
Comprehensive Analysis of Antiphage Defense Mechanisms: Serovar-Specific Patterns





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Phylogenetic Analysis
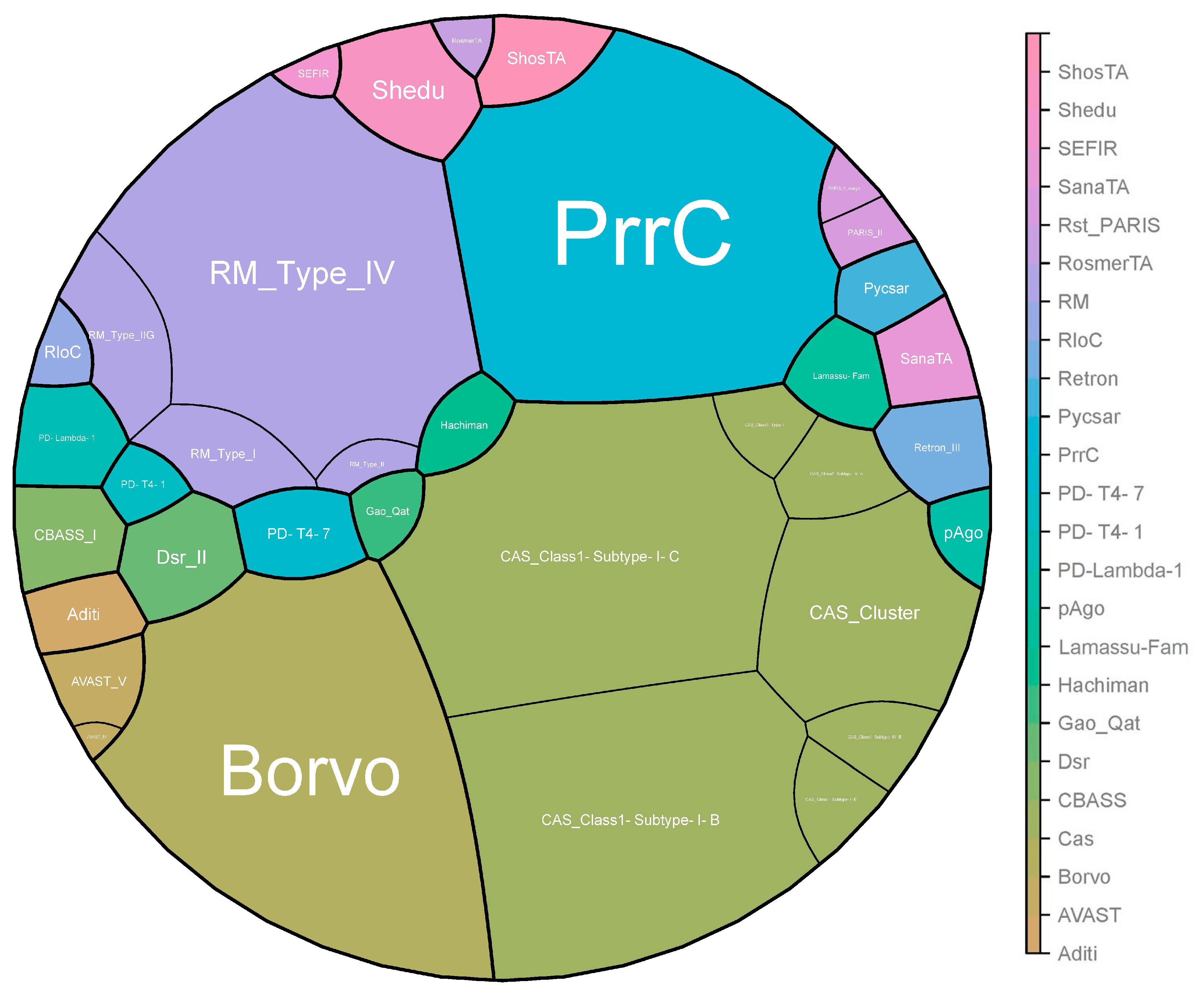
2.2. General Distribution of Antiphage Systems
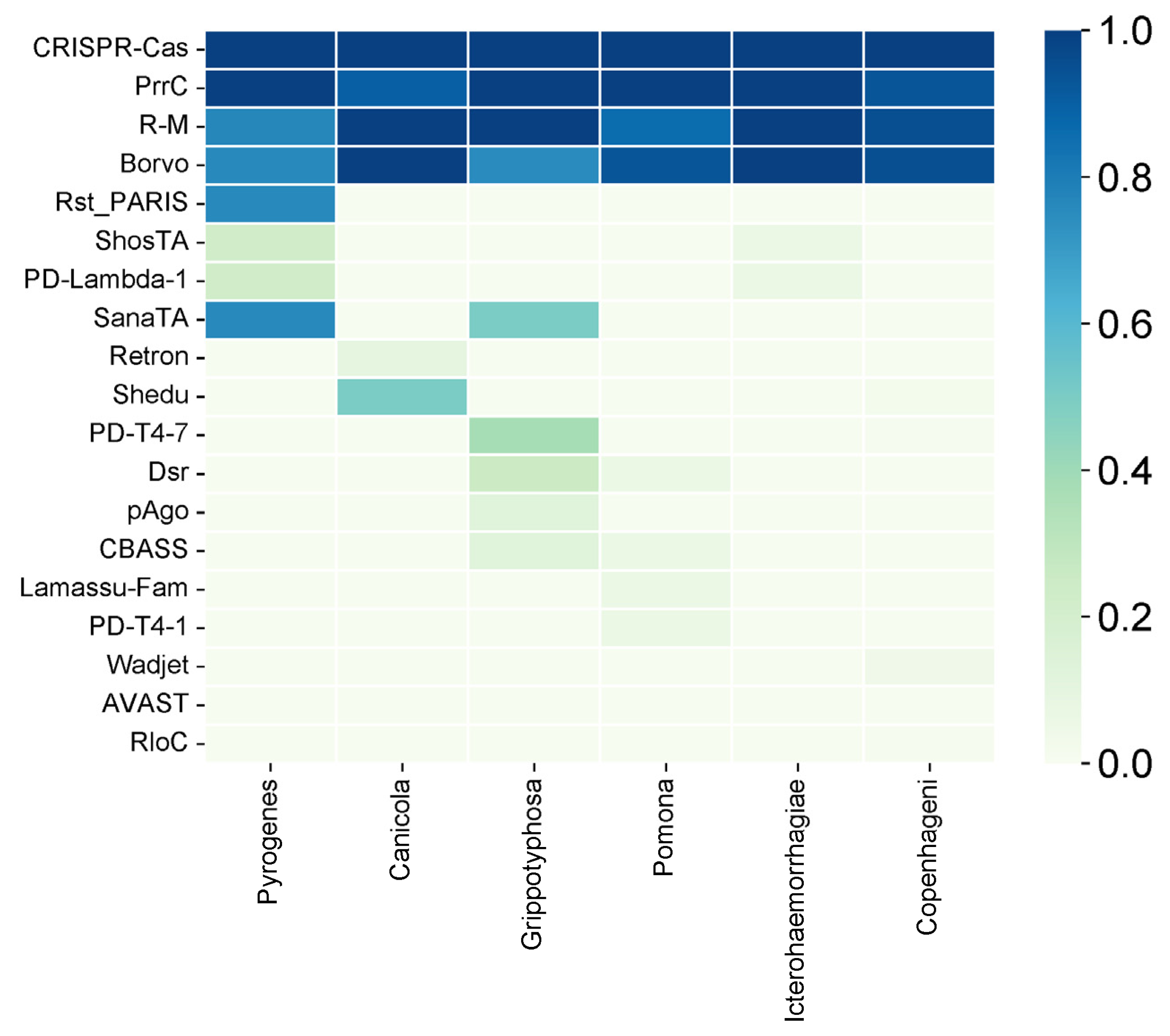
2.3. Serovar-Specific Findings
3. Discussion
4. Materials and Methods
4.1. Antiphage System Analysis
4.2. Phylogenetic Analysis
4.2.1. Determination of Closely Related Type Strains
4.2.2. Pairwise Comparison of Genome Sequences
4.2.3. Phylogenetic Inference
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bharti, A.R.; Nally, J.E.; Ricaldi, J.N.; Matthias, M.A.; Diaz, M.M.; Lovett, M.A.; Levett, P.N.; Gilman, R.H.; Willig, M.R.; Gotuzzo, E. Leptospirosis: A zoonotic disease of global importance. J. Lancet Infect. Dis. 2003, 3, 757–771. [Google Scholar] [CrossRef]
- Petakh, P.; Oksenych, V.; Kamyshna, I.; Boisak, I.; Lyubomirskaya, K.; Kamyshnyi, O. Exploring the complex interplay: Gut microbiome, stress, and leptospirosis. Front. Microbiol. 2024, 15, 1345684. [Google Scholar] [CrossRef]
- Pavlo, P.; Viktoriia, T.; Oleksandr, K. Surveillance of human leptospirosis infections in Ukraine between 2018 and 2023. Front. Public Health 2024, 12, 1394781. [Google Scholar]
- Haake, D.A.; Levett, P.N. Leptospirosis in humans. J. Leptospira Leptospirosis 2015, 387, 65–97. [Google Scholar]
- Adler, B.; de la Peña Moctezuma, A. Leptospira and leptospirosis. Vet. Microbiol. 2010, 140, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Petakh, P.; Behzadi, P.; Oksenych, V.; Kamyshnyi, O. Current treatment options for leptospirosis: A mini-review. Front. Microbiol. 2024, 15, 1403765. [Google Scholar] [CrossRef] [PubMed]
- Petakh, P.; Rostoka, L.; Isevych, V.; Kamyshnyi, A. Identifying risk factors and disease severity in leptospirosis: A meta-analysis of clinical predictors. Trop. Dr. 2023, 53, 464–469. [Google Scholar] [CrossRef]
- Petakh, P.; Isevych, V.; Kamyshnyi, A.; Oksenych, V. Weil’s Disease-Immunopathogenesis, Multiple Organ Failure, and Potential Role of Gut Microbiota. Biomolecules 2022, 12, 1830. [Google Scholar] [CrossRef]
- Petakh, P.; Isevych, V.; Mohammed, I.B.; Nykyforuk, A.; Rostoka, L. Leptospirosis: Prognostic Model for Patient Mortality in the Transcarpathian Region, Ukraine. Vector Borne Zoonotic Dis. 2022, 22, 584–588. [Google Scholar] [CrossRef]
- Petakh, P.; Kamyshnyi, O. AMR mechanisms in L. interrogans serovars: A comprehensive study. Front. Cell. Infect. Microbiol. 2024, 14, 1384427. [Google Scholar] [CrossRef]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef] [PubMed]
- Neuman, H.; Forsythe, P.; Uzan, A.; Avni, O.; Koren, O. Antibiotics in early life: Dysbiosis and the damage done. FEMS Microbiol. Rev. 2018, 42, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Strati, F.; Pujolassos, M.; Burrello, C.; Giuffrè, M.R.; Lattanzi, G.; Caprioli, F.; Troisi, J.; Facciotti, F. Antibiotic-associated dysbiosis affects the ability of the gut microbiota to control intestinal inflammation upon fecal microbiota transplantation in experimental colitis models. Microbiome 2021, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.; Faine, S.; Christopher, W.L.; Chappel, R.J. Development of an improved selective medium for isolation of leptospires from clinical material. Vet. Microbiol. 1986, 12, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Vinod Kumar, K.; Lall, C.; Raj, R.V.; Vedhagiri, K.; Sunish, I.P.; Vijayachari, P. In Vitro Antimicrobial Susceptibility of Pathogenic Leptospira Biofilm. Microb. Drug Resist. 2016, 22, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Schönberg, A. Studies on the effect of antibiotic substances on leptospires and their cultivation from material with a high bacterial count. Zentralblatt Fur Bakteriol. 1 Abt Orig. A Med. Mikrobiol. Infekt. Und Parasitol. 1981, 249, 400–406. [Google Scholar] [CrossRef]
- Hyman, P.; Abedon, S.T. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar]
- Girons, I.S.; Margarita, D.; Amouriaux, P.; Baranton, G. First isolation of bacteriophages for a spirochaete: Potential genetic tools for Leptospira. Res. Microbiol. 1990, 141, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Schiettekatte, O.; Vincent, A.T.; Malosse, C.; Lechat, P.; Chamot-Rooke, J.; Veyrier, F.J.; Picardeau, M.; Bourhy, P. Characterization of LE3 and LE4, the only lytic phages known to infect the spirochete Leptospira. Sci. Rep. 2018, 8, 11781. [Google Scholar] [CrossRef]
- Schiettekatte, O.; Bourhy, P. Isolation, Purification, and Characterization of Leptophages. In Leptospira spp.: Methods and Protocols; Koizumi, N., Picardeau, M., Eds.; Springer: New York, NY, USA, 2020; pp. 67–75. [Google Scholar]
- Bernheim, A.; Sorek, R. The pan-immune system of bacteria: Antiviral defence as a community resource. Nat. Rev. Microbiol. 2020, 18, 113–119. [Google Scholar] [CrossRef]
- Xiao, G.; Yi, Y.; Che, R.; Zhang, Q.; Imran, M.; Khan, A.; Yan, J.; Lin, X. Characterization of CRISPR-Cas systems in Leptospira reveals potential application of CRISPR in genotyping of Leptospira interrogans. APMIS Acta Pathol. Microbiol. Et. Immunol. Scand. 2019, 127, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Huang, Z.; Zhu, Z.; Zhang, J.; Wu, Q.; Xue, L.; Wang, J.; Ding, Y. Recent advances in phage defense systems and potential overcoming strategies. Biotechnol. Adv. 2023, 65, 108152. [Google Scholar] [CrossRef] [PubMed]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 2018, 359, eaar4120. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 2013, 41, 4360–4377. [Google Scholar] [CrossRef] [PubMed]
- Hochhauser, D.; Millman, A.; Sorek, R. The defense island repertoire of the Escherichia coli pan-genome. PLoS Genet. 2023, 19, e1010694. [Google Scholar] [CrossRef] [PubMed]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Yuan, M.M.; Chen, J.; Zheng, X.; Wong, D.; Alvarez, P.J.J.; Yu, P. The association of prokaryotic antiviral systems and symbiotic phage communities in drinking water microbiomes. ISME Commun. 2023, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Vasu, K.; Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. MMBR 2013, 77, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Lu, S.; Wang, Y.; He, L.; Wang, M.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Zhao, X.; et al. Bacterial DNA methyltransferase: A key to the epigenetic world with lessons learned from proteobacteria. Front. Microbiol. 2023, 14, 1129437. [Google Scholar] [CrossRef]
- Mucke, M.; Kruger, D.H.; Reuter, M. Diversity of type II restriction endonucleases that require two DNA recognition sites. Nucleic Acids Res. 2003, 31, 6079–6084. [Google Scholar] [CrossRef]
- Nussenzweig, P.M.; Marraffini, L.A. Molecular Mechanisms of CRISPR-Cas Immunity in Bacteria. Annu. Rev. Genet. 2020, 54, 93–120. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Bernheim, A.; Bikard, D.; Touchon, M.; Rocha, E.P.C. Atypical organizations and epistatic interactions of CRISPRs and cas clusters in genomes and their mobile genetic elements. Nucleic Acids Res. 2019, 48, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Senavirathna, I.; Jayasundara, D.; Warnasekara, J.; Matthias, M.A.; Vinetz, J.M.; Agampodi, S. Complete genome sequences of twelve strains of Leptospira interrogans isolated from humans in Sri Lanka. Infect. Genet. Evol. 2023, 113, 105462. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, G.; David, M.; Borasio, G.D.; Teichmann, A.; Paz, A.; Amitsur, M.; Green, R.; Snyder, L. Phage and host genetic determinants of the specific anticodon loop cleavages in bacteriophage T4-infected Escherichia coli CTr5X. J. Mol. Biol. 1986, 188, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Sirotkin, K.; Cooley, W.; Runnels, J.; Snyder, L.R. A role in true-late gene expression for the T4 bacteriophage 5′ polynucleotide kinase 3′ phosphatase. J. Mol. Biol. 1978, 123, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Huiting, E.; Bondy-Denomy, J. Defining the expanding mechanisms of phage-mediated activation of bacterial immunity. Curr. Opin. Microbiol. 2023, 74, 102325. [Google Scholar] [CrossRef] [PubMed]
- Penner, M.; Morad, I.; Snyder, L.; Kaufmann, G. Phage T4-coded Stp: Double-Edged Effector of Coupled DNA and tRNA-Restriction Systems. J. Mol. Biol. 1995, 249, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Distribution of the PrrC System among Prokaryotes. Available online: https://defensefinder.mdmlab.fr/wiki/defense-systems/prrc (accessed on 20 April 2024).
- Millman, A.; Melamed, S.; Leavitt, A.; Doron, S.; Bernheim, A.; Hör, J.; Garb, J.; Bechon, N.; Brandis, A.; Lopatina, A.; et al. An expanded arsenal of immune systems that protect bacteria from phages. Cell Host Microbe 2022, 30, 1556–1569.e5. [Google Scholar] [CrossRef]
- Stokar-Avihail, A.; Fedorenko, T.; Hör, J.; Garb, J.; Leavitt, A.; Millman, A.; Shulman, G.; Wojtania, N.; Melamed, S.; Amitai, G.; et al. Discovery of phage determinants that confer sensitivity to bacterial immune systems. Cell 2023, 186, 1863–1876.e16. [Google Scholar] [CrossRef]
- DefenseFinder’s Web Page. Available online: https://defensefinder.mdmlab.fr/wiki/defense-systems/borvo#ref-10.1016/j.cell.2023.02.029 (accessed on 20 December 2023).
- Gu, Y.; Li, H.; Deep, A.; Enustun, E.; Zhang, D.; Corbett, K.D. Bacterial Shedu immune nucleases share a common enzymatic core regulated by diverse sensor domains. bioRxiv 2023. [Google Scholar] [CrossRef]
- Vassallo, C.N.; Doering, C.R.; Littlehale, M.L.; Teodoro, G.I.C.; Laub, M.T. A functional selection reveals previously undetected anti-phage defence systems in the E. coli pangenome. Nat. Microbiol. 2022, 7, 1568–1579. [Google Scholar] [CrossRef] [PubMed]
- BV-BRC: Bacterial and Viral Bioinformatics Resource Center. Available online: https://www.bv-brc.org/ (accessed on 10 December 2023).
- Abby, S.S.; Néron, B.; Ménager, H.; Touchon, M.; Rocha, E.P.C. MacSyFinder: A Program to Mine Genomes for Molecular Systems with an Application to CRISPR-Cas Systems. PLoS ONE 2014, 9, e110726. [Google Scholar] [CrossRef] [PubMed]
- Néron, B.; Denise, R.; Coluzzi, C.; Touchon, M.; Rocha, E.P.C.; Abby, S.S. MacSyFinder v2: Improved modelling and search engine to identify molecular systems in genomes. Peer Community J. 2023, 3, e28. [Google Scholar] [CrossRef]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed]
- Kreft, Ł.; Botzki, A.; Coppens, F.; Vandepoele, K.; Van Bel, M. PhyD3: A phylogenetic tree viewer with extended phyloXML support for functional genomics data visualization. Bioinformatics 2017, 33, 2946–2947. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petakh, P.; Oksenych, V.; Khovpey, Y.; Kamyshnyi, O. Comprehensive Analysis of Antiphage Defense Mechanisms: Serovar-Specific Patterns. Antibiotics 2024, 13, 522. https://doi.org/10.3390/antibiotics13060522
Petakh P, Oksenych V, Khovpey Y, Kamyshnyi O. Comprehensive Analysis of Antiphage Defense Mechanisms: Serovar-Specific Patterns. Antibiotics. 2024; 13(6):522. https://doi.org/10.3390/antibiotics13060522
Chicago/Turabian StylePetakh, Pavlo, Valentyn Oksenych, Yevheniya Khovpey, and Oleksandr Kamyshnyi. 2024. "Comprehensive Analysis of Antiphage Defense Mechanisms: Serovar-Specific Patterns" Antibiotics 13, no. 6: 522. https://doi.org/10.3390/antibiotics13060522
APA StylePetakh, P., Oksenych, V., Khovpey, Y., & Kamyshnyi, O. (2024). Comprehensive Analysis of Antiphage Defense Mechanisms: Serovar-Specific Patterns. Antibiotics, 13(6), 522. https://doi.org/10.3390/antibiotics13060522