1. Introduction
Tuberculosis (TB), caused by
M. tuberculosis, is an ancient infectious disease. Recent data released by the World Health Organization show that around 10 million people fell in with the disease every year worldwide [
1]. The increasing spread of drug-resistant
M. tuberculosis makes TB treatment more difficult, and drug resistance has become one of the major challenges. The best way to solve the above problem is to introduce new anti-TB drugs. However, no new first-line drug has been introduced in clinical TB treatment for more than 50 years, since rifampicin [
2]. Therefore, rational use of existing anti-tuberculosis drugs is necessary. In addition, researchers also have made efforts in using phages as an individual or supplementary therapy to treat
M. tuberculosis infections [
3].
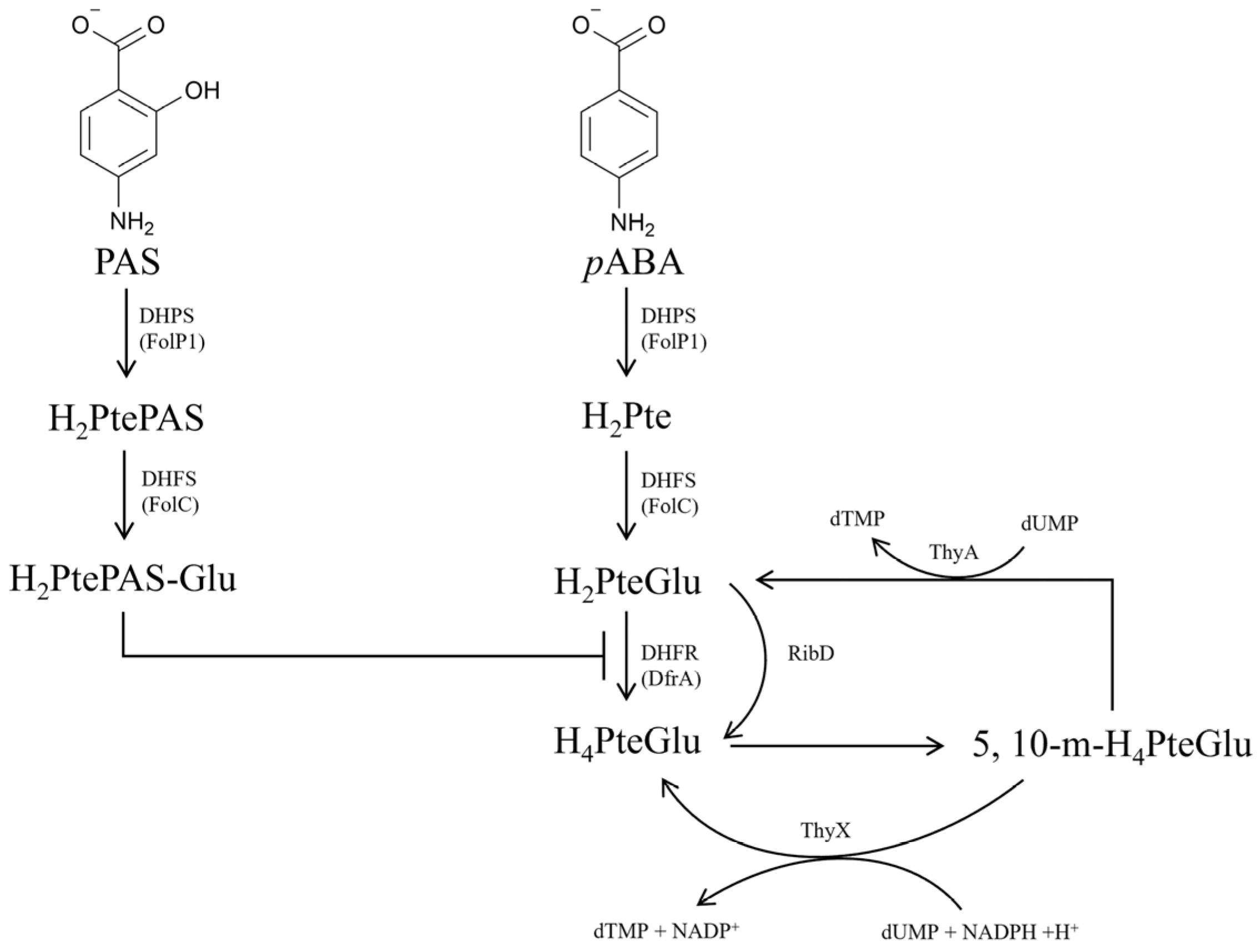
Folate is an essential nutrient for all sorts of life. Bacteria need to synthesize folate de novo, but mammals are unable to synthesize it, which makes the bacterial de novo folate biosynthesis pathway an ideal target for developing new antibacterial drugs [
4]. As is well known, dihydropteroate (H
2Pte) is synthesized by dihydropteroate synthetase (DHPS, FolP) using
para-aminobenzoic acid (
pABA) and 7,8-dihydropterin pyrophosphate (H
2PtePP) as substrates, which is further converted into dihydrofolate (H
2PteGlu) by FolC (
Figure 1) [
5]. Dihydrofolate reductase (DHFR, DfrA or RibD) and thymidylate synthase (ThyA or ThyX) maintain the interconversion and balance between H
2PteGlu, H
4PteGlu and 5, 10-methylenetetrahydrofolate (5, 10-m-H
4PteGlu) (
Figure 1). PAS was first used as a first-line anti-TB drug in 1946 [
6], and is presently still used for treating multiple drug-resistant TB [
7]. The mechanism of action of PAS had been gradually discovered over 70 years of clinical utilization. As a structural analogue of
pABA, PAS is firstly catalyzed by the FolP1 of
M. tuberculosis to form H
2PtePAS, an analogue of H
2Pte. Subsequently, H
2PtePAS was further catalyzed by the FolC, yielding H
2PtePAS-Glu [
5] (
Figure 1). Ultimately, H
2PtePAS-Glu inhibited the activity of
M. tuberculosis DfrA (
Figure 1), resulting in bacterial growth inhibition and cell death [
8].
Although the mechanism of PAS action has been elucidated, its mechanisms of resistance still await investigation. Until the present, confirmed molecular markers associated with PAS resistance in
M. tuberculosis clinical isolates included mutations of
folC [
9,
10,
11],
thyA [
9,
11,
12,
13], and
ribD [
8,
9,
11]. Among them,
folC or
thyA gene mutations were the main reasons for PAS resistance, accounting for two-thirds of the PAS resistant clinical isolates [
9,
11,
14]. Molecular mechanisms of PAS resistance caused by
folC and
ribD mutations have been elucidated [
8,
10]. Our previous research showed that H
2Pte binding pocket variants of FolC failed to activate H
2PtePAS to H
2PtePAS-Glu, hindering the activation of PAS and hence conferring resistance to PAS [
10]. On the other hand,
ribD could serve as an alternative for DHFR, as mutations in the promoter region of the gene could cause over-expression of
ribD, and thus lead to PAS resistance [
8]. However, the molecular mechanism of PAS resistance caused by
thyA mutations still remains unclear, though the association between
thyA mutations and PAS resistance has been established for nearly two decades [
13]. According to the data of epidemiological analysis,
thyA mutations were identified in about 1/3 of the PAS resistant
M. tuberculosis clinical isolates [
9,
11,
12]. Thus, unravelling the mechanism of PAS resistance caused by
thyA mutations will broaden our understanding of folate metabolism in
M. tuberculosis and be useful for guiding the clinical administration of PAS. To elucidate how
thyA mutations caused PAS resistance in
M. tuberculosis, the
thyA gene was deleted in H37Ra using the phage-mediated allelic exchange method, and a clinical PAS resistant isolate F461 harboring the
thyA R235P mutation was selected [
14]. Subsequently, the effect of
thyA deletion on bacterial H
4PteGlu content was determined by UPLC-MS/MS. Then, the competition for catalysis of FolC between H
4PteGlu and H
2PtePAS was analyzed by in vitro enzymatic activity assays. Meanwhile,
folC was over-expressed in the
thyA deletion mutant and the selected PAS resistant clinical isolate, PAS susceptibilities of these two strains were tested. The level of FolC in ThyA deficiency strain was explored by RNA-seq and Western blot assays. The results are presented herein.
3. Discussion
Folates, especially derivatives of H
4PteGlu, are one carbon carriers required by the biosynthesis of purines, thymidylate, methionine, serine, and glycine, thus making them essential for all sorts of lives [
20,
21]. Bacteria must synthesize these essential cofactors
de novo, while mammal can intake them from their diet [
4]. This difference makes the bacterial
de novo folate biosynthesis pathway an ideal target for developing new antibacterial drugs [
4]. Although thousands of folates antagonists have been designed for folate biosynthesis pathway heretofore, PAS is the only one used for TB treatment with a unique mode of action only observed in
M. tuberculosis complex. Thus, better understanding the mechanisms of PAS resistance in
M. tuberculosis will benefit the development of new antifolates against this bacterium.
As the first molecular marker for PAS resistance in
M. tuberculosis clinical isolates,
thyA gene mutations have been identified for nearly two decades [
13], but the molecular mechanism of how these mutations lead to PAS resistance remains unknown. Ten years later, when probing the molecular mechanism of PAS resistance caused by
folC mutation [
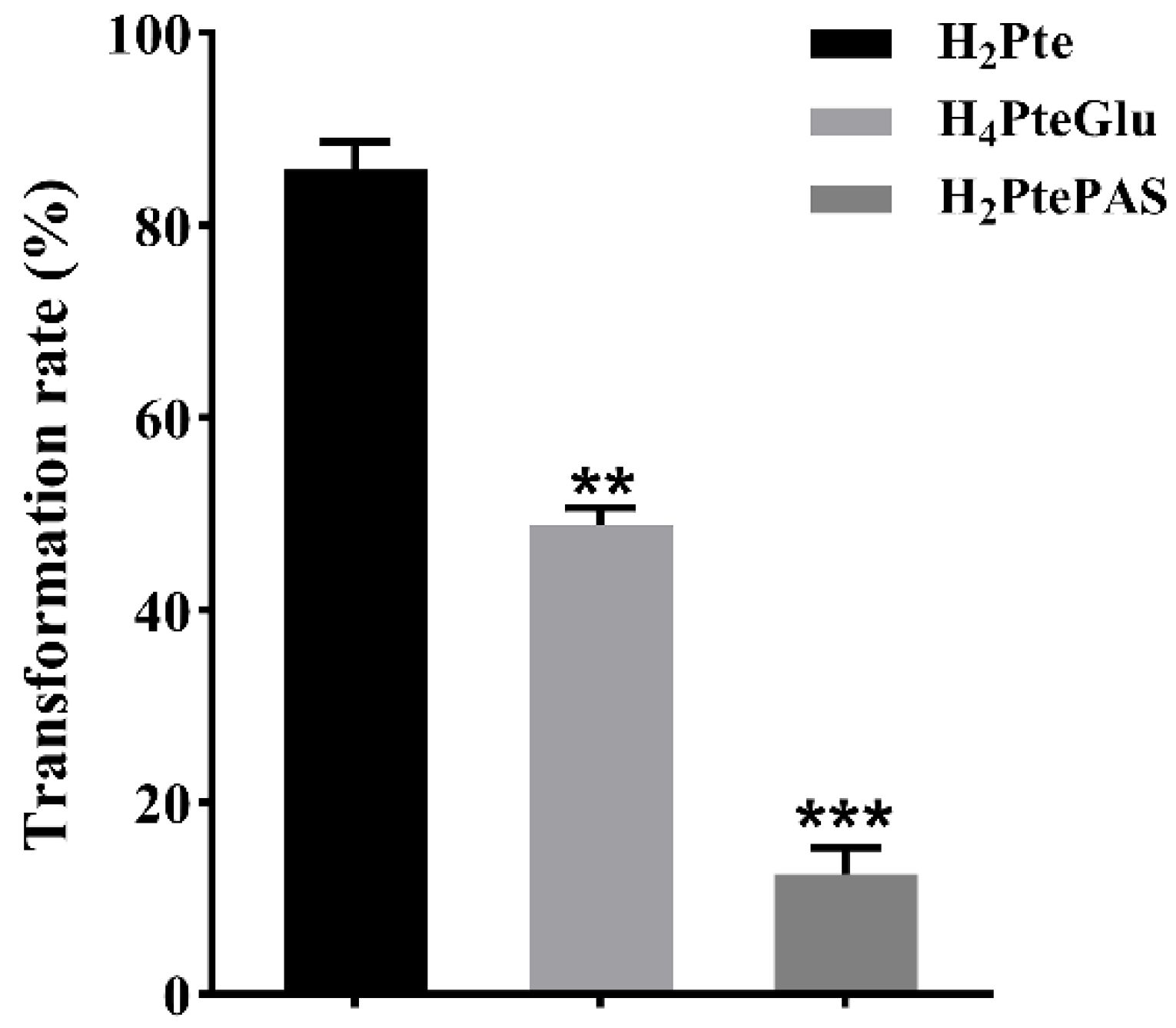
10], we noticed that though FolC could also catalyze the conversion of H
2PtePAS to H
2PtePAS-Glu, but the catalytic efficiency was much lower than that of the natural substrate H
2Pte, implying that the bio-activation process of PAS might be vulnerable to interference of natural metabolite of folate biosynthesis. Indeed, exogenous H
2Pte made
M. tuberculosis more resistant to PAS [
10]. Previous studies have showed that FolC could not only convert H
2Pte into H
2PteGlu, but also add glutamic acid tail to H
4PteGlu in
E. coli [
18,
19]. In this study, we found that
M. tuberculosis FolC is also bifunctional. In addition, its catalytic efficiency for H
2PtePAS is remarkably lower than that for H
4PteGlu (
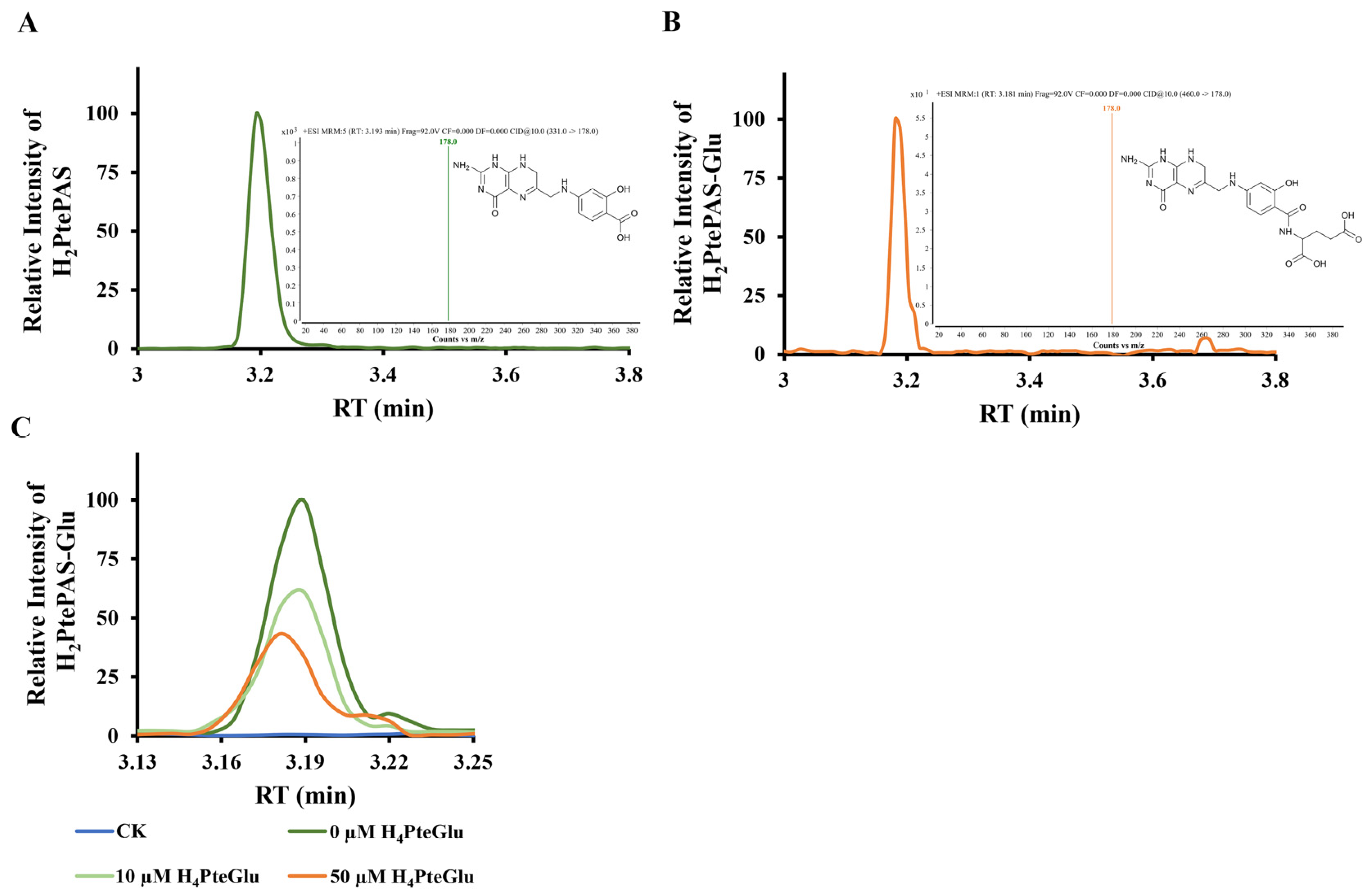
Figure 4), implying intracellular H
4PteGlu may interfere the activation of PAS by FolC. As expected, the in vitro biochemical experiments showed that H
4PteGlu hinders the conversion of H
2PtePAS to H
2PtePAS-Glu in a concentration-dependent manner (
Figure 5C). Since
M. tuberculosis is not able to intake exogenous H
4PteGlu, it is not possible to test the effect of exogenous H
4PteGlu on PAS susceptibility. Alternatively, we compared the H
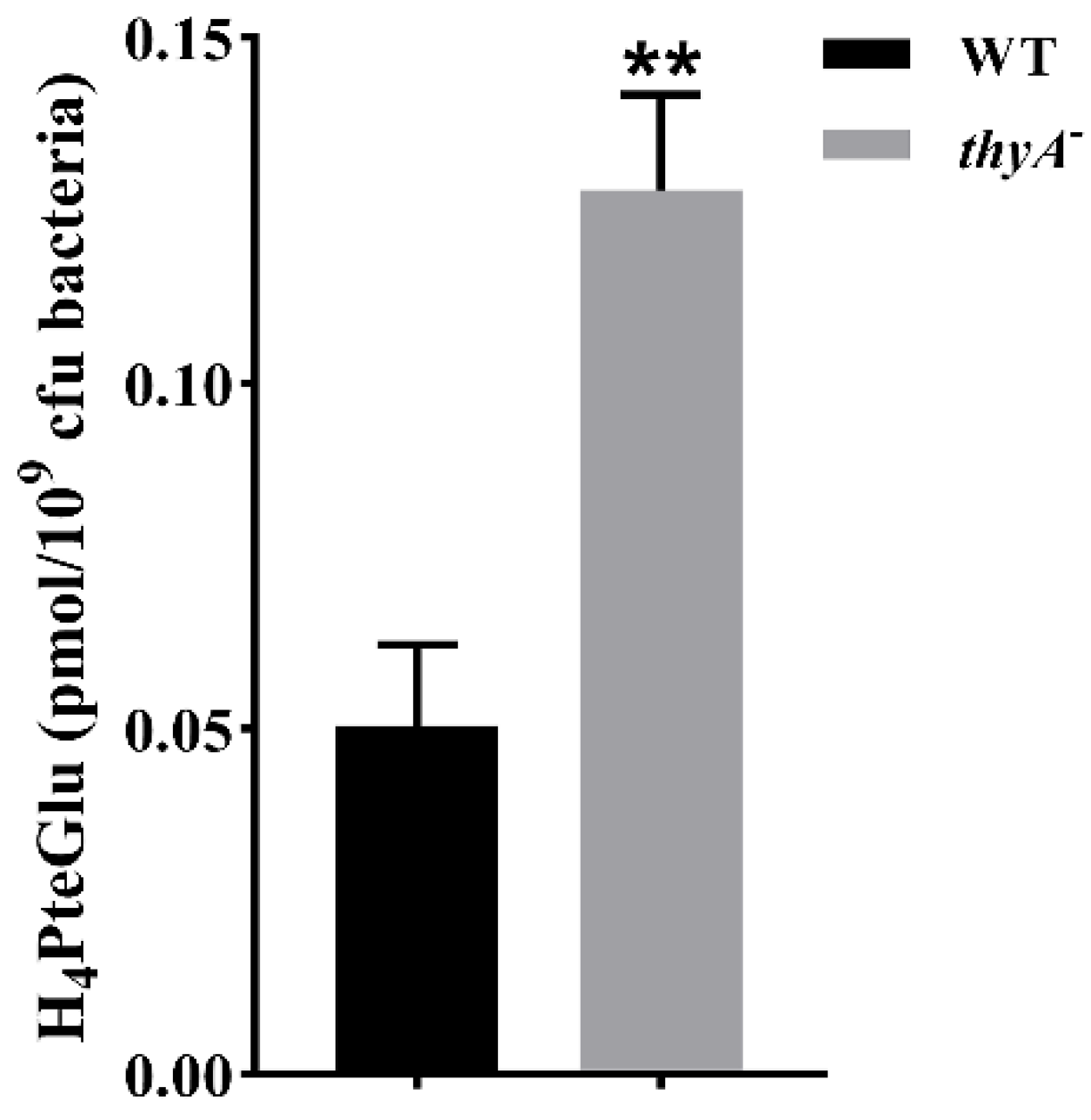
4PteGlu content between H37Ra and the
thyA deletion mutant, and found that the H
4PteGlu content in the
thyA deletion mutant was significantly higher than that of the wild-type strain (
Figure 3). This is not surprising since the bacterium has to solely rely on ThyX to synthesize thymidylate in the absence of ThyA, and utilization of the former yields H
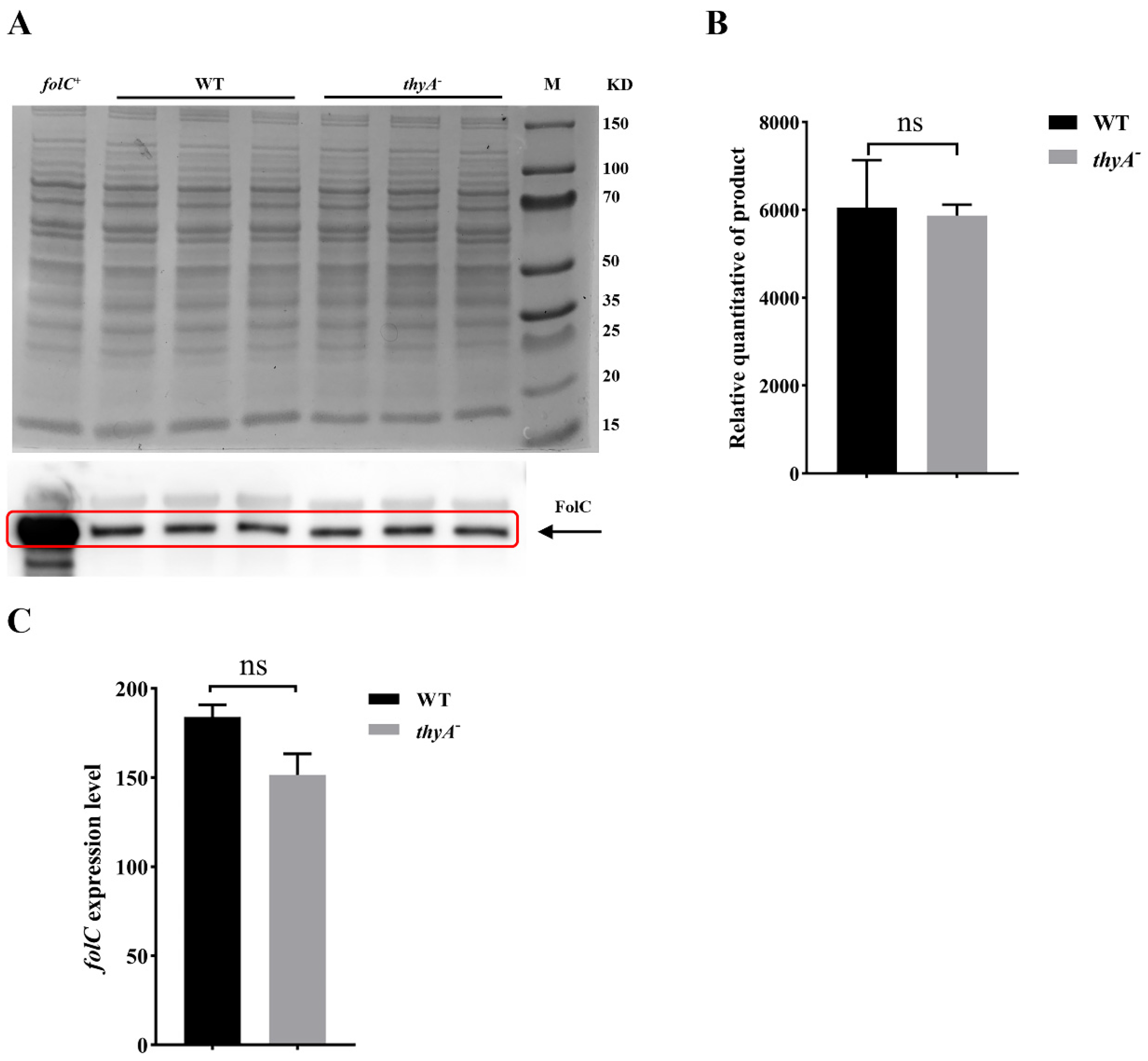
4PteGlu. Since the expression level of FolC remained unchanged in the
thyA deletion mutant, increased H
4PteGlu content could hinder the conversion of H
2PtePAS since they compete for the same protein. Correspondingly, this competition could be mitigated by over-expression of the target protein FolC. As expected, over-expression of
folC could reverse the PAS resistance phenotype caused by
thyA deletion or clinical
thyA R235P mutation (
Table 2). We noticed that the PAS resistance phenotype caused by
thyA deletion or mutation could only be partially restored by
folC over-expression, suggesting the existence of other mechanisms for PAS resistance caused by functional deficiency of ThyA.
When assessing whether the resistance to PAS of the
thyA deletion mutant was related to the efficiency of PAS activation, we over-expressed
folP1,
folC, and
dfrA in H37Ra and H37Ra Δ
thyA. To our surprise, over-expression of
dfrA in the
thyA deletion mutant did not affect the susceptibility to PAS (
Table 2), suggesting either the lack of DfrA protein or loss of function of DfrA in the
thyA deletion mutant. Previous works also showed that
thyA and
dfrA double deletion mutants had been identified in
M. tuberculosis clinical isolates from different countries [
11,
22]. Thus, in the absence of
thyA,
M. tuberculosis discards the commonly used DHFR Rv2763c (DfrA), and switches to another alternative to synthesize H
4PteGlu. Although RibD was shown to be an alternative DHFR in
M. tuberculosis, previous research revealed that RibD could only replace DfrA when it was highly over-expressed in a multi-copy plasmid [
8], suggesting that the dihydrofolate reductase activity of RibD is quite low, which was confirmed by subsequent biochemical analysis [
23]. Zheng et al. found that mutations in the promoter region of
ribD could cause over-expression of
ribD [
8]. To determine whether RibD is the alternative DHFR in the absence of ThyA in
M. tuberculosis, we further analyzed genome sequences of isolates with frameshift or deletion mutations in
thyA or
dfrA genes from previous studies and NCBI database. The results showed that there was no mutation in either the promoter region (300 bp upstream start codon) or the coding sequence (CDS) of the
ribD gene in ThyA or DfrA deficient clinical isolates (
Supplementary Table S2). Moreover, our RNA-seq data also showed that the expression level of
ribD remained unchanged in the
thyA deletion mutant (
Figure S2). Therefore, RibD is not the alternative DHFR in the absence of ThyA. What the alternative DHFR is in the absence of ThyA requires further investigation. It will be important to test if the alternative DHFR would be more resistant to the inhibition of H
2PtePAS-Glu, since over-expressing
folC could only partially restore PAS sensitivity to the
thyA deletion mutant.
Previous studies already showed that the C
−16T mutation in the upstream regulatory region of
thyX could lead to increased expression of
thyX and PAS resistance in
M. tuberculosis [
24,
25]. Thus, it is not surprising to see that over-expressing
thyX led to PAS resistance in H37Ra. The fact that over-expressing
thyX in the
thyA deletion mutant did not affect PAS susceptibility of the latter indicated that over-expressing
thyX and deleting
thyA in
Mtb might share the same mechanism of PAS resistance. In addition, over-expressing
thyA in H37Ra also led to low level PAS resistance. Considering the role of ThyA in folate salvage, we speculated that the intracellular H
2PteGlu content might be increased when over-expressing
thyA; this would in turn reduce the demand for dihydrofolate biosynthesis through FolC. Previous studies showed that FolC was critical for the bio-activation of PAS, and decreased FolC enzymatic activity caused PAS resistance [
8,
10].
In conclusion, our results showed that functional deficiency of ThyA led to increased H4PteGlu content of the bacterial cells, which competed with H2PtePAS for FolC catalysis, thus hindered the activation of PAS and conferred PAS resistance in M. tuberculosis. Meanwhile, our study also suggested that M. tuberculosis could switch from Rv2763c to a yet unknown alternative DHFR in the absence of thyA, and further investigation is required to identify the protein and elucidate its role on PAS resistance caused by ThyA functional deficiency. Our study broadens the understanding of folate metabolism in M. tuberculosis and might be useful for guiding the clinical administration of PAS.
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, and Growth Conditions
Clinical isolate F461,
M. tuberculosis H37Ra and its derivative strains were cultured at 37 °C in 7H9 broth (Difco, St. Louis, MO, USA) supplemented with 10% (
v/
v) oleic acid-albumin-dextrose-catalase (OADC, Difco), 0.5% (
v/
v) glycerol, and 0.05% (
v/
v) Tween 80 (Sigma-Aldrich, St. Louis, MO, USA), or on 7H10 agar medium (Difco) supplemented with 10% (
v/
v) OADC and 0.5% (
v/
v) glycerol.
Mycobacterium smegmatis mc
2155 was grown in Middlebrook 7H9 medium or 7H10 agar medium.
E. coli strains HB101 and BL21 (DE3) were cultured in Luria-Bertani (LB) medium (Difco), or on LB agar plates at 37 °C. Plasmids pMAL-c2X (New England BioLabs, Beverly, MA, USA), pET-28a (Novagen, Madison, WI, USA), and pMV261 were used for the construction of expression plasmids. All bacteria strains, plasmids, and primers used in this study are described in detail in
Supplementary Table S3.
4.2. Antibiotics and Chemicals
These concentrations of antibiotics (75 μg mL−1 and 150 μg mL−1 hygromycin (Sigma-Aldrich), 25 μg mL−1 and 100 μg mL−1 Kanamycin (MD Bio, Inc., Qingdao, China), and 150 μg mL−1 ampicillin (MD Bio, Inc.)) were used to culture bacteria, unless otherwise indicated. H2Pte and H4PteGlu were purchased from Schircks Laboratories. PAS (Sigma-Aldrich) was used at indicated concentrations.
4.3. Genetic Manipulation of Mycobacterial Strains
folC,
thyA,
thyX,
dfrA, and
folP1 were amplified from wild-type
M. tuberculosis H37Ra genomic DNA using PCR with the primers (
Supplementary Table S3). The purified amplicon was digested and ligated to pMV261, generating pMV261-
folC, pMV261-
thyA, pMV261-
dfrA, pMV261-
folP1, and pMV261-
thyX.
M. tuberculosis strain was transformed with sequence-confirmed pMV261 recombinant plasmid, then plated on 7H10 medium containing 25 μg mL
−1 kanamycin. After 3 weeks of incubation at 37 °C, single colonies were purified and liquid cultures were prepared for the extraction of genomic DNA and determination of PAS MICs, separately. The presence of pMV261 recombinant plasmid was verified by PCR amplification using primers specific for pMV261-JDFP and pMV261-JDRP (
Supplementary Table S3).
A modified strategy for phage-mediated allelic exchange [
26] was used to construct
M. tuberculosis H37Ra Δ
thyA mutant. Briefly, the native copy of
thyA was deleted by specialized transduction using phAE159 containing a hygromycin resistance cassette. All primers used are listed in
Supplementary Table S3.
4.4. Purification of Recombinant FolP1 and FolC
FolP1 and FolC proteins were purified as previously reported [
10]. Briefly,
folP1 and
folC were amplified from
M. tuberculosis H37Ra genomic DNA using specific primers (
Supplementary Table S3) and separately cloned into pET28a to yield pET28a::
folP1 to introduce an N-terminal hexa-histidine tag and into pMAL-c2X to yield pMAL-c2X::
folC to introduce an N-terminal maltose-binding protein (MBP) tag linked with a factor Xa cleavage site. The sequence-confirmed recombinant plasmids were transformed into
E. coli BL21 (DE3). The cells were grown at 37 °C in LB broth containing 150 μg mL
−1 ampicillin or 100 μg mL
−1 kanamycin to an OD
600 of ~0.6. Isopropyl-β-D-thiogalactopyranoside (IPTG, Acmec, China) was added to 0.25 mM, then the cells were incubated further at 16 °C for 20 h. The bacterial cells were harvested by centrifugation, disrupted by sonication, and clarified by centrifugation.
Recombinant FolP1 protein was purified over prewashed nickel–nitrilotriacetic acid HisTrap HP affinity resin (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Nonspecifically bound protein was removed by washing the resin with 50 mM Tris-HCl, 0.5 M NaCl, and 60 mM imidazole (pH 8.0). Recombinant FolP1 was eluted with 50 mM Tris-HCl, 0.5 M NaCl, and 400 mM imidazole (pH 8.0), and analyzed by SDS-PAGE.
Recombinant FolC proteins were first purified over an amylose resin column (New England BioLabs). The FolC protein obtained from the first purification contains MBP tag. To remove the MBP tag, the purified samples were incubated with factor Xa at 4 °C overnight in reaction buffer (20 mM HEPES (pH 8.0), 100 mM NaCl, 2 mM CaCl2, and 10% glycerol). Then, the cleavage mixtures were dialyzed against 50 mM phosphate buffer (pH 8.0). The samples were loaded on a HiTrap DEAE FF column (GE Healthcare), and a step gradient from 50 mM to 1 M NaCl in phosphate buffer was applied to elute FolC. The fractions were then analyzed by SDS-PAGE. Recombinant FolC was eluted with 300 mM NaCl.
4.5. Western Blot Assay
The H37Ra and H37Ra ΔthyA strains were cultured at 37 °C in 10 mL of 7H9 medium and harvested at logarithmic phase by centrifugation. For Western blot analysis, bacterial cells were resuspended in phosphate buffer saline (PBS, pH 7.0), then lysed using zirconium beads. Protein samples acquired from the supernatant after centrifugation. The protein concentration of the supernatant was determined using the NanoDrop2000 (Thermo, Waltham, MA, USA). Then, the protein samples were separated by SDS-PAGE and immediately transferred to a polyvinylidene difluoride membrane (Merck Millipore, Darmstadt, Germany) by a Bio-Rad SD device (Bio-Rad Laboratories, Hercules, CA, USA) at 15 V for 30 min. Finally, the proteins were probed with rabbit FolC polyclonal antibody (ABclonal biotechnology, Wuhan, China, Cat. No. WG-00133D).
4.6. RNA-Seq Analysis
Mycobacterial strains were grown in 7H9 to mid logarithmic phase and were collected by centrifugation. Total RNA was extracted using RNeasy mini kit (Qiagen, Hilden, Germany). Library constructions were prepared using TruSeq Stranded Total RNA Sample Preparation kit (Illumina, San Diego, CA, USA), and RNA sequencing was conducted on Illumina NovaSeq6000 at Beijing Novogene Corporation. The insert size conformation of purified libraries was validated by an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Bowtie2 was used to map the cleaned reads to the M. tuberculosis H37Ra genome acquired from the National Center for Biotechnology Information (NCBI) (
https://www.ncbi.nlm.nih.gov/nuccore/CP000611.1) (Accessed on 25 May 2023). Then, HTSeqv0.6.1 was run with a reference annotation to generate fragments per kilobase of exon model per million mapped reads values for estimation of fold changes. Three biological replicates were used in RNA-seq and the
p- and q-values were calculated. The differentially expressed genes were selected using the following filter criteria: q-value < 0.005 and |log2 (fold change)| > 1. Raw RNA sequencing data have been deposited at NCBI Sequence Read Archive, Accession PRJNA1005084.
4.7. In Vitro Enzymatic Activity Assays
The dihydrofolate synthase activities of FolC using H
2Pte, H
2PtePAS, and H
4PteGlu as substrates were measured and H
2PtePAS was enzymatically synthesized as previously described [
5,
10]. Briefly, the reaction mixture contained 1.2 µM FolP1, 40 mM Tris-20 mM glycine (pH 9.5), 5 mM MgCl
2, 1 mM DTT, 200 mM NaCl, appropriate amounts of 6-hydroxymethyl-7,8-pterin pyrophosphate (H
2PtePP), and 250 µM PAS. The reaction mixture was incubated at 37 °C until no increment of H
2PtePAS accumulation was detected by UPLC-MS/MS. FolP1 was removed by passing through a 10-kDa Microcon centrifugal filter, and 325 µL of the remaining reaction mixture was used as a substrate for FolC. The FolC reaction mixture contained 0.5 µM FolC protein, 2.5 mM ATP, and 0.5 mM L-glutamate in 100 mM Tris-50 mM glycine (pH 9.5), 10 mM MgCl
2, 5 mM DTT, 100 mM KCl, 50 mM NaCl, 10% glycerol, appropriate amounts H
2PtePAS, and the presence/absence of H
4PteGlu. The mixture was incubated at 37 °C for 15 min. H
2Pte, H
2PtePAS, and H
4PteGlu were identified by UPLC-MS/MS. UPLC column was Waters ACQUITY UPLC HSS T3 Column (2.1 × 100 mm, 1.8 μm particles) using a flow rate of 0.4 mL/min at 40 °C during a 6 min gradient (0–1 min from 2% B to 1% B, 1–3.5 min from 1% B to 50% B, 3.5–3.8 min from 50% B to 95% B, 3.8–6 min 95% B), while using the solvents A (water containing 20 mM ammonium acetate) and B (methanol). Electrospray ionization was performed using the positive ion mode, the pressure of the nebulizer was 30 psi, the dry gas temperature was 325 °C with a flow rate of 11 L/min, the sheath gas temperature was 350 °C with a flow rate of 10 L/min, and the capillary was set at 4000 V. Multiple reaction monitoring (MRM) was used for the quantification of screening fragment ions. Peak determination and peak area integration were performed using Mass Hunter Workstation software (Agilent, Version B.08.00).
p-values (
p) were calculated using
t-tests. The graphs for the transformation rate of H
2Pte, H
4PteGlu, and H
2PtePAS were prepared using GraphPad Prism.
4.8. Drug Susceptibility Testing
Mycobacterial cells were cultured to mid-log phase (OD
600: 0.5–1.0) and diluted to about 10
5 cfu mL
−1 using 10-fold serial dilutions in fresh 7H9 medium with or without 10% OADC. Then, bacterial cells were plated on 7H10 agar solid plates containing various concentrations of PAS (0, 0.00125, 0.0025, 0.005, 0.01, 0.02, 0.04, 0.08, 0.16, 0.32, 0.64, 1.28, 2.56, 5.12, 10.24, 20.48, 40.96, and 81.92 μg mL
−1). PAS was purchased from Sigma-Aldrich and solubilized according to the manufacture’s recommendations. Plates were then incubated at 37 °C for 21 days. The MIC was defined as the lowest concentration of antibiotics required to inhibit 99% of CFUs after this culture period. The MICs were performed through two technical repetitions using three biological replicates. All of the bacteria strains used are listed in
Supplementary Table S3.
4.9. Determination of H4PteGlu Content In Vivo
Bacteria samples (~5 × 109 cfu) were re-suspended in 0.4 mL pre-cooled 20 mM HEPES (containing 2% vitamin C and 1% dithiothreitol, pH 7.0) and subjected to three liquid nitrogen freeze–thaw cycles and zirconia bead grinding before sonication in an ice bath for 15 cycles (1 min pulse followed by 1 min pause). The above extraction procedure was repeated three times. The mixture was then centrifuged for 10 min at 12,000× g at 4 °C, and each supernatant was filtered using a 0.22 µm membrane filter before UPLC-MS/MS analysis. The samples were detected as above with some changes. Briefly, the samples (5 μL) were individually injected on an UPLC column (Agilent ZORBAX Eclipse Plus C18 column, 2.1 × 100 mm, 1.8 μm particles) using a flow rate of 0.4 mL/min at 50 °C using the solvents A (water containing 0.1% (v/v) formic acid) and B (methanol containing 0.1% (v/v) formic acid). The bacterial biomasses of the individual samples were determined by colony counting method. All data obtained by metabolomics were averaged from the independent sextuplicates. p-values (p) were calculated using t-tests. The graphs for the determination of H4PteGlu in vivo were prepared using GraphPad Prism.
4.10. Comparative Analysis of Variants in M. tuberculosis Genomes
M. tuberculosis clinical isolates with complete or partial deletion of
thyA or
dfrA were extensively collected from previous studies [
11,
22,
27] and the NCBI database (
https://www.ncbi.nlm.nih.gov/genome/browse#!/prokaryotes/mycobacterium%20tuberculosis) (Accessed on 7 December 2022). A total of 31
M. tuberculosis genomes from clinical isolates were obtained, and the mutations in the promoter region (300 bp upstream start codon) or the CDS of
ribD were analyzed in these isolates (
Supplemental Table S2). All of the raw reads were available. The acquired reads were subjected to quality assessment using FastQC v.0.11.9. Subsequently, low-quality sequences were removed and trimmed using fastp. Reads shorter than 50 bp were discarded, the last 10 bp were trimmed, and bases with an average quality below 25 were removed using a sliding window of 20 bp. Finally, variant calling against the
M. tuberculosis H37Rv (NC_000962.3) genome was performed using the Snippy pipeline.
4.11. Statistical Analysis
GraphPad Prism 8.0.1 was used to analyze all experimental data, adopting the two-tailed unpaired t-test method. Mean ± standard deviation (SD) was adopted to express the experimental data.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}