as a Transformative Tool for Antibiotic Resistance Surveillance: Highlighting the Impact of Mobile Genetic Elements with a Focus on the Complex Role of Phages" Antibiotics 14, no. 3: 296. https://doi.org/10.3390/antibiotics14030296
APA StyleOlsen, N. S., & Riber, L. (2025). Metagenomics as a Transformative Tool for Antibiotic Resistance Surveillance: Highlighting the Impact of Mobile Genetic Elements with a Focus on the Complex Role of Phages. Antibiotics, 14(3), 296. https://doi.org/10.3390/antibiotics14030296