
Helicobacter pylori in Inflammatory Bowel Diseases: Active Protagonist or Innocent Bystander?


 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epidemiological Insights on H. pylori and IBDs
3. Association between H. pylori and IBDs
4. Inverse Association between H. pylori Infection and IBDs
5. Variability in H. pylori Strains and IBD Risk
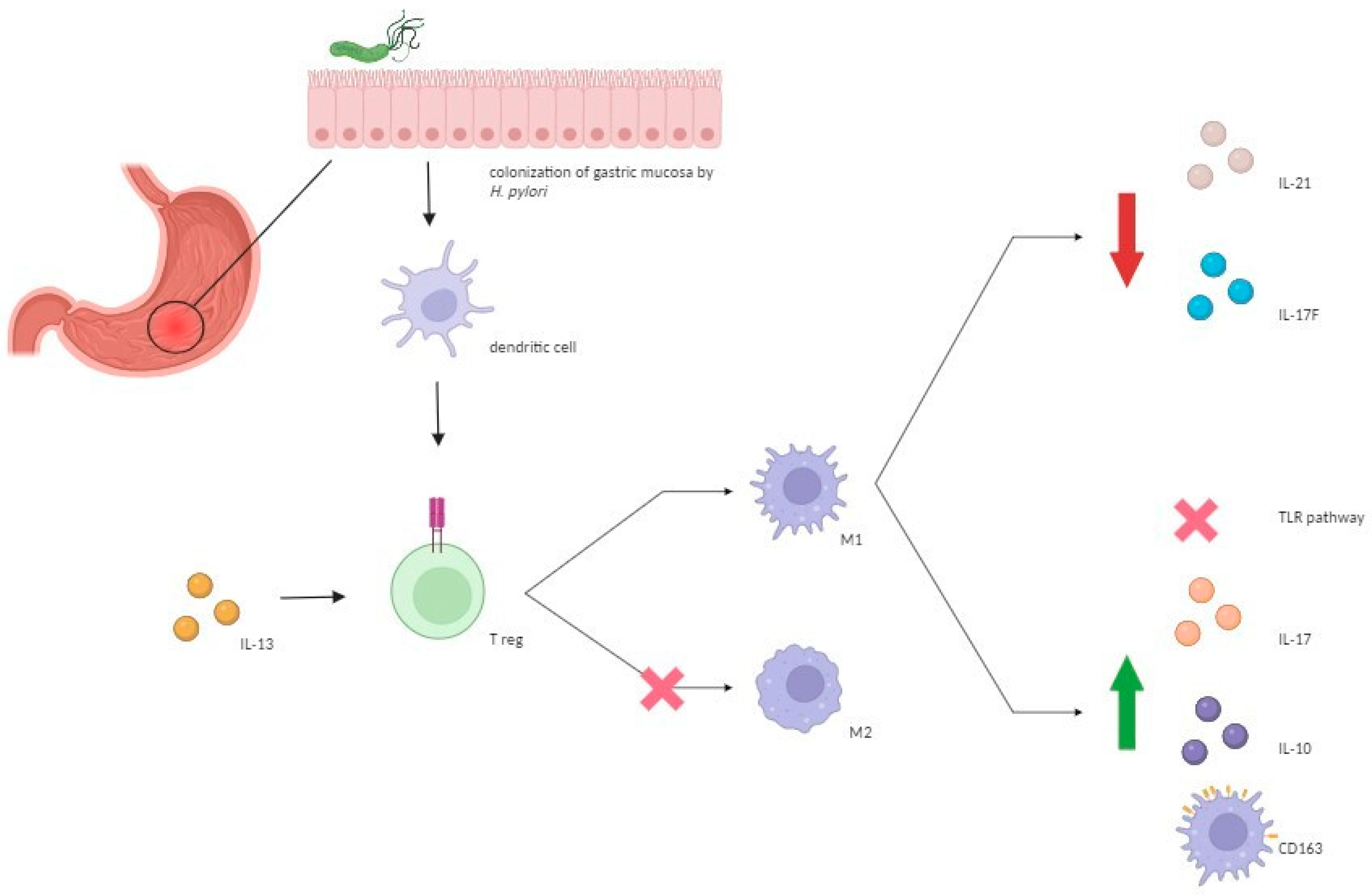
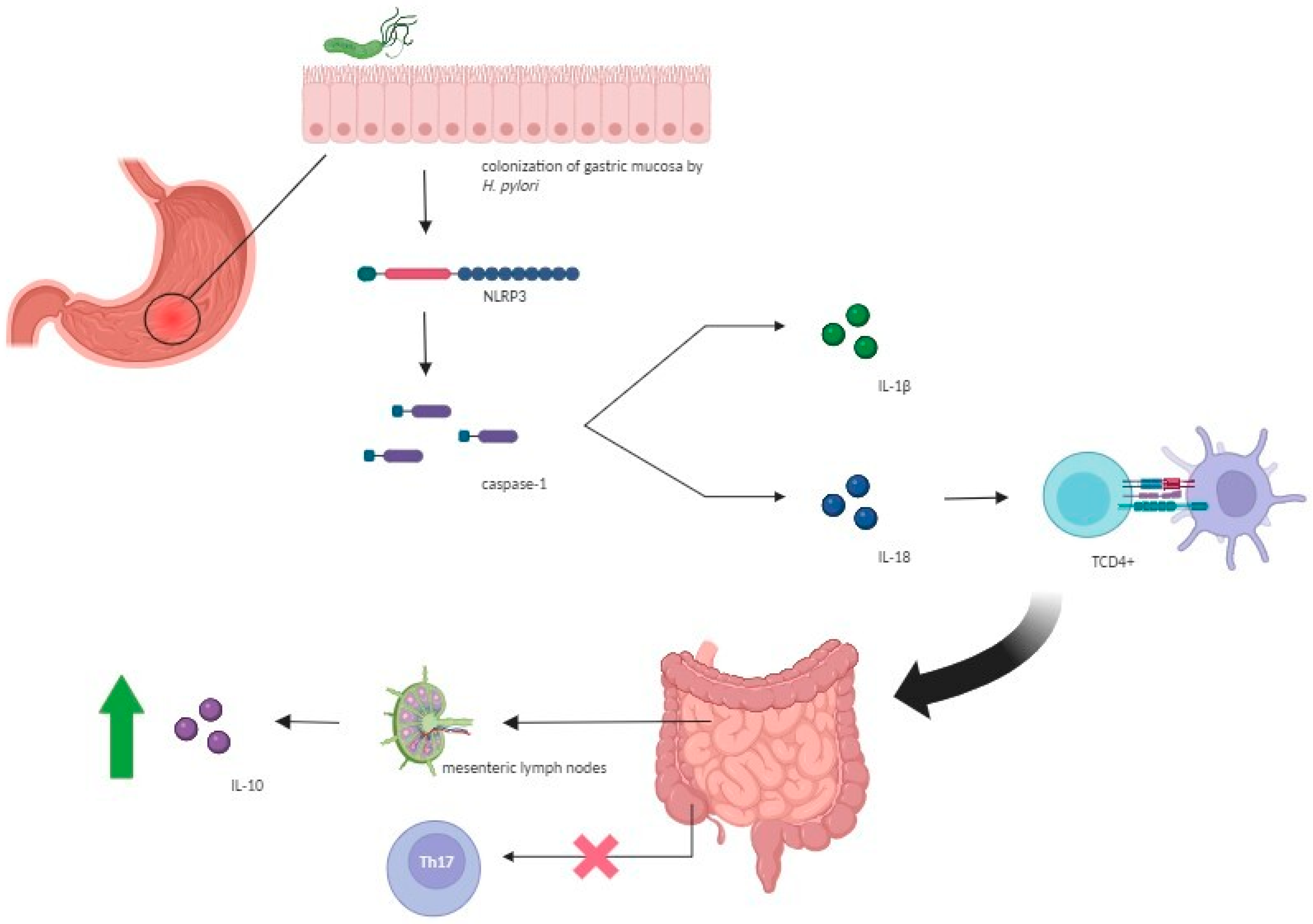
6. Pathogenetic Mechanisms Underlying the Protective Role of H. pylori against IBDs
7. Controversial Opinions on the Role of H. pylori in IBDs: Protective or Provocateur Agent?
8. H. pylori and IBD Course
9. Potential Implications of H. pylori Eradication on the Course of IBDs
10. Effect of Common Medical Treatments Used in IBDs on H. pylori Infection
11. H. pylori, IBDs, and Colorectal Cancer
12. Conclusions
- -
- Fecal calprotectin prior to initiating eradication treatment, following the onset of any gastrointestinal disorder and by default at 3–6 months after eradication (this is the time interval by which CD occurred after H. pylori eradication in the two cases reported in the literature) should be monitored. Fecal calprotectin is believed to be a reliable biomarker, even in these patients, for early screening of CD because it is not found to be significantly increased during chronic H. pylori infection.
- -
- Patients, educated about CD symptoms, should be followed-up once a month for the first six months after eradication [39].
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Axelrad, J.E.; Cadwell, K.H.; Colombel, J.; Shah, S.C. Systematic Review: Gastrointestinal Infection and Incident Inflammatory Bowel Disease. Aliment. Pharmacol. Ther. 2020, 51, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Castaño-Rodríguez, N.; Kaakoush, N.O.; Lee, W.S.; Mitchell, H.M. Dual Role of Helicobacter and Campylobacter Species in IBD: A Systematic Review and Meta-Analysis. Gut 2017, 66, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E.V. Clinical Epidemiology of Inflammatory Bowel Disease: Incidence, Prevalence, and Environmental Influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- M’Koma, A.E. Inflammatory Bowel Disease: Clinical Diagnosis and Surgical Treatment-Overview. Medicina 2022, 58, 567. [Google Scholar] [CrossRef] [PubMed]
- Axelrad, J.E.; Cadwell, K.H.; Colombel, J.-F.; Shah, S.C. The Role of Gastrointestinal Pathogens in Inflammatory Bowel Disease: A Systematic Review. Ther. Adv. Gastroenterol. 2021, 14, 17562848211004493. [Google Scholar] [CrossRef]
- Tepler, A.; Narula, N.; Peek, R.M.; Patel, A.; Edelson, C.; Colombel, J.-F.; Shah, S.C. Systematic Review with Meta-Analysis: Association between Helicobacter pylori CagA Seropositivity and Odds of Inflammatory Bowel Disease. Aliment. Pharmacol. Ther. 2019, 50, 121–131. [Google Scholar] [CrossRef]
- Kong, G.; Liu, Z.; Lu, Y.; Li, M.; Guo, H. The Association between Helicobacter pylori Infection and Inflammatory Bowel Disease in Children: A Systematic Review with Meta-Analysis. Medicine 2023, 102, e34882. [Google Scholar] [CrossRef]
- Püspök, A.; Dejaco, C.; Oberhuber, G.; Waldhör, T.; Hirschl, A.M.; Vogelsang, H.; Gasche, C. Influence of Helicobacter pylori Infection on the Phenotype of Crohn’s Disease. Am. J. Gastroenterol. 1999, 94, 3239–3244. [Google Scholar] [CrossRef]
- Kayali, S.; Gaiani, F.; Manfredi, M.; Minelli, R.; Nervi, G.; Nouvenne, A.; Leandro, G.; Di Mario, F.; de’ Angelis, G.L. Inverse Association between Helicobacter pylori and Inflammatory Bowel Disease: Myth or Fact? Acta Bio Medica Atenei Parm. 2018, 89, 81–86. [Google Scholar] [CrossRef]
- Mak, J.W.Y.; Sun, Y.; Limsrivilai, J.; Abdullah, M.; Kaibullayeva, J.; Balderramo, D.; Vergara, B.I.; Paudel, M.S.; Banerjee, R.; Hilmi, I.; et al. Development of the Global Inflammatory Bowel Disease Visualization of Epidemiology Studies in the 21st Century (GIVES-21). BMC Med. Res. Methodol. 2023, 23, 129. [Google Scholar] [CrossRef]
- Tohidpour, A. CagA-Mediated Pathogenesis of Helicobacter pylori. Microb. Pathog. 2016, 93, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Feilstrecker Balani, G.; Dos Santos Cortez, M.; Picasky Da Silveira Freitas, J.E.; Freire De Melo, F.; Zarpelon-Schutz, A.C.; Teixeira, K.N. Immune Response Modulation in Inflammatory Bowel Diseases by Helicobacter pylori Infection. World J. Gastroenterol. 2023, 29, 4604–4615. [Google Scholar] [CrossRef] [PubMed]
- Korzenik, J.R.; Podolsky, D.K. Evolving Knowledge and Therapy of Inflammatory Bowel Disease. Nat. Rev. Drug Discov. 2006, 5, 197–209. [Google Scholar] [CrossRef]
- Choy, M.C.; Visvanathan, K.; De Cruz, P. An Overview of the Innate and Adaptive Immune System in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2017, 23, 2–13. [Google Scholar] [CrossRef]
- Ahluwalia, B.; Moraes, L.; Magnusson, M.K.; Öhman, L. Immunopathogenesis of Inflammatory Bowel Disease and Mechanisms of Biological Therapies. Scand. J. Gastroenterol. 2018, 53, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased Expression of Interleukin 17 in Inflammatory Bowel Disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef]
- Giuffrida, P.; Di Sabatino, A. Targeting T Cells in Inflammatory Bowel Disease. Pharmacol. Res. 2020, 159, 105040. [Google Scholar] [CrossRef]
- Fantini, M.C.; Rizzo, A.; Fina, D.; Caruso, R.; Sarra, M.; Stolfi, C.; Becker, C.; Macdonald, T.T.; Pallone, F.; Neurath, M.F.; et al. Smad7 Controls Resistance of Colitogenic T Cells to Regulatory T Cell-Mediated Suppression. Gastroenterology 2009, 136, 1308–1316.e1–3. [Google Scholar] [CrossRef]
- Gerlach, K.; Hwang, Y.; Nikolaev, A.; Atreya, R.; Dornhoff, H.; Steiner, S.; Lehr, H.-A.; Wirtz, S.; Vieth, M.; Waisman, A.; et al. TH9 Cells That Express the Transcription Factor PU.1 Drive T Cell-Mediated Colitis via IL-9 Receptor Signaling in Intestinal Epithelial Cells. Nat. Immunol. 2014, 15, 676–686. [Google Scholar] [CrossRef]
- Weigmann, B.; Neurath, M.F. Th9 Cells in Inflammatory Bowel Diseases. Semin. Immunopathol. 2017, 39, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Maizels, R.M.; Smits, H.H.; McSorley, H.J. Modulation of Host Immunity by Helminths: The Expanding Repertoire of Parasite Effector Molecules. Immunity 2018, 49, 801–818. [Google Scholar] [CrossRef] [PubMed]
- Holck, S.; Nørgaard, A.; Bennedsen, M.; Permin, H.; Norn, S.; Andersen, L.P. Gastric Mucosal Cytokine Responses in Helicobacter pylori-Infected Patients with Gastritis and Peptic Ulcers. Association with Inflammatory Parameters and Bacteria Load. FEMS Immunol. Med. Microbiol. 2003, 36, 175–180. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arnold, I.C.; Müller, A. Helicobacter pylori: Does Gastritis Prevent Colitis? Inflamm. Intest. Dis. 2016, 1, 102–112. [Google Scholar] [CrossRef]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory T Cells Suppress Systemic and Mucosal Immune Activation to Control Intestinal Inflammation. Immunol. Rev. 2006, 212, 256–271. [Google Scholar] [CrossRef]
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting Edge: Cure of Colitis by CD4+CD25+ Regulatory T Cells. J. Immunol. 2003, 170, 3939–3943. [Google Scholar] [CrossRef]
- Zhang, H.; Dai, Y.; Liu, Y.; Wu, T.; Li, J.; Wang, X.; Wang, W. Helicobacter pylori Colonization Protects against Chronic Experimental Colitis by Regulating Th17/Treg Balance. Inflamm. Bowel Dis. 2018, 24, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Marotti, B.; Rocco, A.; De Colibus, P.; Compare, D.; de Nucci, G.; Staibano, S.; Tatangelo, F.; Romano, M.; Nardone, G. Interleukin-13 Mucosal Production in Helicobacter pylori-Related Gastric Diseases. Dig. Liver Dis. 2008, 40, 240–247. [Google Scholar] [CrossRef]
- Etzerodt, A.; Moestrup, S.K. CD163 and Inflammation: Biological, Diagnostic, and Therapeutic Aspects. Antioxid. Redox Signal 2013, 18, 2352–2363. [Google Scholar] [CrossRef]
- Skytthe, M.K.; Graversen, J.H.; Moestrup, S.K. Targeting of CD163+ Macrophages in Inflammatory and Malignant Diseases. Int. J. Mol. Sci. 2020, 21, 5497. [Google Scholar] [CrossRef]
- Hou, J.; Wang, X.; Zhang, M.; Wang, M.; Gao, P.; Jiang, Y. Circulating CD14+CD163+CD209+ M2-like Monocytes Are Associated with the Severity of Infection in Helicobacter pylori-Positive Patients. Mol. Immunol. 2019, 108, 13–22. [Google Scholar] [CrossRef]
- Hornsby, M.J.; Huff, J.L.; Kays, R.J.; Canfield, D.R.; Bevins, C.L.; Solnick, J.V. Helicobacter pylori Induces an Antimicrobial Response in Rhesus Macaques in a Cag Pathogenicity Island-Dependent Manner. Gastroenterology 2008, 134, 1049–1057. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Mueller, O.; Herrlinger, K.R.; Fellermann, K.; Schroeder, J.M.; Stange, E.F. Inducible and Constitutive Beta-Defensins Are Differentially Expressed in Crohn’s Disease and Ulcerative Colitis. Inflamm. Bowel Dis. 2003, 9, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Fellermann, K.; Stange, E.F. Human Defensins in Crohn’s Disease. Chem. Immunol. Allergy 2005, 86, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Hitzler, I.; Sayi, A.; Kohler, E.; Engler, D.B.; Koch, K.N.; Hardt, W.-D.; Müller, A. Caspase-1 Has Both Proinflammatory and Regulatory Properties in Helicobacter Infections, Which Are Differentially Mediated by Its Substrates IL-1β and IL-18. J. Immunol. 2012, 188, 3594–3602. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-J.; Park, J.-H.; Franchi, L.; Backert, S.; Núñez, G. The Cag Pathogenicity Island and Interaction between TLR2/NOD2 and NLRP3 Regulate IL-1β Production in Helicobacter pylori Infected Dendritic Cells. Eur. J. Immunol. 2013, 43, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Engler, D.B.; Leonardi, I.; Hartung, M.L.; Kyburz, A.; Spath, S.; Becher, B.; Rogler, G.; Müller, A. Helicobacter pylori-Specific Protection against Inflammatory Bowel Disease Requires the NLRP3 Inflammasome and IL-18. Inflamm. Bowel Dis. 2015, 21, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhu, S.; Li, P.; Min, L.; Zhang, S. Helicobacter pylori Infection and Inflammatory Bowel Disease: A Crosstalk between Upper and Lower Digestive Tract. Cell Death Dis. 2018, 9, 961. [Google Scholar] [CrossRef]
- Murad, H.A. Does Helicobacter pylori Eradication Therapy Trigger or Protect against Crohn’s Disease? Acta Gastroenterol. Belg. 2016, 79, 349–354. [Google Scholar]
- Papamichael, K. Helicobacter pylori Infection and Inflammatory Bowel Disease: Is There a Link? WJG 2014, 20, 6374. [Google Scholar] [CrossRef]
- Oliveira, A.G.; Rocha, G.A.; Rocha, A.M.C.; Sanna, M.d.G.P.; Moura, S.B.; Dani, R.; Marinho, F.P.; Moreira, L.S.; Ferrari, M.d.L.A.; Castro, L.P.F.; et al. Isolation of Helicobacter pylori from the Intestinal Mucosa of Patients with Crohn’s Disease. Helicobacter 2006, 11, 2–9. [Google Scholar] [CrossRef]
- Duchmann, R.; Märker-Hermann, E.; Meyer zum Büschenfelde, K.H. Bacteria-Specific T-Cell Clones Are Selective in Their Reactivity towards Different Enterobacteria or H. pylori and Increased in Inflammatory Bowel Disease. Scand. J. Immunol. 1996, 44, 71–79. [Google Scholar] [CrossRef]
- Tursi, A. Onset of Crohn’s Disease after Helicobacter pylori Eradication: Inflamm. Bowel Dis. 2006, 12, 1008–1009. [Google Scholar] [CrossRef] [PubMed]
- Guslandi, M.; Fanti, L.; Testoni, P.A. Helicobacter pylori Seroprevalence in Crohn’s Disease: Lack of Influence by Pharmacological Treatment. Hepatogastroenterology 2002, 49, 1296–1297. [Google Scholar] [PubMed]
- el-Omar, E.; Penman, I.; Cruikshank, G.; Dover, S.; Banerjee, S.; Williams, C.; McColl, K.E. Low Prevalence of Helicobacter pylori in Inflammatory Bowel Disease: Association with Sulphasalazine. Gut 1994, 35, 1385–1388. [Google Scholar] [CrossRef]
- Triantafillidis, J.K.; Gikas, A.; Apostolidiss, N.; Merikas, E.; Mallass, E.; Peros, G. The Low Prevalence of Helicobacter Infection in Patients with Inflammatory Bowel Disease Could Be Attributed to Previous Antibiotic Treatment. Am. J. Gastroenterol. 2003, 98, 1213–1214. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Tsujinaka, S.; Miura, T.; Kitamura, Y.; Suzuki, H.; Shibata, C. Inflammatory Bowel Disease and Colorectal Cancer: Epidemiology, Etiology, Surveillance, and Management. Cancers 2023, 15, 4154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-S.; Wang, F.; Chang, D.; Han, B.; You, D.-Y. Meta-Analysis of Different Test Indicators: Helicobacter pylori Infection and the Risk of Colorectal Cancer. Int. J. Color. Dis. 2008, 23, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, A.; Genta, R.M. Helicobacter pylori Is a Risk Factor for Colonic Neoplasms. Am. J. Gastroenterol. 2013, 108, 208–215. [Google Scholar] [CrossRef]
- Kapetanakis, N.; Kountouras, J.; Zavos, C.; Anastasiadou, K.; Tsarouchas, G.; Michael, S.; Gavalas, E.; Tsiaousi, E.; Polyzos, S.A.; Venizelos, I.; et al. Potential Oncogenic Properties of Mobilized Stem Cells in a Subpopulation of Inflammatory Bowel Disease Patients Infected with Helicobacter pylori. Inflamm. Bowel Dis. 2013, 19, E27–E29. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bretto, E.; Frara, S.; Armandi, A.; Caviglia, G.P.; Saracco, G.M.; Bugianesi, E.; Pitoni, D.; Ribaldone, D.G. Helicobacter pylori in Inflammatory Bowel Diseases: Active Protagonist or Innocent Bystander? Antibiotics 2024, 13, 267. https://doi.org/10.3390/antibiotics13030267
Bretto E, Frara S, Armandi A, Caviglia GP, Saracco GM, Bugianesi E, Pitoni D, Ribaldone DG. Helicobacter pylori in Inflammatory Bowel Diseases: Active Protagonist or Innocent Bystander? Antibiotics. 2024; 13(3):267. https://doi.org/10.3390/antibiotics13030267
Chicago/Turabian StyleBretto, Elisabetta, Simone Frara, Angelo Armandi, Gian Paolo Caviglia, Giorgio Maria Saracco, Elisabetta Bugianesi, Demis Pitoni, and Davide Giuseppe Ribaldone. 2024. "Helicobacter pylori in Inflammatory Bowel Diseases: Active Protagonist or Innocent Bystander?" Antibiotics 13, no. 3: 267. https://doi.org/10.3390/antibiotics13030267
APA StyleBretto, E., Frara, S., Armandi, A., Caviglia, G. P., Saracco, G. M., Bugianesi, E., Pitoni, D., & Ribaldone, D. G. (2024). Helicobacter pylori in Inflammatory Bowel Diseases: Active Protagonist or Innocent Bystander? Antibiotics, 13(3), 267. https://doi.org/10.3390/antibiotics13030267