Abstract
Pseudomonas aeruginosa (P. aeruginosa) with multi-drug resistance (MDR) is a major cause of serious healthcare-associated infections, leading to high morbidity and mortality. This opportunistic pathogen is responsible for various infectious diseases, such as those seen in cystic fibrosis, ventilator-associated pneumonia, urinary tract infection, otitis externa, and burn and wound injuries. Due to its relatively large genome, P. aeruginosa has great diversity and can use various molecular mechanisms for antimicrobial resistance. For example, outer membrane permeability can contribute to antimicrobial resistance and is determined by lipopolysaccharide (LPS) and porin proteins. Recent findings on the regulatory interaction between peptidoglycan and LPS synthesis provide additional clues against pathogenic P. aeruginosa. This review focuses on recent advances in antimicrobial agents and inhibitors targeting LPS and porin proteins. In addition, we explore current and emerging treatment strategies for MDR P. aeruginosa, including phages, vaccines, nanoparticles, and their combinatorial therapies. Novel strategies and their corresponding therapeutic agents are urgently needed for combating MDR pathogens.
1. Introduction
Pseudomonas aeruginosa (P. aeruginosa) is a common opportunistic human pathogen. It often causes various complicated acute and chronic infections in immunocompromised hosts. P. aeruginosa can multiply and become the main pathogen in cystic fibrosis (CF) patients, ventilator-associated pneumonia, urinary tract infection, otitis externa, burn and wound injuries, bone and joint infections, and systemic infections. Antimicrobial resistance (AMR) is an urgent global public health threat resulting in the death of at least 1.27 million people worldwide and was associated with nearly 5 million deaths in 2019 [1]. In the US alone, AMR underlines 2.8 million infections and 35,000 deaths per year according to the Centers for Diseases Control and Prevention (CDC)’s 2019 Antibiotic Resistance Threats Report. Multidrug-resistant (MDR) and extensively drug-resistant (XDR) P. aeruginosa isolates are frequent causes of serious nosocomial infections and major sources of morbidity and mortality. Data from the CDC show that 8.9% of P. aeruginosa isolates were MDR in 2021 [2]. A detailed report from the National Healthcare Safety Network shows that 18.6% of MDR isolates were from intensive care unit patients, 29.9% from long-term acute-care hospitals, and 11.6% from hospital oncology units [3]. P. aeruginosa is one of the six MDR ESKAPE pathogens including Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, and Enterobacter spp., which cause life-threatening nosocomial infections. In 2017, the World Health Organization (WHO) listed carbapenem-resistant P. aeruginosa as a critical-priority bacterium requiring the urgent development of new antimicrobials to counter a growing global public health crisis [4]. During the COVID-19 pandemic, P. aeruginosa was identified as the second most common bacterial co-infection in patients with COVID-19 [5]. The occurrence of severe infections by MDR P. aeruginosa further increased the complexity of the clinical management of COVID-19 patients [5,6]. In January 2023, an outbreak of a rare strain of XDR P. aeruginosa linked to eye drops was reported by the CDC. As of 15 May 2023, 81 patients were identified in the US as part of the outbreak [7]. The outbreak involved a carbapenem-resistant P. aeruginosa strain carrying the Verona Integron-encoded Metallo-beta-lactamase (VIM) and Guiana Extended-Spectrum beta-lactamase (GES) genes.
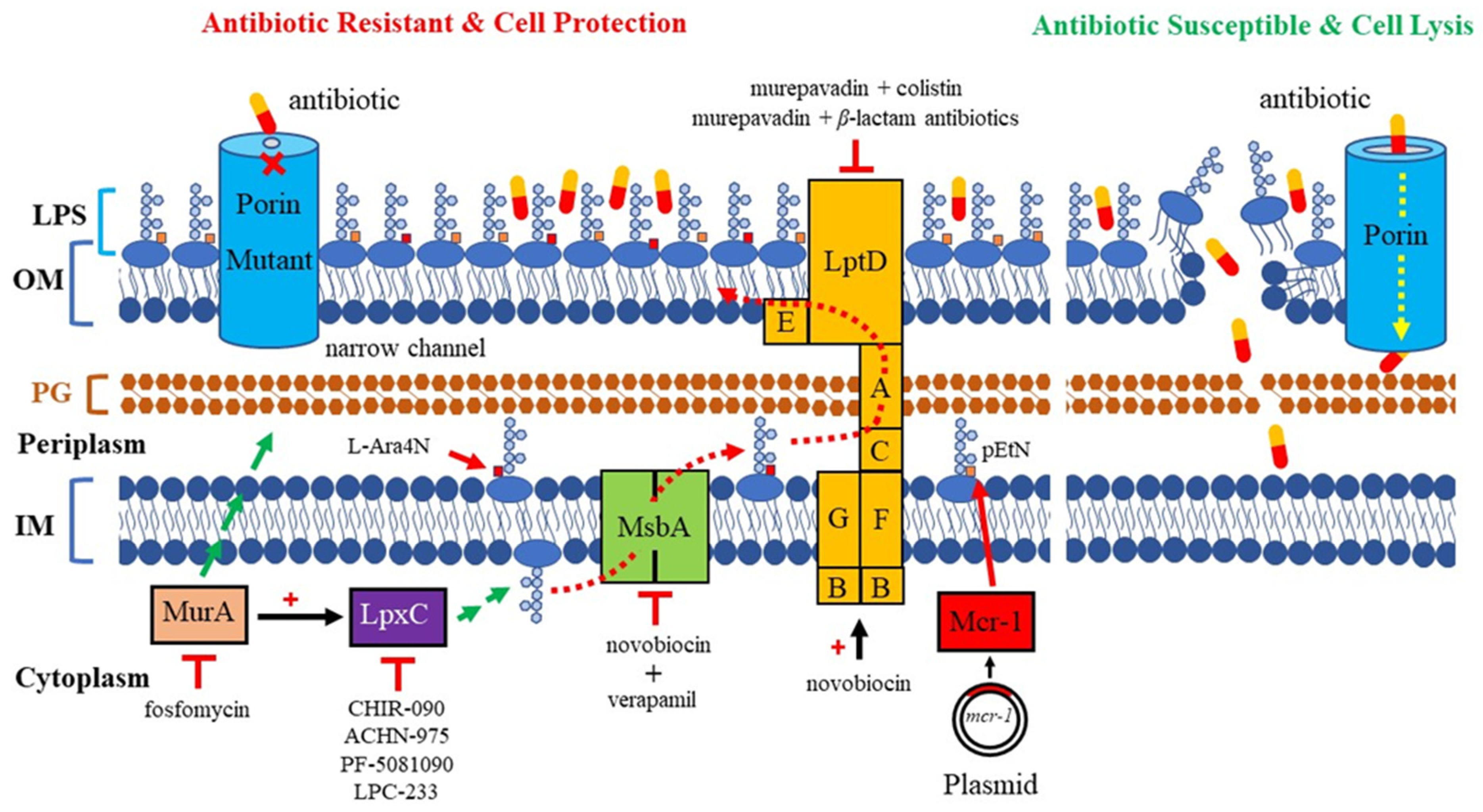
The molecular mechanisms of AMR are complex. The continuous use of various antibiotics over the years has led bacteria to accumulate various AMR mechanisms [8,9]. AMR in P. aeruginosa can be caused by low outermembrane (OM) permeability, drug-resistant efflux pumps, the presence of antibioticresistance genes, the formation of biofilms, etc. Noticeably, multiple mechanisms are simultaneously present, resulting in resistance to nearly all antibiotics available against P. aeruginosa [10]. AMR mostly depends on the structure and composition of the bacterial cell surface, especially alterations in the OM of Gram-negative bacteria. The OM acts as a frontline defense against hostile environments. The XDR P. aeruginosa is highly related to low OM permeability. The characterization of the OM is essential for understanding how antibiotics penetrate this barrier, and for the subsequent development of new therapeutic strategies. In this review, we focus on the major pathogenic components of LPS and porins, which play a significant role in OM permeation, and their relevant therapeutic strategies in development against MDR P. aeruginosa (Figure 1).
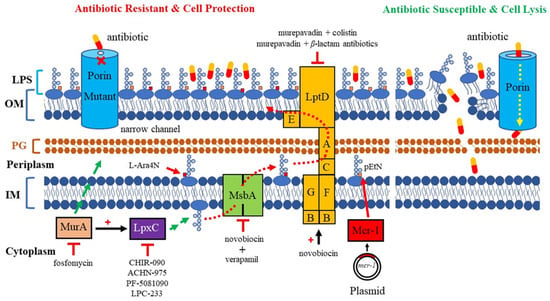
Figure 1.
Mechanism of antimicrobial resistance related to LPS and porin in P. aeruginosa. Red dashed line arrows: LPS is transported from its synthesis site to the outer membrane. Yellow dashed line arrow: porins are channels that allow the entry of antibiotics. Red cross sign: narrow-channel porin is a barrier that blocks antibiotics from passing through the outer membrane. Red arrows: addition of a pEtN or L-Ara4N positively charged moiety to the lipid A of LPS. Red flat-headed arrows: inhibition of pathway. Red plus signs: upregulation of enzymatic activity; MurA stimulates LpxC activity, and novobiocin directly binds the ATPase LptB and increases the activity of the LPS transporter. MurA commits the first step of PG biosynthesis (long green arrow). LpxC commits the catalysis step of LPS biosynthesis (short green arrow).
2. Mechanisms of Antimicrobial Resistance Targeting LPS and Porins
P. aeruginosa has an asymmetric OM composed of phospholipid in the inner leaflet and lipopolysaccharide (LPS) glycolipid in the outer leaflet, compared to a cytoplasmic inner membrane (IM) with a symmetric phospholipid bilayer. LPS has received much attention because of its ability to stimulate the host immune system as an endotoxin [11]. It is now known that OM provides a highly selective permeability barrier against many toxic compounds such as host antimicrobial peptides and antibiotics, as well as both lipophilic and hydrophilic compounds, including nutrients. The high selectivity of the OM in P. aeruginosa is mainly attributed to the presence of LPS [12].
2.1. LPS Biosynthesis
LPS is a major surface molecule of Gram-negative bacteria and consists of three distinct domains: lipid A, the hydrophobic portion of LPS that anchors the molecule in the OM; core oligosaccharide; and O antigen, an extended polysaccharide chain extending into the extracellular environment. Lipid A is essential for bacteria growth. Mutants with reduced lipid A biosynthesis grow slowly and are sensitive to a wide range of antibiotics [13]. The absence of lipid A impedes the aggregation of LPS, leading to bacteria cell death [14]. The biosynthesis of lipid A relies on a zinc-dependent metalloamidase, UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC). Owing to the critical role of LpxC in lipid A biosynthesis and its lack of homology with mammalian proteins, LpxC inhibitors are expected to be potential antibiotics for the treatment of Gram-negative bacterial infections [15,16,17].
An extensive research effort has focused on the discovery of novel LpxC inhibitors against P. aeruginosa. CHIR-090 was the first reported LpxC inhibitor in 2005 [18]. Another two compounds, ACHN-975 [19,20] and PF-5081090 [21] (Figure 1 and Table 1), discovered later, exhibit extensive antimicrobial activity. These two LpxC inhibitors are more active against P. aeruginosa with lower minimal inhibitory concentration MIC90 (0.5~1 μg/mL) and IC50 (1.1 nM) than CHIR-090 [22,23]. However, few inhibitors of LpxC have reached clinical trials, largely due to the limited efficacy and unfavorable cardiovascular toxicity of the candidate inhibitors tested [24]. ACHN-975 is the first LpxC inhibitor to be evaluated in phase I clinical trials. Zhao et al. [25] characterized a new inhibitor (LPC-233) of LpxC, which can specifically inhibit lipid A synthesis. This slow, tight-binding LpxC inhibitor contains a difluoromethyl-allo-threonyl hydroxamate head (Table 1) and has shown outstanding antibiotic activity against a wide range of Gram-negative bacteria, including MDR/XDR P. aeruginosa (MIC90 = 1 μg/mL). This oral compound is bioavailable and efficiently eliminates infections caused by susceptible and MDR Gram-negative bacterial pathogens in various murine models. In addition, it displays exceptional in vitro and in vivo safety profiles, and no cardiovascular toxicity is detected in vivo. These results establish the feasibility of developing oral LpxC-targeting antibiotics for clinical use.
LPS biosynthesis requires both UDP-N-acetylglucosamine (UDP-GlcNAc) and acyl-ACP molecules. Both are also necessary for the biosynthesis of peptidoglycan (PG) and phospholipid, respectively, though LPS and PG have individual synthesis pathways [26]. The coordination between these essential surface layers of the OM has not been made clear. It was not until recently that the discovery of a regulatory interaction between the dedicated enzyme involved in the PG and LPS synthesis pathways in P. aeruginosa was made. Hummels et al. [27] found that the PG synthesis enzyme MurA interacted directly and specifically with LpxC. P. aeruginosa treated with the MurA inhibitor fosfomycin had a PG synthesis inhibition phenotype involving membrane bleb formation and lysis; however, MurA-depleted P. aeruginosa instead adopted an enlarged, ovoid shape. Moreover, MurA was shown to stimulate LpxC activity in cells and in a purified system [27,28]. MurA is the target of antibiotic fosfomycin, and LpxC is an attractive target for developing antibacterial agents against Gram-negative bacteria [17,23]. These imply a potential to develop dual-target drugs that alter both MurA and LpxC activities while simultaneously disrupting PG and OM assembly. Thereby, the combinatorial antibiotic-inhibiting treatment will be more effective in killing P. aeruginosa and/or sensitizing it to other antibiotics that were made ineffective by the barrier function of its envelope.
2.2. LPS Modification
P. aeruginosa has intrinsic, acquired, and adaptive resistance to a variety of antimicrobials. Antimicrobials are becoming increasingly ineffective, as MDR spreads quickly, leading to infections that are difficult and sometimes impossible to treat. Polymyxins have been revived as the last-line defense against infections by MDR Gram-negative bacteria. Colistin (polymyxin E) targets LPS through the modification of lipid A, which competitively interacts with the anionic lipid A, displacing divalent Ca2+ and Mg2+ ions to destabilize the OM, subsequently disrupting the IM, leading to cell death [29]. Colistin resistance in Gram-negative bacteria can be chromosomal or plasmidmediated. The first case of plasmid-mediated colistin resistance conferred by the mcr-1 gene in Enterobacteriaceae was reported in 2016. It was observed that P. aeruginosa transformed with mcr-1 demonstrated elevated colistin MIC from 0.5 mg/L to 8 mg/L and polymyxin B MIC from 0.5 mg/L to 4 mg/L [30], suggesting the potential emergence and spread of plasmid-borne colistin resistance threating human health. Most common mechanisms conferring resistance to colistin are directed against modifications of the lipid A moiety of LPS with the addition of positively charged moieties, such as phosphoethanolamine (pEtN) and 4-amino-4-deoxy-L-arabinose (L-Ara4N) (Figure 1), resulting in a reduction in colistin affinity [31,32]. Most Gram-negative bacteria utilize lipid A modifications to evade the host immune response [33]. Changes in P. aeruginosa growth conditions can induce the extensive remodeling of lipid A, including the addition or removal of phosphate groups and acyl chains [34]. P. aeruginosa clinical isolates showed that polymyxin resistance is associated with the addition of L-Ara4N to phosphate groups within the lipid A and core oligosaccharide moieties of LPS. This pathway refers to the aminoarabinosylation of lipid A [35,36], which a critical prerequisite for the acquisition of colistin resistance in P. aeruginosa. Researchers identified a promising inhibitor of the enzyme responsible for colistinresistancemediating lipid A aminoarabinosylation in P. aeruginosa. In addition, ent-beyerane skeleton (Table 1) may hold promise for the further development of colistin resistance inhibitors [37].
2.3. LPS Transport
LPS synthesis and transport pathways are attractive targets for the development of new antimicrobial therapeutics. LPS transport requires MsbA and Lpt proteins. MsbA is a member of the ABC transporter superfamily and performs the first step of LPS transport by flipping core LPS across the IM [38]. LPS is then transported to the cell surface via the Lpt pathway [39]. Two inhibitors targeting MsbA are reported. Inhibitor G907, a quinolone-like compound, binds to the transmembrane pocket of MsbA and locks the protein in an inward-facing LPS-bound conformation [40]. However, this inhibitor is less effective in P. aeruginosa. Another MsbA inhibitor, tetrahydrobenzothiopene, stimulates the ATPase activity of MsbA and causes the decoupling of ATP hydrolysis from LPS [41]. Recently, a study on tetrahydrobenzothiopene derivatives (Table 1) was reported. The in vitro evaluation showed that most of the target compounds exhibited a great potency in inhibiting the growth of bacteria. One candidate showed MIC values of 1.1 μM against E. coli, 1.0 μM against P. aeruginosa, 0.5 μM against Salmonella, and 1.1 μM against S. aureus. This demonstrates a promising lead compound for the development of new antimicrobial agents against MDR and persistent bacteria [42,43].

Table 1.
Compounds targeting LPS in P. aeruginosa.
Table 1.
Compounds targeting LPS in P. aeruginosa.
| Name | Structure | Mechanism of Action | Stage of Development | References | |
|---|---|---|---|---|---|
| LPS Biosynthesis | |||||
| 1 | CHIR-090 | 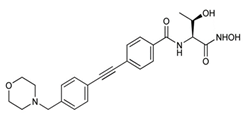 | Slow, tight-binding LpxC | pre-clinical development | [18,22] |
| 2 | ACHN-975 | 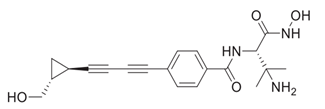 | Inhibits LpxC | Clinical phase I trial, terminateded due to inflammation | [19,20] |
| 3 | PF-5081090 | 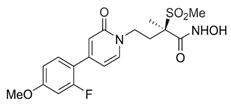 | Inhibits LpxC | pre-clinical development | [21,22] |
| 4 | LPC-233 | 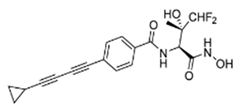 | Slow, tight-binding LpxC | pre-clinical development | [25] |
| LPS Modification | |||||
| 5 | ent-beyerane skeleton | 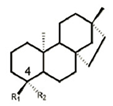 | ArnT inhibitor, potential colistin resistance inhibitor | prepclinical discovery | [37] |
| LPS Transport | |||||
| 6 | G907 | 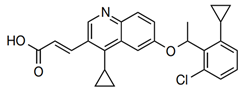 | Inhibits MsbA | pre-clinical development | [40] |
| 7 | Tetrahydrobenzothiophene derivatives | 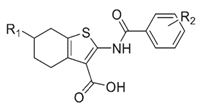 | Inhibits MsbA | pre-pclinical discovery | [42,43] |
| 8 | Novobiocin | 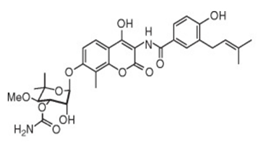 | Inhibits MsbA, enhances ATPase of LptB | pre-clinical development | [44] |
| 9 | Murepavadin, POL7080 | 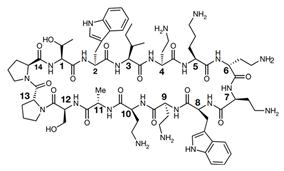 | Inhibits LptD | Clinical phase III trial, terminateded due to kidney problem | [45] |
Recently, the biological effects of verapamil, an inhibitor of ABC transporters, were investigated in P. aeruginosa. It was noticed that IC50 for novobiocin decreased 34% in the presence of verapamil [44]. Verapamil did not inhibit P. aeruginosa growth but increased its sensitivity to novobiocin. The molecular modulization of protein MsbA followed by a docking analysis revealed that novobiocin and verapamil interacted at a common site on MsbA protein. The result indicates that both novobiocin and verapamil act as MsbA potential competitive inhibitors.
Lpt proteins represent another promising target for developing new classes of antibiotics. Murepavadin (POL0780) is a peptidomimetic antibiotic that interacts with the LPS transporter LptD to block LPS assembly and insertion into the OM [45] (Figure 1 and Table 1). As the first OM protein-targeting antibiotic to enter late-stage clinical development, murepavadin displays both potent activities in vitro against P. aeruginosa, including MDR clinical isolates with MIC90 at 0.12 mg/L, and in vivo pharmacokinetics assays in mouse models of infection. Regrettably, the phase III trial, testing murepavadin in 150 enrolled patients with hospital-acquired pneumonia who required mechanical respiratory support, was halted due to safety concerns. Fifty-six percent of patients reported kidney problems in one of the trials [46].
It is known that colistin kills P. aeruginosa through the modification of lipid A on the OM. However, how it disrupts the inner membrane is not clear. Sabnis et al. [47] designed a new therapeutic approach and exposed P. aeruginosa to colistin and/or murepavadin. An MIC assay showed that murepavadin sensitized P. aeruginosa to colistin by increasing LPS abundance in the IM. In addition, in both in vitro clinical MDR isolates and an in vivo mouse model of lung infection, the combination therapy of colistin and murepavadin demonstrated enhanced efficacy in the killing and clearance of P. aeruginosa. These results also demonstrated that colistin exerts bactericidal activity by targeting LPS in the IM. A recent study demonstrated that murepavadin impaired bacterial OM integrity, which induced the envelope stress response in P. aeruginosa. The combination of murepavadin with ceftazidime/avibactam slowed down the resistance development in a mouse model of acute pneumonia by P. aeruginosa infection [48]. Taken together, these experiments provide robust evidence that colistin targets LPS directly and can be used in a combination of murepavadin and other antibiotics for the treatment of serious infections caused by P. aeruginosa.
2.4. Porins
The molecular characterization of the OM is essential for understanding how antibiotics penetrate this barrier, both for the development of new therapeutic strategies and for rational drug design. Porins are beta barrel pore proteins contained in the OM of Gram-negative bacteria. These proteins possess an internal hydrophilic channel that generally restricts the entry of most lipophilic molecules and only permits the passage of certain small hydrophilic molecules from the external environment to the interior cell [49,50]. A study on antibiotic properties using high-throughput screening of more than 8 million compounds targeting Gram-negative infections confirmed that the successful compounds were mostly hydrophilic small molecules. This points toward size exclusion by porins [51,52]. In contrast to Enterobacteria, Pseudomonas lack large diffusion porins, such as OmpF and OmpC [53]. The OM permeability of P. aeruginosa was 100-fold lower than E. coli [54]. In addition, the low permeability in P. aeruginosa is attributed to a large family of OprD (also termed OccD1) porins comprising 19 members. These porins act together as specific channel proteins involved in the uptake of different nutrients [50,55,56,57].
OprF is the most abundant non-lipoprotein on the OM of P. aeruginosa. Owing to its C-terminal containing a PG-binding domain, OprF is mainly involved in maintaining the OM structure [58]. Mutations on OprF confirmed that its N-terminal is responsible for protein production and membrane insertion, while its C-terminal is liable for stable interaction with PG anchoring on the OM [59]. As OprF and OprI are highly conserved and induce a cross protective immunity across all P. aeruginosa strains, they become promising vaccine candidates for the control of P. aeruginosa infection. A phase III clinical trial of IC43, a hybrid OprF/I vaccine, has been completed [60]. OprF involves adhesion, biofilm formation, and virulence. In comparison to the wild-type, isogenic OprF mutant, and an OprF-complement strain, OprF is required for P. aeruginosa virulence as the OprF mutant strain displays reduced cytotoxicity in cells [61]. A study on the OprF mutant in a vertebrate model showed that OprF protected P. aeruginosa against macrophage clearance by avoiding bacterial elimination in acidified phagosomes [62]. Using scanning electron microscopy, a comparative study was conducted on biofilm formation, examining both a wild-type and an isogenic OprF mutant of P. aeruginosa. The results showed that OprF played a dynamic role in P. aeruginosa virulence. The absence of OprF resulted in a slow growth rate corresponding to an elongated lag phase and reduced biofilm production [63].
The high stability of P. aeruginosa OM is due to the presence of the OprH, the smallest porin found in P. aeruginosa. Edrington et al. [64] provided the first molecular structure of OprH and evidence for multiple interactions between OprH and LPS that likely contributed to the antibiotic resistance of P. aeruginosa. Lee et al. [65] built various simulation systems to investigate the impact of different LPS molecules on OprH structure and dynamics. The results showed that the OprHLPS interactions mainly depended on the secondary structure of OprH and the chemical structure of LPS, which may contribute to bacterial AMR. Similar results were validated using a solution nuclear magnetic resonance spectroscopy system [66]. As polymyxin-resistant P. aeruginosa clinical isolates are capable of lipid A modification, OprH is regarded as a potential target for novel antimicrobial therapies.
Carbapenem is a mainstay therapy for P. aeruginosa infection. In general, carbapenems can efficiently cross the OM by passing through the aqueous channels, such as OprD in P. aeruginosa. Reduced permeability caused by downregulated OprD protein appears to be the most common mechanism of intrinsic resistance to carbapenem [67]. Mutations in OprD [68,69,70,71,72] are associated with imipenem resistance and reduced susceptibility to meropenem through the loss of or change in OprD. One study investigated OprD mutations in locally prevalent MDR P. aeruginosa strains in cystic fibrosis of clinical relevance. An analysis of whole-genome sequencing found shared strain sub-lineages with specific OprD variants pre-existing in the local population before spreading between patients [73].
Wolter et al. [74] were the first to report carbapenem resistance resulting from the insertional inactivation of the oprD gene by insertion sequence elements in seven clinical P. aeruginosa isolates. These isolates exhibited dual resistance to fluoroquinolones and imipenem. In an isogenic pair of MDR P. aeruginosa clinical isolates, we uncovered an extra m6A methylation in the promoter of an endotoxinAregulating gene, toxR, most likely causing the higher expression of OpdQ, a member of the OprD porin family. We proposed an epigenetic regulation of opdQ expression pertinent to the phenotypic change in P. aeruginosa from resistant to susceptible to piperacillin/tazobactam and increases in MIC to meropenem [75].
3. New Strategy Development against P. aeruginosa Infection
New antibiotics are considered ‘drugs of last resort’ against MDR bacteria. The delay in discovering new antibiotics has exacerbated the MDR problem and leaves a gap between the diagnosis of an MDR pathogen and effective treatment. Alternative therapies, especially those differing from traditional antibiotics, are emerging. These non-traditional therapies in the global preclinical antibacterial pipeline are mainly phage therapies, anti-virulence therapies, antibodies (antibody–drug conjugates), vaccines, and nanoparticles [76,77]. Compared to traditional antibiotics, these new methods have the advantage of avoiding negative effects on host commensal bacteria and AMR development.
3.1. Phages
Bacteriophage therapy is one of the promising alternatives against MDR P. aeruginosa. Many research studies have demonstrated the ability of phages to eradicate P. aeruginosa [78,79,80]. Several clinical studies have been conducted to evaluate the effectiveness of phage therapy in treating specific P. aeruginosa infections (https://clinicaltrials.gov) (accessed on 20 December 2023).
A randomized phase 1/2 trial (NCT02116010) was designed to compare the efficacy and tolerability of a cocktail of 12 natural lytic bacteriophages (PP1131) with standard of care for patients with burns. The study showed that PP1131 decreased the bacterial burden in burn wounds at a slower pace than standard of care at low concentrations of phage. It was felt that a higher phage dose in a larger number of participants was warranted for further studies [81].
Trials of AP-PA02, another cocktail of bacteriophages designed to fight P. aeruginosa infections in patients with chronic pulmonary infection and CF bronchiectasis (NCT04596319, phase I/II) and non-CF bronchiectasis (NCT05616221, phase II), are underway. Studies aim to evaluate the safety, tolerability, phage kinetics, and efficacy of inhaled AP-PA02.
Another two clinical trials are being conducted: NCT04323475, a phage cocktail-SPK therapy for second-degree burn wounds in adult patients, and NCT04684641, a bacteriophage therapy YPT-01 for P. aeruginosa infections in adults with CF. To date, no new participants have yet been recruited or enrolled.
Phage therapy has so far failed to translate into positive outcomes in the limited number of clinical trials that have been performed. One problem is the rapid evolution of phage resistance, which limits the clinical efficacy of phage therapy. Yang et al. [82] strategically formulated a cocktail of phages that successfully suppressed the evolution of resistance. After prolonged incubation, phage resistance in P. aeruginosa was observed. Lipid remodeling during phage infection may alter binding and subsequent infection dynamics [83]. One case study described phage therapy for a complex bone and joint infection of XDR P. aeruginosa, in which phage therapy combined with ceftolozane/tazobactam and colistin resulted in rapid wound healing over 2 weeks [84]. Another recent case reported a successful aerosolized bacteriophage with concomitant antibiotic treatment of a chronic lung infection due to MDR P. aeruginosa. The patient showed clinically significant improvement even without the complete eradication of P. aeruginosa lung colonization [85]. Therefore, effective phage therapy strategies including phageantibiotic synergies and optimizing phage administration will likely improve the outcome in future trials [86].
3.2. Vaccines
Extensive research has focused on vaccine development against P. aeruginosa over the last 50 years. Four vaccines have entered phase III trials during these years. While some showed promising results, no anti-P. aeruginosa vaccine has yet been approved. LPS and OM proteins are important antigens of P. aeruginosa, which have been shown to be immunogenic for hosts. However, due to the high diversity of P. aeruginosa serotypes, it is hard to design a vaccine effective for all serotypes [87].
Octavalent O-polysaccharide-exotoxin A conjugate (Aerugen®) is an LPS-based conjugate vaccine. A phase I study showed high affinity IgG response to exotoxin and LPS in healthy volunteers. A phase II study initially showed similar immunoglobulin functions in CF patients not colonized with P. aeruginosa. Unfortunately, it did not have an impact on clinical outcomes. The phase III trial in CF patients was finally stopped as the interim results did not show significant differences between the placebo and control groups [88].
Another vaccine that has reached phase III clinical trial is His-tagged OprF-OprI fusion protein (IC43). This vaccine seems to be an effective candidate due to its better safety and immunogenicity profile in phase I and II trials [89,90]. In phase III, 800 patients on mechanical ventilation were randomized to either IC43 100 μg or saline placebo, given in two vaccinations 7 days apart. The mortality in patients twenty-eight days after the first vaccination and the immunogenicity and safety of IC43 were evaluated. The IC43 vaccine was well tolerated and achieved high immunogenicity in these patients, but the overall mortality rate in the vaccinated patients was not significantly different from that in the placebo group [60].
The U.S. Food and Drug Administration licensed two new pneumococcal conjugate polyvalent vaccines for the prevention of invasive pneumococcal disease in 2021 [91]. It inspired the development of a trivalent vaccine that targets multiple antigens affecting major virulence factors in P. aeruginosa. Recently, a recombinant protein POmT comprising three antigens—the full-length V antigen (PcrV) of the P. aeruginosa type III secretion system (TTSS), the OM domain of OprF, and a non-catalytic mutant of the carboxyl domain of exotoxin A—was created. A significant improvement in acute lung injury and a reduction in the acute mortality of P. aeruginosa-induced acute lung injury were observed in a mouse model [92].
The great success of mRNA-based COVID-19 vaccines during the pandemic attracted more researchers to employ the potential of mRNA as a preventive and therapeutic vaccine for distinct infectious diseases. Compared to conventional vaccines, mRNA vaccines have a favorable safety profile and higher efficiency because mRNA is non-infectious and poses little concern for DNA integration. PcrV protein at the tip of TTSS is an effective target for active and passive immunization against P. aeruginosa [93,94,95]. Recently, researchers reported the development of mRNA vaccines targeting the TTSS of P. aeruginosa. Mice were vaccinated with nucleoside-modified mRNA encapsulated in lipid nanoparticles. Together, the mRNA-immunized mice showed improved survival, decreased lung bacterial loads, and fewer pathological changes in the lung compared to saline controls [96].
Meanwhile, another group of researchers designed two mRNA vaccines targeting PcrV and fusion protein OprF-I, which demonstrated highly immunogenic antigens conserved across different serotypes of P. aeruginosa. Notably, both PcrV and OprF-I mRNA vaccines could induce immunity; the PcrV vaccination elicited a higher level of humoral and cellular immune responses and significantly reduced the colonization of P. aeruginosa in the skin, lung, liver, spleen, and kidney, providing a broad protective effect against P. aeruginosa. Furthermore, the immune response and protection elicited by a single mRNA vaccine and combined ones showed the superiority of mRNA vaccines over PcrV and OprF protein vaccines, respectively [97]. Many Gram-negative bacteria share highly homologous TTSS. mRNA targeting different bacteria can be designed quickly and scaled up with low cost. This new approach can provide a potential application prospective for different Gram-negative pathogens.
3.3. Nanoparticles
Photothermally active nanomaterials are emerging as potent antimicrobial agents [98]. Recently, a selective photothermal therapy based on LPS aptamer functionalized nanorods for MDR P. aeruginosa infection was reported [99]. Animal experiments showed that the nanorods permitted the active targeting of LPS on the surface of Gram-negative bacteria and a specific anti-inflammatory ability in the MDR P. aeruginosa-infected wound murine model. In addition, the nanorods can precisely overcome MDR P. aeruginosa through physical damage and effectively reduce excess M1 inflammatory macrophages to accelerate the healing of infected wounds. Overall, this molecular therapeutic strategy displayed great potential as a prospective antimicrobial treatment for MDR P. aeruginosa infection.
The combination of bacteria-imprinting technology and photothermal therapy has emerged as a potential therapeutic strategy for fighting drug-resistant bacteria [100,101,102,103]. A group of scientists reported a promising material: photothermal molecularly imprinted polymers (PMIPs) [104]. Based on the affinity of P. aeruginosa LPS with boric acid, LPS-imprinted PMIPs were synthesized for the study of the efficient capture and elimination of P. aeruginosa. Fluorescent images demonstrated that the engineered PMIP had low toxicity to normal cells, higher affinity to LPS, and more significant targeting capability toward P. aeruginosa than nonimprinted polymers. Although PMIP alone showed low anti-biofilm activity against P. aeruginosa in established biofilms, most viable cells were effectively eliminated by the combination of PMIP and irradiation near-infrared light therapy. So, this new technology can be used as a potentially powerful tool for the safe and efficient deactivation and removal of P. aeruginosa.
4. Challenges
The rapid growth of AMR/MDR is driven by the misuse and overuse of antibiotics that are commonly and widely used to treat bacterial infections. Antibiotics can wipe out harmful pathogens of concern and save lives. Simultaneously, antibiotics eradicate beneficial microbes, reduce microbiota diversity, and alter metabolic activity with deleterious consequences for human health [105]. Additionally, antibiotics may select bacteria with AMR to overgrow as these bacteria evolve and respond to the selective pressures placed upon them, whereas the combinatorial use of antibiotics can lead to the production and spread of MDR bacteria. Moreover, the source of antibiotics has probably been exhausted and the development of new antibiotics has become slow and expensive [106], resulting in the AMR crisis that we are experiencing now. Developing and implementing novel therapeutic strategies other than traditional antibiotic therapy is thus imperative in combating AMR. Here, we reviewed the recent advances in the development of novel therapeutic strategies against MDR P. aeruginosa.
P. aeruginosa is among the largest of the bacterial genomes with its genome size at a range of 5.5–7.0 million base pairs. This large genome facilitates P. aeruginosa’s evolutionary adaptation to diverse environments and development of resistance to antibiotics, phages, and/or vaccines. Additionally, P. aeruginosa possesses an abundance and diversity of bacteriophages, both lysogenic and lytic, driving bacterial evolution and providing a great source of phage candidates for phage therapy. However, few of these natural phages have been characterized in detail regarding their target specificity and toxic contents, leading to concerns about the safety, reliability, and efficacy of their applications. Recently, an increasing number of MDR P. aeruginosa clinical isolates have been whole-genome sequenced, revealing its high genome diversity, dynamic evolution, and MDR complexity. This diversity and complexity makes the prevention and treatment of MDR P. aeruginosa much more challenging. Notably, PAO1 and PA14 are the most commonly employed reference strains with moderate and hyper-virulent phenotypes, respectively. However, sequencing has demonstrated significant deviations from the clinical isolates of P. aeruginosa infection [107]. In the genomic analysis of four isolates from different environments and CF patients, these isolates had a defective Las quorum-sensing system, but remained virulent when compared to PAO1 and PA14 [108]. This result reminds us that special care should be taken when translating laboratory results into clinical applications, and whole-genome sequencing should be used to reduce variations in the accrued data in P. aeruginosa studies [109].
Most of the aforementioned novel therapeutic strategies are target-specific, making them attractive alternatives to antibiotic treatment in maintaining healthy microbiota diversity. Owing to our limited knowledge of phage-targeting specificity to prevent bacterial development of anti-phage systems [110], phage cocktails, instead of individual phages, are usually designed for phage therapy. A broad-range bacteriophage cocktail was found to be superior to the individual phage in destroying P. aeruginosa biofilm and providing a faster treatment in mice with acute respiratory infection [111]. In addition, synergistic phage–antimicrobial cocktail therapy can improve phage therapy effectiveness. This has been reported in several successful personalized phage therapies for patients suffering from chronic MDR P. aeruginosa life-threatening infection [84,85,112]. In PAO1 and PA14 test models, the evolution of bacterial resistance to a lytic phage attack changed the efflux pump mechanism, causing increased sensitivity to drugs from several antibiotic classes [113]. To effectively combat MDR P. aeruginosa, combinatorial treatment, complementarily or synergistically, may be the better strategy.
Author Contributions
C.Y. conceived the study, conducted the literature review, drew the figure, and drafted the manuscript; M.Z.A., J.T.F. and W.H. critically reviewed and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
LPS, lipopolysaccharide; OM, outer membrane; IM, inner membrane; PG, Peptidoglycan; L-Ara4N, 4-amino-4-deoxy-L-arabinose; pEtN, phosphoethanolamine; MurA, UDP-N-acetylglucosamine enolpyruvyl transferase; LptD, LPS transporter D; LpxC, UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase.
References
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Multidrug-Resistant Pseudomonas aeruginosa; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2021. Available online: https://arpsp.cdc.gov/profile/antibiotic-resistance/mdr-pseudomonas-aeruginosa (accessed on 23 January 2024).
- Weiner-Lastinger, L.M.; Abner, S.; Edwards, J.R.; Kallen, A.J.; Karlsson, M.; Magill, S.S.; Pollock, D.; See, I.; Soe, M.M.; Walters, M.S.; et al. Antimicrobial-resistant pathogens associated with adult healthcare-associated infections: Summary of data reported to the National Healthcare Safety Network, 2015–2017. Infect. Control Hosp. Epidemiol. 2020, 41, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Lansbury, L.; Lim, B.; Baskaran, V.; Lim, W.S. Co-infections in people with COVID-19: A systematic review and meta-analysis. J. Infect. 2020, 81, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, M.; Barda, B. Pseudomonas aeruginosa Bloodstream Infections in SARS-CoV-2 Infected Patients: A Systematic Review. J. Clin. Med. 2023, 12, 2252. [Google Scholar] [CrossRef] [PubMed]
- Outbreak of Extensively Drug-Resistant Pseudomonas aeruginosa Associated with Artificial Tears; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2023; Updated 15 May 2023. Available online: https://www.cdc.gov/hai/outbreaks/crap-artificial-tears.html (accessed on 23 January 2024).
- Darby, E.M.; Trampari, E.; Siasat, P.; Gaya, M.S.; Alav, I.; Webber, M.A.; Blair, J.M.A. Molecular mechanisms of antibiotic resistance revisited. Nat. Rev. Microbiol. 2023, 21, 280–295. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xiao, W.; Zhou, C.; Pu, Q.; Deng, X.; Lan, L.; Liang, H.; Song, X.; Wu, M. Pseudomonas aeruginosa: Pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct. Target. Ther. 2022, 7, 199. [Google Scholar] [CrossRef]
- Froon, A.H.; Dentener, M.A.; Greve, J.W.; Ramsay, G.; Buurman, W.A. Lipopolysaccharide toxicity-regulating proteins in bacteremia. J. Infect. Dis. 1995, 171, 1250–1257. [Google Scholar] [CrossRef]
- Huszczynski, S.M.; Lam, J.S.; Khursigara, C.M. The Role of Pseudomonas aeruginosa Lipopolysaccharide in Bacterial Pathogenesis and Physiology. Pathogens 2019, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Barb, A.W.; Zhou, P. Mechanism and inhibition of LpxC: An essential zinc-dependent deacetylase of bacterial lipid A synthesis. Curr. Pharm. Biotechnol. 2008, 9, 9–15. [Google Scholar] [PubMed]
- Caughlan, R.E.; Jones, A.K.; DeLucia, A.M.; Woods, A.L.; Xie, L.; Ma, B.; Barnes, S.W.; Walker, J.R.; Sprague, E.R.; Yang, X.; et al. Mechanisms decreasing in vitro susceptibility to the LpxC inhibitor CHIR-090 in the gram-negative pathogen Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2012, 56, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Kline, T.; Andersen, N.H.; Harwood, E.A.; Bowman, J.; Malanda, A.; Endsley, S.; Erwin, A.L.; Doyle, M.; Fong, S.; Harriset, A.L.; et al. Potent, novel in vitro inhibitors of the Pseudomonas aeruginosa deacetylase LpxC. J. Med. Chem. 2002, 45, 3112–3129. [Google Scholar] [CrossRef] [PubMed]
- Mdluli, K.E.; Witte, P.R.; Kline, T.; Barb, A.W.; Erwin, A.L.; Mansfield, B.E.; McClerren, A.L.; Pirrung, M.C.; Tumey, L.N.; Warrener, P.; et al. Molecular validation of LpxC as an antibacterial drug target in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 2178–2184. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, D.V.; Holl, R. LpxC inhibitors: A patent review (2010–2016). Expert Opin. Ther. Pat. 2017, 27, 1227–1250. [Google Scholar] [CrossRef]
- McClerren, A.L.; Endsley, S.; Bowman, J.L.; Andersen, N.H.; Guan, Z.; Rudolph, J.; Raetz, C.R.H. A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciprofloxacin. Biochemistry 2005, 44, 16574–16583. [Google Scholar] [CrossRef]
- Krause, K.M.; Haglund, C.M.; Hebner, C.; Serio, A.W.; Lee, G.; Nieto, V.; Cohen, F.; Kane, T.R.; Machajewski, T.D.; Hildebrandt, D.; et al. Potent LpxC Inhibitors with In Vitro Activity against Multidrug-Resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e00977-19. [Google Scholar] [CrossRef]
- Kalinin, D.V.; Holl, R. Insights into the Zinc-Dependent Deacetylase LpxC: Biochemical Properties and Inhibitor Design. Curr. Top. Med. Chem. 2016, 16, 2379–2430. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, A.P.; McPherson, C.J.; Kuhn, M.; Carifa, A.; Mullins, L.; George, D.; Desbonnet, C.; Eidem, T.M.; Montgomery, J.I.; Brown, M.F.; et al. LpxC inhibitors as new antibacterial agents and tools for studying regulation of lipid A biosynthesis in Gram-negative pathogens. mBio 2014, 5, e01551-14. [Google Scholar] [CrossRef] [PubMed]
- Alkatheri, A.H.; Yap, P.S.-X.; Abushelaibi, A.; Lai, K.-S.; Cheng, W.-H.; Lim, S.-H.E. Microbial Genomics: Innovative Targets and Mechanisms. Antibiotics 2023, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Erwin, A.L. Antibacterial Drug Discovery Targeting the Lipopolysaccharide Biosynthetic Enzyme LpxC. Cold Spring Harb. Perspect. Med. 2016, 6, a025304. [Google Scholar] [CrossRef]
- Cohen, F.; Aggen, J.B.; Andrews, L.D.; Assar, Z.; Boggs, J.; Choi, T.; Dozzo, P.; Easterday, A.N.; Haglund, C.M.; Hildebrandt, D.J.; et al. Optimization of LpxC Inhibitors for Antibacterial Activity and Cardiovascular Safety. ChemMedChem 2019, 14, 1560–1572. [Google Scholar] [CrossRef]
- Zhao, J.; Cochrane, C.S.; Najeeb, J.; Gooden, D.; Sciandra, C.; Fan, P.; Lemaitre, N.; Newns, K.; Nicholas, R.A.; Guan, Z.; et al. Preclinical safety and efficacy characterization of an LpxC inhibitor against Gram-negative pathogens. Sci. Transl. Med. 2023, 15, eadf5668. [Google Scholar] [CrossRef]
- King, J.D.; Kocíncová, D.; Westman, E.L.; Lam, J.S. Review: Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 2009, 15, 261–312. [Google Scholar] [CrossRef] [PubMed]
- Hummels, K.R.; Berry, S.P.; Li, Z.; Taguchi, A.; Min, J.K.; Walker, S.; Marks, D.S.; Bernhardt, T.G. Coordination of bacterial cell wall and outer membrane biosynthesis. Nature 2023, 615, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Eschenburg, S.; Priestman, M.; Schönbrunn, E. Evidence that the fosfomycin target Cys115 in UDP-N-acetylglucosamine enolpyruvyl transferase (MurA) is essential for product release. J. Biol. Chem. 2005, 280, 3757–3763. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Kasiakou, S.K.; Saravolatz, L.D. Colistin: The revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Baron, S.; Hadjadj, L.; Rolain, J.-M.; Olaitan, A.O. Molecular mechanisms of polymyxin resistance: Knowns and unknowns. Int. J. Antimicrob. Agents 2016, 48, 583–591. [Google Scholar] [CrossRef]
- Nang, S.C.; Azad, M.A.K.; Velkov, T.; Zhou, Q.; Li, J. Rescuing the Last-Line Polymyxins: Achievements and Challenges. Pharmacol. Rev. 2021, 73, 679–728. [Google Scholar] [CrossRef] [PubMed]
- Steimle, A.; Autenrieth, I.B.; Frick, J.-S. Structure and function: Lipid A modifications in commensals and pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Needham, B.D.; Trent, M.S. Fortifying the barrier: The impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 2013, 11, 467–481. [Google Scholar] [CrossRef]
- Lo Sciuto, A.; Imperi, F. Aminoarabinosylation of Lipid A is Critical for the Development of Colistin Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, e01820-17. [Google Scholar] [CrossRef] [PubMed]
- Cervoni, M.; Sposato, D.; Sciuto, A.L.; Imperi, F. Regulatory Landscape of the Pseudomonas aeruginosa Phosphoethanolamine Transferase Gene eptA in the Context of Colistin Resistance. Antibiotics 2023, 12, 200. [Google Scholar] [CrossRef]
- Quaglio, D.; Mangoni, M.L.; Stefanelli, R.; Corradi, S.; Casciaro, B.; Vergine, V.; Lucantoni, F.; Cavinato, L.; Cammarone, S.; Loffredo, M.R.; et al. ent-Beyerane Diterpenes as a Key Platform for the Development of ArnT-Mediated Colistin Resistance Inhibitors. J. Org. Chem. 2020, 85, 10891–10901. [Google Scholar] [CrossRef]
- Alexander, M.K.; Miu, A.; Oh, A.; Reichelt, M.; Ho, H.; Chalouni, C.; Labadie, S.; Wang, L.; Liang, J.; Nickerson, N.N.; et al. Disrupting Gram-Negative Bacterial Outer Membrane Biosynthesis through Inhibition of the Lipopolysaccharide Transporter MsbA. Antimicrob. Agents Chemother. 2018, 62, e01142-18. [Google Scholar] [CrossRef]
- Sperandeo, P.; Martorana, A.M.; Polissi, A. The lipopolysaccharide transport (Lpt) machinery: A nonconventional transporter for lipopolysaccharide assembly at the outer membrane of Gram-negative bacteria. J. Biol. Chem. 2017, 292, 17981–17990. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.; Miu, A.; Alexander, M.K.; Garcia, N.K.; Oh, A.; Zilberleyb, I.; Reichelt, M.; Austin, C.D.; Tam, C.; Shriver, S.; et al. Structural basis for dual-mode inhibition of the ABC transporter MsbA. Nature 2018, 557, 196–201. [Google Scholar] [CrossRef]
- Zhang, G.; Baidin, V.; Pahil, K.S.; Moison, E.; Tomasek, D.; Ramadoss, N.S.; Chatterjee, A.K.; McNamara, C.W.; Young, T.S.; Schultz, P.G.; et al. Cell-based screen for discovering lipopolysaccharide biogenesis inhibitors. Proc. Natl. Acad. Sci. USA 2018, 115, 6834–6839. [Google Scholar] [CrossRef]
- Pei, S.; Lai, L.; Sun, W.; Lu, Z.; Hao, J.; Liu, Y.; Wu, W.; Guan, S.; Su, X. Discovery of novel tetrahydrobenzothiophene derivatives as MSBA inhibitors for antimicrobial agents. Bioorg Chem. 2023, 142, 106932. [Google Scholar] [CrossRef]
- Lai, L.; Yang, J.; Sun, W.; Su, X.; Chen, J.; Chen, X.; Pei, S. Design, synthesis and antibacterial evaluation of a novel class of tetrahydrobenzothiophene derivatives. RSC Med. Chem. 2023, 14, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Hulen, C.; Racine, P.J.; Feuilloley, M.; Elomri, A.; Lomri, N.E. Effects of Verapamil and Two Bisbenzylisoquinolines, Curine and Guattegaumerine Extracted from Isolona hexaloba, on the Inhibition of ABC Transporters from Pseudomonas aeruginosa. Antibiotics 2022, 11, 700. [Google Scholar] [CrossRef] [PubMed]
- Luther, A.; Urfer, M.; Zahn, M.; Muller, M.; Wang, S.Y.; Mondal, M.; Vitale, A.; Hartmann, J.B.; Sharpe, T.; Monte, F.L.; et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 2019, 576, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Temporarily Halt Enrollment for the Pivotal Phase III Trials PRISM-MDR and PRISM-UDR Evaluating Murepavadin in Patients with Nosocomial Pneumonia. 2019. Available online: https://www.swissbiotech.org/listing/polyphor-temporarily-halts-phase-iii-studyor-the-treatment-of-patients-with-nosocomial-pneumonia/ (accessed on 23 January 2014).
- Sabnis, A.; Hagart, K.L.; Klockner, A.; Becce, M.; Evans, L.E.; Furniss, R.C.D.; Mavridou, D.A.; Murphy, R.; Stevens, M.M.; Davies, J.C.; et al. Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane. eLife 2021, 10, e65836. [Google Scholar] [CrossRef]
- Wei, X.; Gao, J.; Xu, C.; Pan, X.; Jin, Y.; Bai, F.; Cheng, Z.; Lamont, L.; Pletzer, D.; Wu, W. Murepavadin induces envelope stress response and enhances the killing efficacies of beta-lactam antibiotics by impairing the outer membrane integrity of Pseudomonas aeruginosa. Microbiol. Spectr. 2023, 11, e0125723. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, J.D.; Kleinekathöfer, U.; Winterhalter, M. How to Enter a Bacterium: Bacterial Porins and the Permeation of Antibiotics. Chem. Rev. 2021, 121, 5158–5192. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, S.; Bouffartigues, E.; Bodilis, J.; Maillot, O.; Lesouhaitier, O.; Feuilloley, M.G.J.; Orange, N.; Dufour, A.; Cornelis, P. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 2017, 41, 698–722. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.; Hassan, K.A. The Gram-negative permeability barrier: Tipping the balance of the in and the out. mBio 2023, 2023, e0120523. [Google Scholar] [CrossRef]
- Brown, D.G.; May-Dracka, T.L.; Gagnon, M.M.; Tommasi, R. Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J. Med. Chem. 2014, 57, 10144–10161. [Google Scholar] [CrossRef]
- Pratt, L.A.; Hsing, W.; Gibson, K.E.; Silhavy, T.J. From acids to osmZ: Multiple factors influence synthesis of the OmpF and OmpC porins in Escherichia coli. Mol. Microbiol. 1996, 20, 911–917. [Google Scholar] [CrossRef]
- Yoshimura, F.; Nikaido, H. Permeability of Pseudomonas aeruginosa outer membrane to hydrophilic solutes. J. Bacteriol. 1982, 152, 636–642. [Google Scholar] [CrossRef]
- Eren, E.; Vijayaraghavan, J.; Liu, J.; Cheneke, B.R.; Touw, D.S.; Lepore, B.W.; Indic, M.; Movileanu, L.; Berg, B.v.D. Substrate specificity within a family of outer membrane carboxylate channels. PLoS Biol. 2012, 10, e1001242. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Brinkman, F.S.L. Function of pseudomonas porins in uptake and efflux. Annu. Rev. Microbiol. 2002, 56, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Bodrenko, I.; Acosta-Gutiérrez, S.; D’agostino, T.; Pathania, M.; Ghai, I.; Schleberger, C.; Bumann, D.; Wagner, R.; Winterhalter, M.; et al. Getting Drugs through Small Pores: Exploiting the Porins Pathway in Pseudomonas aeruginosa. ACS Infect. Dis. 2018, 4, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, G.; Gayet, L.; Liguori, L.; Odier, M.; Martin, D.K.; Cortès, S.; Schaack, B.; Lenormand, J.-L. Cell-free expression of the outer membrane protein OprF of Pseudomonas aeruginosa for vaccine purposes. Life Sci. Alliance 2021, 4, e202000958. [Google Scholar] [CrossRef] [PubMed]
- Adlbrecht, C.; Wurm, R.; Depuydt, P.; Spapen, H.; Lorente, J.A.; Staudinger, T.; Creteur, J.; Zauner, C.; Meier-Hellmann, A.; Eller, P.; et al. Efficacy, immunogenicity, and safety of IC43 recombinant Pseudomonas aeruginosa vaccine in mechanically ventilated intensive care patients-a randomized clinical trial. Crit. Care 2020, 24, 74. [Google Scholar] [CrossRef] [PubMed]
- Fito-Boncompte, L.; Chapalain, A.; Bouffartigues, E.; Chaker, H.; Lesouhaitier, O.; Gicquel, G.; Bazire, A.; Madi, A.; Connil, N.; Véron, W.; et al. Full virulence of Pseudomonas aeruginosa requires OprF. Infect. Immun. 2011, 79, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Moussouni, M.; Berry, L.; Sipka, T.; Nguyen-Chi, M.; Blanc-Potard, A.-B. Pseudomonas aeruginosa OprF plays a role in resistance to macrophage clearance during acute infection. Sci. Rep. 2021, 11, 359. [Google Scholar] [CrossRef]
- Bukhari, S.I.; Aleanizy, F.S. Association of OprF mutant and disturbance of biofilm and pyocyanin virulence in Pseudomonas aeruginosa. Saudi Pharm. J. 2020, 28, 196–200. [Google Scholar] [CrossRef]
- Edrington, T.C.; Kintz, E.; Goldberg, J.B.; Tamm, L.K. Structural basis for the interaction of lipopolysaccharide with outer membrane protein H (OprH) from Pseudomonas aeruginosa. J. Biol. Chem. 2011, 286, 39211–39223. [Google Scholar] [CrossRef]
- Lee, J.; Patel, D.S.; Kucharska, I.; Tamm, L.K.; Im, W. Refinement of OprH-LPS Interactions by Molecular Simulations. Biophys. J. 2017, 112, 346–355. [Google Scholar] [CrossRef]
- Kucharska, I.; Liang, B.; Ursini, N.; Tamm, L.K. Molecular Interactions of Lipopolysaccharide with an Outer Membrane Protein from Pseudomonas aeruginosa Probed by Solution NMR. Biochemistry 2016, 55, 5061–5072. [Google Scholar] [CrossRef]
- Skurnik, D.; Roux, D.; Cattoir, V.; Danilchanka, O.; Lu, X.; Yoder-Himes, D.R.; Han, K.; Guillard, T.; Jiang, D.; Gaultier, C.; et al. Enhanced in vivo fitness of carbapenem-resistant oprD mutants of Pseudomonas aeruginosa revealed through high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 20747–20752. [Google Scholar] [CrossRef] [PubMed]
- Ocampo-Sosa, A.A.; Cabot, G.; Rodriguez, C.; Roman, E.; Tubau, F.; Macia, M.D.; Moya, B.; Zamorano, L.; Suárez, C.; Peña, C.; et al. Alterations of OprD in carbapenem-intermediate and -susceptible strains of Pseudomonas aeruginosa isolated from patients with bacteremia in a Spanish multicenter study. Antimicrob. Agents Chemother. 2012, 56, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Pan, Y.; Fang, Y. Role of the Outer Membrane Protein OprD2 in Carbapenem-Resistance Mechanisms of Pseudomonas aeruginosa. PLoS ONE 2015, 10, e0139995. [Google Scholar] [CrossRef]
- Kiani, M.; Astani, A.; Eslami, G.; Khaledi, M.; Afkhami, H.; Rostami, S.; Zarei, M.; Khozani, N.R.; Zandi, H. Upstream region of OprD mutations in imipenem-resistant and imipenem-sensitive Pseudomonas isolates. AMB Express 2021, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.-C.; Kuo, A.-J.; Su, L.-H.; Liu, T.-P.; Lee, M.-H.; Su, I.-N.; Wu, T.-L. Development of carbapenem resistance in Pseudomonas aeruginosa is associated with OprD polymorphisms, particularly the amino acid substitution at codon 170. J. Antimicrob. Chemother. 2017, 72, 2489–2495. [Google Scholar] [CrossRef] [PubMed]
- Rostami, S.; Farajzadeh Sheikh, A.; Shoja, S.; Farahani, A.; Tabatabaiefar, M.A.; Jolodar, A.; Sheikhi, R. Investigating of four main carbapenem-resistance mechanisms in high-level carbapenem resistant Pseudomonas aeruginosa isolated from burn patients. J. Chin. Med. Assoc. 2018, 81, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Sherrard, L.J.; Wee, B.A.; Duplancic, C.; Ramsay, K.A.; Dave, K.A.; Ballard, E.; Wainwright, C.E.; Grimwood, K.; Sidjabat, H.E.; Whiley, D.M.; et al. Emergence and impact of oprD mutations in Pseudomonas aeruginosa strains in cystic fibrosis. J. Cyst. Fibros. 2022, 21, e35–e43. [Google Scholar] [CrossRef] [PubMed]
- Wolter, D.J.; Hanson, N.D.; Lister, P.D. Insertional inactivation of oprD in clinical isolates of Pseudomonas aeruginosa leading to carbapenem resistance. FEMS Microbiol. Lett. 2004, 236, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Hamouche, J.E.; Wang, G.; Smith, M.; Yin, C.; Dhand, A.; Dimitrova, N.; Fallon, J.T. Integrated Genome-Wide Analysis of an Isogenic Pair of Pseudomonas aeruginosa Clinical Isolates with Differential Antimicrobial Resistance to Ceftolozane/Tazobactam, Ceftazidime/Avibactam, and Piperacillin/Tazobactam. Int. J. Mol. Sci. 2020, 21, 1026. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Petersen, F.C.; Shekhar, S. Commensal Bacteria: An Emerging Player in Defense Against Respiratory Pathogens. Front. Immunol. 2019, 10, 1203. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Rezk, N.; Abdelsattar, A.S.; Elzoghby, D.; Agwa, M.M.; Abdelmoteleb, M.; Aly, R.G.; Fayez, M.S.; Essam, K.; Zaki, B.M.; El-Shibiny, A. Bacteriophage as a potential therapy to control antibiotic-resistant Pseudomonas aeruginosa infection through topical application onto a full-thickness wound in a rat model. J. Genet. Eng. Biotechnol. 2022, 20, 133. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.A.; Drilling, A.; Morales, S.; Cornet, M.E.; Woodworth, B.A.; Fokkens, W.J.; Psaltis, A.J.; Vreugde, S.; Wormald, P.-J. Activity of Bacteriophages in Removing Biofilms of Pseudomonas aeruginosa Isolates from Chronic Rhinosinusitis Patients. Front. Cell Infect. Microbiol. 2017, 7, 418. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.M.; Neill, D.R.; Kaman, B.; Sahota, J.S.; Clokie, M.R.J.; Winstanley, C.; Kadioglu, A. Phage therapy is highly effective against chronic lung infections with Pseudomonas aeruginosa. Thorax 2017, 72, 666–667. [Google Scholar] [CrossRef]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Yang, Y.; Shen, W.; Zhong, Q.; Chen, Q.; He, X.; Baker, J.L.; Xiong, K.; Jin, X.; Wang, J.; Hu, F.; et al. Development of a Bacteriophage Cocktail to Constrain the Emergence of Phage-Resistant Pseudomonas aeruginosa. Front. Microbiol. 2020, 11, 327. [Google Scholar] [CrossRef]
- Lyon, R.; Jones, R.A.; Shropshire, H.; Aberdeen, I.; Scanlan, D.J.; Millard, A.; Chen, Y. Membrane lipid renovation in Pseudomonas aeruginosa—implications for phage therapy? Environ. Microbiol. 2022, 24, 4533–4546. [Google Scholar] [CrossRef]
- Ferry, T.; Boucher, F.; Fevre, C.; Perpoint, T.; Chateau, J.; Petitjean, C.; Josse, J.; Chidiac, C.; L’hostis, G.; Leboucher, G.; et al. Innovations for the treatment of a complex bone and joint infection due to XDR Pseudomonas aeruginosa including local application of a selected cocktail of bacteriophages. J. Antimicrob. Chemother. 2018, 73, 2901–2903. [Google Scholar] [CrossRef]
- Kohler, T.; Luscher, A.; Falconnet, L.; Resch, G.; McBride, R.; Mai, Q.A.; Simonin, J.L.; Chanson, M.; Maco, B.; Galiotto, R.; et al. Personalized aerosolised bacteriophage treatment of a chronic lung infection due to multidrug-resistant Pseudomonas aeruginosa. Nat. Commun. 2023, 14, 3629. [Google Scholar] [CrossRef]
- Vaitekenas, A.; Tai, A.S.; Ramsay, J.P.; Stick, S.M.; Kicic, A. Pseudomonas aeruginosa Resistance to Bacteriophages and Its Prevention by Strategic Therapeutic Cocktail Formulation. Antibiotics 2021, 10, 145. [Google Scholar] [CrossRef]
- Hoggarth, A.; Weaver, A.; Pu, Q.; Huang, T.; Schettler, J.; Chen, F.; Yuan, X.; Wu, M. Mechanistic research holds promise for bacterial vaccines and phage therapies for Pseudomonas aeruginosa. Drug Des. Devel Ther. 2019, 13, 909–924. [Google Scholar] [CrossRef]
- Döring, G.; Pier, G.B. Vaccines and immunotherapy against Pseudomonas aeruginosa. Vaccine 2008, 26, 1011–1024. [Google Scholar] [CrossRef]
- Westritschnig, K.; Hochreiter, R.; Wallner, G.; Firbas, C.; Schwameis, M.; Jilma, B. A randomized, placebo-controlled phase I study assessing the safety and immunogenicity of a Pseudomonas aeruginosa hybrid outer membrane protein OprF/I vaccine (IC43) in healthy volunteers. Hum. Vaccin. Immunother. 2014, 10, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Rello, J.; Krenn, C.G.; Locker, G.; Pilger, E.; Madl, C.; Balica, L.; Dugernier, T.; Laterre, P.F.; Spapen, H.; Depuydt, P.; et al. A randomized placebo-controlled phase II study of a Pseudomonas vaccine in ventilated ICU patients. Crit. Care 2017, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Farrar, J.L.; Gierke, R.; Britton, A.; Childs, L.; Leidner, A.J.; Campos-Outcalt, D.; Morgan, R.L.; Long, S.S.; Talbot, H.K.; et al. Use of 15-Valent Pneumococcal Conjugate Vaccine and 20-Valent Pneumococcal Conjugate Vaccine Among U.S. Adults: Updated Recommendations of the Advisory Committee on Immunization Practices—United States, 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Kinoshita, M.; Muranishi, K.; Ohara, J.; Sudo, K.; Kawaguchi, K.; Shimizu, M.; Naito, Y.; Moriyama, K.; Sawa, T. Effect of a Novel Trivalent Vaccine Formulation against Acute Lung Injury Caused by Pseudomonas aeruginosa. Vaccines 2023, 11, 1088. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Ito, E.; Nguyen, V.H.; Haight, M. Anti-PcrV antibody strategies against virulent Pseudomonas aeruginosa. Hum. Vaccin. Immunother. 2014, 10, 2843–2852. [Google Scholar] [CrossRef]
- Wan, C.; Zhang, J.; Zhao, L.; Cheng, X.; Gao, C.; Wang, Y.; Xu, W.; Zou, Q.; Gu, J. Rational Design of a Chimeric Derivative of PcrV as a Subunit Vaccine Against Pseudomonas aeruginosa. Front. Immunol. 2019, 10, 781. [Google Scholar] [CrossRef]
- Naito, Y.; Hamaoka, S.; Kinoshita, M.; Kainuma, A.; Shimizu, M.; Katoh, H.; Moriyama, K.; Ishii, K.J.; Sawa, T. The protective effects of nasal PcrV-CpG oligonucleotide vaccination against Pseudomonas aeruginosa pneumonia. Microbiol. Immunol. 2018, 62, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Kinoshita, M.; Sudo, K.; Inoue, K.; Naito, Y.; Oba, M.; Uchida, S.; Sawa, T. Mrna Vaccine Induces Prot. Immun. Against Type III Secret. Virulence Pseudomonas aeruginosa. biorXiv 2023. Available online: https://www.biorxiv.org/content/10.1101/2023.06.09.544431v1 (accessed on 23 January 2024).
- Wang, X.; Liu, C.; Rcheulishvili, N.; Papukashvili, D.; Xie, F.; Zhao, J.; Hu, X.; Yu, K.; Yang, N.; Pan, X.; et al. Strong immune responses and protection of PcrV and OprF-I mRNA vaccine candidates against Pseudomonas aeruginosa. NPJ Vaccines 2023, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Reddy, S.; Barathe, P.; Shriram, V.; Anand, U.; Proćków, J.; Kumar, V. Combating Drug-Resistant Bacteria Using Photothermally Active Nanomaterials: A Perspective Review. Front. Microbiol. 2021, 12, 747019. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Li, B.; Luo, T.; Nie, C.; Pan, W.; Ge, X.; Zheng, J.; Rui, Y.; Zheng, L. Selective Photothermal Therapy Based on Lipopolysaccharide Aptamer Functionalized MoS(2) Nanosheet-Coated Gold Nanorods for Multidrug-Resistant Pseudomonas aeruginosa Infection. Adv. Healthc. Mater. 2023, 12, e2202794. [Google Scholar] [CrossRef] [PubMed]
- Naskar, A.; Kim, K.S. Friends against the Foe: Synergistic Photothermal and Photodynamic Therapy against Bacterial Infections. Pharmaceutics 2023, 15, 1116. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, Y.; Chen, Y.; Liu, L.; Mo, A.; Peng, Q. Nanomaterials-based photothermal therapy and its potentials in antibacterial treatment. J. Control Release 2020, 328, 251–262. [Google Scholar] [CrossRef]
- Fan, S.; Lin, W.; Huang, Y.; Xia, J.; Xu, J.F.; Zhang, J.; Pi, J. Advances and Potentials of Polydopamine Nanosystem in Photothermal-Based Antibacterial Infection Therapies. Front. Pharmacol. 2022, 13, 829712. [Google Scholar] [CrossRef]
- Nam, J.; Son, S.; Ochyl, L.J.; Kuai, R.; Schwendeman, A.; Moon, J.J. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat. Commun. 2018, 9, 1074. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, M.; Huang, Z.; Sun, Y.; Ye, L. Molecularly Imprinted Polymers for Targeting Lipopolysaccharides and Photothermal Inactivation of Pseudomonas aeruginosa. ACS Appl. Polym. Mater. 2023, 5, 3055–3064. [Google Scholar] [CrossRef]
- Patangia, D.V.; Ryan, C.A.; Dempsey, E.; Ross, R.P.; Stanton, C. Impact of antibiotics on the human microbiome and consequences for host health. Microbiologyopen 2022, 11, e1260. [Google Scholar] [CrossRef]
- Lewis, K. The Science of Antibiotic Discovery. Cell 2020, 181, 29–45. [Google Scholar] [CrossRef]
- Grace, A.; Sahu, R.; Owen, D.R.; Dennis, V.A. Pseudomonas aeruginosa reference strains PAO1 and PA14: A genomic, phenotypic, and therapeutic review. Front. Microbiol. 2022, 13, 1023523. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Carranza, E.; García-Reyes, S.; González-Valdez, A.; Soberón-Chávez, G. Tracking the genome of four Pseudomonas aeruginosa isolates that have a defective Las quorum-sensing system, but are still virulent. Access Microbiol. 2020, 2, acmi000132. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, L.B.; Brüggemann, H.; de Sales, R.O.; Koga, P.C.M.; de Souza, A.V.; Martino, M.D.V.; Galhardo, R.S.; Severino, P. Mutagenesis Induced by Sub-Lethal Doses of Ciprofloxacin: Genotypic and Phenotypic Differences Between the Pseudomonas aeruginosa Strain PA14 and Clinical Isolates. Front. Microbiol. 2019, 10, 1553. [Google Scholar] [CrossRef]
- Macdonald, E.; Wright, R.; Connolly, J.P.R.; Strahl, H.; Brockhurst, M.; van Houte, S.; Blower, T.R.; Palmer, T.; Mariano, G. The novel anti-phage system Shield co-opts an RmuC domain to mediate phage defense across Pseudomonas species. PLoS Genet. 2023, 19, e1010784. [Google Scholar] [CrossRef] [PubMed]
- Forti, F.; Roach, D.R.; Cafora, M.; Pasini, M.E.; Horner, D.S.; Fiscarelli, E.V.; Rossitto, M.; Cariani, L.; Briani, F.; Debarbieux, L.; et al. Design of a Broad-Range Bacteriophage Cocktail That Reduces Pseudomonas aeruginosa Biofilms and Treats Acute Infections in Two Animal Models. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Chan, B.K.; Turner, P.E.; Kim, S.; Mojibian, H.R.; Elefteriades, J.A.; Narayan, D. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evol. Med. Public. Health 2018, 2018, 60–66. [Google Scholar] [CrossRef]
- Chan, B.K.; Sistrom, M.; Wertz, J.E.; Kortright, K.E.; Narayan, D.; Turner, P.E. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 26717. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).