
Genomic Analysis of Mycobacterium tuberculosis Strains Resistant to Second-Line Anti-Tuberculosis Drugs in Lusaka, Zambia


 ,
,  , , ,
, , ,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Results
2.1. Study Samples
2.2. Resistance Profiles
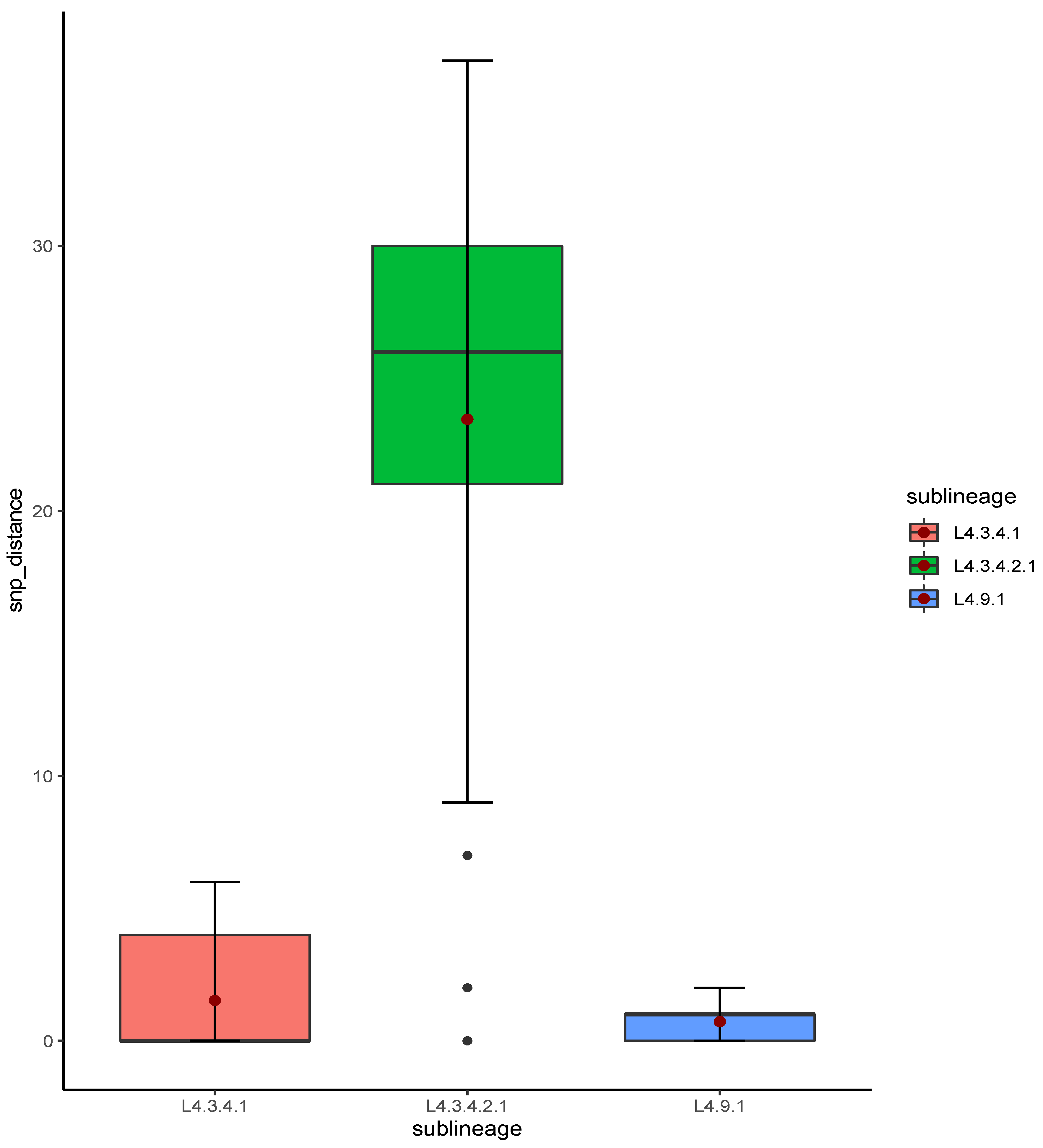
2.3. Genotypes, Phylogeny and Clustering
Phylogenetic Analysis of Global Strains Belonging to Sub-Lineages L4.3.4.1 and L4.3.4.2.1
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Drug Susceptibility Testing and DNA Extraction
4.3. Targeted Sequencing of Drug-Resistance Associated Genes
4.4. Whole Genome Sequencing
- Library preparation and sequencing
- b.
- Data Analysis: Clustering, phylogenetic analysis and resistance patterns
- c.
- Phylogenetic analysis of global strains belonging to sub-lineages L4.3.4.1 and L4.3.4.2.1
4.5. Drug Susceptibility Testing to Second Line Drugs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Global Tuberculosis Report 2021. 2021. Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 2 February 2022).
- WHO Annex 2: Country Profiles for 30 High-TB Burden Countries. 2017, 1–61. Available online: www.who.int/tb/data (accessed on 22 November 2021).
- WHO Annex 2 Country Profiles for 30 High-TB Burden Countries. 2016. Available online: www.who.int/tb/data (accessed on 10 October 2021).
- WHO Annex 2 Country Profiles for 30 High-TB Burden Countries. 2018. Available online: www.who.int/tb/data (accessed on 10 March 2023).
- Palomino, J.C.; Martin, A. Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics 2014, 3, 317–340. [Google Scholar] [CrossRef] [PubMed]
- Seung, K.J.; Keshavjee, S.; Rich, M.L. Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb. Perspect. Med. 2015, 5, a017863. [Google Scholar] [CrossRef] [PubMed]
- Alipanah, N.; Jarlsberg, L.; Miller, C.; Linh, N.N.; Falzon, D.; Jaramillo, E.; Nahid, P. Adherence interventions and outcomes of tuberculosis treatment: A systematic review and meta-analysis of trials and observational studies. PLoS Med. 2018, 15, e1002595. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Frequently Asked Questions about the Implementation of the New WHO Recommendation on the Use of the Shorter MDR-TB Regimen under Programmatic Conditions; WHO: Geneva, Switzerland, 2016; Volume 2015, pp. 1–14. [Google Scholar]
- Kolyva, A.S.; Karakousis, P.C. Old and New TB Drugs: Mechanisms of Action and Resistance. In Understanding Tuberculosis-New Approaches to Fighting against Drug Resistance; Johns Hopkins University Center for Tuberculosis Research: Baltimore, MD, USA, 2012. [Google Scholar] [CrossRef]
- Ministry of Health Zambia Zambia Consolidated Guidelines for Treatment and Prevention of HIV Infection. 2020, 1–57. Available online: https://www.moh.gov.zm/wp-content/uploads/filebase/Zambia-Consolidated-Guidelines-for-Treatment-and-Prevention-of-HIV-Infection-2020.pdf (accessed on 1 March 2023).
- Torres Ortiz, A.; Coronel, J.; Vidal, J.R.; Bonilla, C.; Moore, D.A.J.; Gilman, R.H.; Balloux, F.; Kon, O.M.; Didelot, X.; Grandjean, L. Genomic signatures of pre-resistance in Mycobacterium tuberculosis. Nat. Commun. 2021, 12, 7312. [Google Scholar] [CrossRef] [PubMed]
- Zaman, K. Tuberculosis: A global health problem. J. Health. Popul. Nutr. 2010, 28, 111–113. [Google Scholar] [CrossRef]
- Silva, M.L.; Cá, B.; Osório, N.S.; Rodrigues, P.N.S.; Maceiras, A.R.; Saraiva, M. Tuberculosis caused by Mycobacterium africanum: Knowns and unknowns. PLoS Pathog. 2022, 18, e1010490. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.D. The immunological life cycle of tuberculosis. Nat. Rev. Immunol. 2012, 12, 581–591. [Google Scholar] [CrossRef]
- Coscolla, M.; Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444. [Google Scholar] [CrossRef]
- Gagneux, S. Host-pathogen coevolution in human tuberculosis. Philos. Trans. R. Soc. London. Ser. B, Biol. Sci. 2012, 367, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Napier, G.; Campino, S.; Merid, Y.; Abebe, M.; Woldeamanuel, Y.; Aseffa, A.; Hibberd, M.L.; Phelan, J.; Clark, T.G. Robust barcoding and identification of Mycobacterium tuberculosis lineages for epidemiological and clinical studies. Genome Med. 2020 121 2020, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Solo, E.S.; Suzuki, Y.; Kaile, T.; Bwalya, P.; Lungu, P.; Chizimu, J.Y.; Shah, Y.; Nakajima, C. Characterization of Mycobacterium tuberculosis genotypes and their correlation to multidrug resistance in Lusaka, Zambia. Int. J. Infect. Dis. 2021, 102, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Chizimu, J.Y.; Solo, E.S.; Bwalya, P.; Kapalamula, T.F.; Akapelwa, M.L.; Lungu, P.; Shrestha, D.; Fukushima, Y.; Mukinka, V.; Thapa, J.; et al. Genetic Diversity and Transmission of Multidrug Resistant Mycobacterium tuberculosis strains in Lusaka, Zambia. Int. J. Infect. Dis. 2021, 114, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Mulenga, C.; Shamputa, I.C.; Mwakazanga, D.; Kapata, N.; Portaels, F.; Rigouts, L. Diversity of Mycobacterium tuberculosis genotypes circulating in Ndola, Zambia. BMC Infect. Dis. 2010, 10, 177. [Google Scholar] [CrossRef]
- Coll, F.; McNerney, R.; Guerra-Assunção, J.A.; Glynn, J.R.; Perdigão, J.; Viveiros, M.; Portugal, I.; Pain, A.; Martin, N.; Clark, T.G. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 2014, 5, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Monde, N.; Zulu, M.; Tembo, M.; Handema, R.; Munyeme, M.; Malama, S. Drug Resistant Tuberculosis in the Northern Region of Zambia: A Retrospective Study. Front. Trop. Dis. 2021, 2, 1–10. [Google Scholar] [CrossRef]
- Sagonda, T.; Mupfumi, L.; Manzou, R.; Makamure, B.; Tshabalala, M.; Gwanzura, L.; Mason, P.; Mutetwa, R. Prevalence of Extensively Drug Resistant Tuberculosis among Archived Multidrug Resistant Tuberculosis Isolates in Zimbabwe. Tuberc. Res. Treat. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Daniel, O.J.; Osman, E.; Oladimeji, O.; Dairo, O.G. Pre-Extensive Drug Resistant Tuberculosis (Pre-XDR-TB) among MDR-TB Patents in Nigeria. Glob. Adv. Res. J. Microbiol. 2013, 2, 1–4. [Google Scholar]
- Diriba, G.; Alemu, A.; Tola, H.H.; Yenew, B.; Amare, M.; Eshetu, K.; Sinshaw, W.; Abebaw, Y.; Meaza, A.; Seid, G.; et al. Pre-extensively drug-resistant tuberculosis among multidrug-resistant tuberculosis patients in Ethiopia: A laboratory-based surveillance study. IJID Reg. 2022, 5, 39–43. [Google Scholar] [CrossRef]
- Chien, J.-Y.; Chiu, W.-Y.; Chien, S.-T.; Chiang, C.-J.; Yu, C.-J.; Hsueh, P.-R. Mutations in gyrA and gyrB among Fluoroquinolone- and Multidrug-Resistant Mycobacterium tuberculosis Isolates. Antimicrob. Agents Chemother. 2016, 60, 2090–2096. [Google Scholar] [CrossRef]
- Maruri, F.; Sterling, T.R.; Kaiga, A.W.; Blackman, A.; van der Heijden, Y.F.; Mayer, C.; Cambau, E.; Aubry, A. A systematic review of gyrase mutations associated with fluoroquinolone-resistant Mycobacterium tuberculosis and a proposed gyrase numbering system. J. Antimicrob. Chemother. 2012, 67, 819–831. [Google Scholar] [CrossRef]
- Meehan, C.J.; Goig, G.A.; Kohl, T.A.; Verboven, L.; Dippenaar, A.; Ezewudo, M.; Farhat, M.R.; Guthrie, J.L.; Laukens, K.; Miotto, P.; et al. Whole genome sequencing of Mycobacterium tuberculosis: Current standards and open issues. Nat. Rev. Microbiol. 2019, 17, 533–545. [Google Scholar] [CrossRef]
- Langendam, M.W.; van der Werf, M.J.; Huitric, E.; Manissero, D. Prevalence of inappropriate tuberculosis treatment regimens: A systematic review. Eur. Respir. J. 2012, 39, 1012–1020. [Google Scholar] [CrossRef]
- Caminero, J.A.; Sotgiu, G.; Zumla, A.; Migliori, G.B. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet. Infect. Dis. 2010, 10, 621–629. [Google Scholar] [CrossRef] [PubMed]
- NTLP Manual for Management of Tuberculosis and Leprosy in Tanzania. MoHCDGEC-HPS-Printing Press DSM: 2019; pp. 285–287. Available online: https://ntlp.go.tz/site/assets/files/1081/ntlp_manual_2020_2021_1.pdf (accessed on 1 March 2023).
- Ministry of Health Zambia the National Tuberculosis and Leprosy Control Program Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis in Zambia. 2017. Available online: https://www.afro.who.int/sites/default/files/2019-06/DR-TB%20Guidelines%20Zambia%20updated%20%2028_2_2018-FINA%20%282%29%20%282%29.pdf (accessed on 19 October 2022).
- Botswana Ministry of Health National Tuberculosis Programme Manual. 2007. Available online: https://www.medbox.org/document/botswana-national-tuberculosis-programme-manual#GO (accessed on 28 September 2022).
- WHO WHO Consolidated Guidelines on Tuberculosis Module 4: Treatment Drug-Resistant Tuberculosis Treatment. 2020. Available online: https://www.who.int/publications/i/item/9789240007048 (accessed on 28 September 2022).
- Devasia, R.A.; Blackman, A.; May, C.; Eden, S.; Smith, T.; Hooper, N.; Maruri, F.; Stratton, C.; Shintani, A.; Sterling, T.R. Fluoroquinolone resistance in Mycobacterium tuberculosis: An assessment of MGIT 960, MODS and nitrate reductase assay and fluoroquinolone cross-resistance. J. Antimicrob. Chemother. 2009, 63, 1173–1178. [Google Scholar] [CrossRef]
- Imperiale, B.R.; Di Giulio, Á.B.; Adrián Cataldi, Á.; Morcillo, N.S. Evaluation of Mycobacterium tuberculosis cross-resistance to isoniazid, rifampicin and levofloxacin with their respective structural analogs. J. Antibiot. (Tokyo) 2014, 67, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Kalungia, A.C.; Burger, J.; Godman, B.; Costa, J.d.O.; Simuwelu, C. Non-prescription sale and dispensing of antibiotics in community pharmacies in Zambia. Expert Rev. Anti. Infect. Ther. 2016, 14, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Jelfs, P.; Sintchenko, V. Fluoroquinolone resistance in non-multidrug-resistant tuberculosis-a surveillance study in New South Wales, Australia, and a review of global resistance rates. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2014, 26, 149–153. [Google Scholar] [CrossRef][Green Version]
- Yasraba, R.; Kauser, J.; Rumina, H.; Sana, J.; Rabia, L.; Faisal, M.; Rafique, M.; Abid, C.; Zahra, H. Fluoroquinolone Resistance among Mycobacterium tuberculosis Strains from Karachi, Pakistan: Data from Community-Based Field Clinics. Antimicrob. Agents Chemother. 2011, 55, 929–930. [Google Scholar] [CrossRef]
- Lai, C.-C.; Tan, C.-K.; Huang, Y.-T.; Liao, C.-H.; Hsueh, P.-R. Fluoroquinolone-resistant tuberculosis at a medical centre in Taiwan, 2005–10. J. Antimicrob. Chemother. 2011, 66, 2437–2438. [Google Scholar] [CrossRef][Green Version]
- Cambaco, O.; Alonso Menendez, Y.; Kinsman, J.; Sigaúque, B.; Wertheim, H.; Do, N.; Gyapong, M.; John-Langba, J.; Sevene, E.; Munguambe, K. Community knowledge and practices regarding antibiotic use in rural Mozambique: Where is the starting point for prevention of antibiotic resistance? BMC Public Health 2020, 20, 1183. [Google Scholar] [CrossRef]
- Sheikh, K.; Uplekar, M. What can we learn about the processes of regulation of tuberculosis medicines from the experiences of health policy and system actors in India, Tanzania, and Zambia? Int. J. Health Policy Manag. 2016, 5, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Chizimu, J.Y.; Solo, E.S.; Bwalya, P.; Tanomsridachchai, W.; Chambaro, H.; Shawa, M.; Kapalamula, T.F.; Lungu, P.; Fukushima, Y.; Mukonka, V.; et al. Whole-Genome Sequencing Reveals Recent Transmission of Multidrug-Resistant Mycobacterium tuberculosis CAS1-Kili Strains in Lusaka, Zambia. Antibiotics 2022, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Solo, E.S.; Nakajima, C.; Kaile, T.; Bwalya, P.; Fukushima, Y.; Chila, S.; Kapata, N.; Mbulo, G.; Fukushima, Y.; Chila, S.; et al. Mutations of rpoB, katG and inhA genes in multidrug-resistant Mycobacterium tuberculosis isolates from Zambia. J. Glob. Antimicrob. Resist. 2020, 22, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 June 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Seemann, T. Snippy: Rapid Haploid Variant Calling and Core Genome Alignment. Available online: https://github.com/tseemann/snippy (accessed on 23 August 2021).
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2014, 43, e15. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinforma. 2020, 69, e96. [Google Scholar] [CrossRef]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Team, Rs. RStudio: Integrated Development for R. RStudio, PBC, Boston, MA. Available online: https://www.rstudio.com/ (accessed on 27 July 2021).
- Walker, T.M.; Ip, C.L.C.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.W.; Wilson, D.J.; Hawkey, P.M.; Crook, D.W.; et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet. Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Lalor, M.; Casali, N.; Walker, T.; Anderson, L.; Davidson, J.; Ratna, N.; Mullarkey, C.; Gent, M.; Foster, K.; Brown, T.; et al. The use of whole-genome sequencing in cluster investigation of an MDR-TB outbreak. Eur. Respir. J. 2018, 51, 1702313. [Google Scholar] [CrossRef] [PubMed]
- Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutations | Number | Percentage (%) | ||
|---|---|---|---|---|
| Rifampicin | Asp435Val a | 8 | 13 | |
| His445Asp a | 7 | 11 | ||
| Ser450Leu a | 34 | 54 | ||
| Rifampicin mutation only (His445Tyr a) | 2 | 3 | ||
| No mutation | 1 | 2 | ||
| Other rifampicin resistance mutations | 11 | 17 | ||
| Isoniazid | Ser315Thr b | 60 | 95 | |
| 15 C>T c | 1 | 2 | ||
| No mutation | 2 | 3 | ||
| MDR | Asp435Tyr a, Ser315Thr b | 2 | 3 | |
| Asp435Val a, Ser315Thr b | 8 | 13 | ||
| His445Asp a, Ser315Thr b | 7 | 11 | ||
| Ser450Leu a, Ser315Thr b | 30 | 48 | ||
| Other MDR mutation combinations | 9 | 14 | ||
| Others (mono and polyresistant) | 3 | 5 | ||
| Ser450Leu a, Ser315Thr b, Asp94His d | 1 | 2 | ||
| Pre-XDR | Ser450Leu a, Ser315Thr b, Asp94Gly d | 1 | 2 | |
| His445Leu a, Ser315Thr b, Asp94Gly d | 2 | 3 | ||
| Polyresistant | 15 C>T c, Gly88Cys d | 1 | 2 | |
| Non polyresistant | 62 | 98 | ||
| Drug Susceptibility Pattern | Gene Mutations | Treatment | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Age | Sex | I | R | E | S | Bdq | Lfx | Mfx | Cfz | Phenotype | gryA | rpoB | katG | inh_promoter | pncA | embB | rpsL | ethA | folC | Sublineage | |||||
| A | 41 | M | X | √ | √ | √ | √ | X | X | √ | poly-resistant | G88C | - | - | 15C>T | R154G | - | - | 15C > T | - | L4.3.4.2.1 | Lfx | I | R | E | Z |
| B | 29 | M | X | X | √ | √ | √ | X | X | √ | pre-XDR | D94H | S450L | S315T | - | - | M306I | - | - | - | L4.1.2 | Lfx | Bdq | Lzd | Cs | - |
| C | 48 | F | X | X | √ | √ | √ | X | X | √ | pre-XDR | D94G | S450L | S315T | - | R154G | Y319S | K43R | 1412_1413insGG | E40G | L4.3.4.2.1 | - | - | - | - | - |
| D | 24 | F | X | X | √ | √ | √ | X | X | √ | pre-XDR | D94G | H445L | S315T | - | - | M306V | - | - | - | L4.3.4.2.1 | Lfx | Km | Eto | Cs | - |
| E | - | - | - | - | - | - | - | - | - | - | * pre-XDR | D94G | H445L | S315T | - | - | M306V | - | - | - | L4.3.4.2.1 | - | - | - | - | - |
| F | 42 | M | X | X | X | X | - | - | - | - | MDR | - | S450L | S315T | - | H43P | M306V | - | 1215_1224del | - | L4.6.1.2 | Lfx | Km | Eto | Cs | Z |
| G | 27 | F | X | X | √ | √ | - | - | - | - | MDR | - | S450L | S315T | - | - | - | - | T61M | - | L4.3.4.2.1 | - | - | - | - | - |
| Drug | Mutations | Number | Percentage (%) | |
|---|---|---|---|---|
| Non-MDR | Polyresistant | Gly88Cys, 15 C>T, Arg154Gly | 1 | 2 |
| Pyrazinamide (Z) | Arg154Gly | 1 | 2 | |
| Trp119Cys | 1 | 2 | ||
| His43Pro | 1 | 2 | ||
| Val128Gly | 1 | 2 | ||
| MDR plus | Other pyrazinamide mutations | 6 | 12 | |
| No mutations | 40 | 80 | ||
| Ethambutol (E) | Met306Val | 2 | 4 | |
| Met306Val | 4 | 8 | ||
| Met306Ile | 3 | 6 | ||
| Tyr319Ser | 3 | 6 | ||
| Other ethambutol mutations | 8 | 16 | ||
| No mutations | 30 | 60 | ||
| Streptomycin (S) | Lys88Arg | 6 | 12 | |
| Other streptomycin mutations | 9 | 18 | ||
| No mutations | 35 | 70 | ||
| Ethionamide (Eto) | Thr61Met | 1 | 2 | |
| Ser450Leu, Ser315Thr, 15C>T | 1 | 2 | ||
| Other ethionamide mutations | 2 | 4 | ||
| No mutations | 46 | 92 | ||
| Para-amino salicylic acid (PAS) | Glu40Gly | 1 | 2 | |
| No mutations | 49 | 98 |
| Gene | Primer Set | |
|---|---|---|
| rpoB | Foward | CAGGACGTGGAGGCGATCAC |
| Reverse | GAGCCGATCAGACCGATGTTGG | |
| katG | Foward | ATGGCCATGAACGACGTCGAAAC |
| Reverse | CGCAGCGAGAGGTCAGTGGCCAG | |
| inhA | Foward | TCACACCGACAAACGTCACGAGC |
| Reverse | AGCCAGCCGCTGTGCGATCGCCA | |
| gyrA | Foward | AGCGCAGCTACATCGACTATGCG |
| Reverse | CTTCGGTGTACCTCATCGCCGCC | |
| gryB | Foward | CGGCACGTAAGGCACGAGAG |
| Reverse | GAACCGGAACAACAACGTCAAC | |
| rrs | Foward | CGGATCGGGGTCTGCAACTCGAC |
| Reverse | CAAGAACCCCTCACGGCCTACG | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chizimu, J.Y.; Solo, E.S.; Bwalya, P.; Kapalamula, T.F.; Mwale, K.K.; Squarre, D.; Shawa, M.; Lungu, P.; Barnes, D.A.; Yamba, K.; et al. Genomic Analysis of Mycobacterium tuberculosis Strains Resistant to Second-Line Anti-Tuberculosis Drugs in Lusaka, Zambia. Antibiotics 2023, 12, 1126. https://doi.org/10.3390/antibiotics12071126
Chizimu JY, Solo ES, Bwalya P, Kapalamula TF, Mwale KK, Squarre D, Shawa M, Lungu P, Barnes DA, Yamba K, et al. Genomic Analysis of Mycobacterium tuberculosis Strains Resistant to Second-Line Anti-Tuberculosis Drugs in Lusaka, Zambia. Antibiotics. 2023; 12(7):1126. https://doi.org/10.3390/antibiotics12071126
Chicago/Turabian StyleChizimu, Joseph Yamweka, Eddie Samuneti Solo, Precious Bwalya, Thoko Flav Kapalamula, Kaemba Kunkuta Mwale, David Squarre, Misheck Shawa, Patrick Lungu, David Atomanyi Barnes, Kaunda Yamba, and et al. 2023. "Genomic Analysis of Mycobacterium tuberculosis Strains Resistant to Second-Line Anti-Tuberculosis Drugs in Lusaka, Zambia" Antibiotics 12, no. 7: 1126. https://doi.org/10.3390/antibiotics12071126
APA StyleChizimu, J. Y., Solo, E. S., Bwalya, P., Kapalamula, T. F., Mwale, K. K., Squarre, D., Shawa, M., Lungu, P., Barnes, D. A., Yamba, K., Mufune, T., Chambaro, H., Kamboyi, H., Munyeme, M., Hang’ombe, B. M., Kapata, N., Mukonka, V., Chilengi, R., Thapa, J., ... Suzuki, Y. (2023). Genomic Analysis of Mycobacterium tuberculosis Strains Resistant to Second-Line Anti-Tuberculosis Drugs in Lusaka, Zambia. Antibiotics, 12(7), 1126. https://doi.org/10.3390/antibiotics12071126