
Genomic Characterization of Drug-Resistant Mycobacterium tuberculosis L2/Beijing Isolates from Astana, Kazakhstan
 ,
,
Abstract
1. Introduction
2. Results
2.1. Sample Selection and Sequencing
2.2. Population Structure
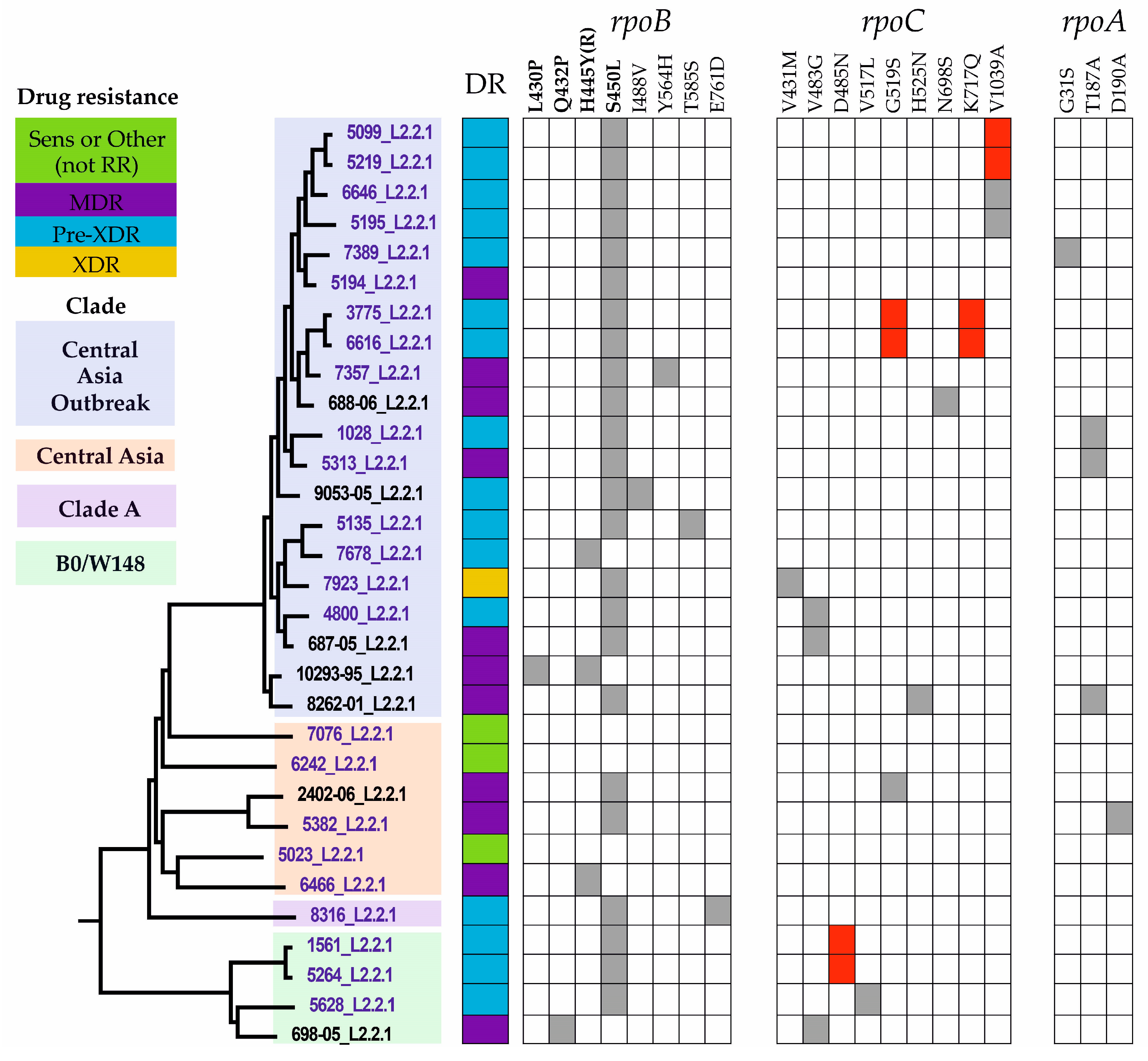
2.3. Drug Resistance-Associated Mutations
2.4. Comparison of Phenotypic Drug Susceptibility Testing and Genotypic Prediction
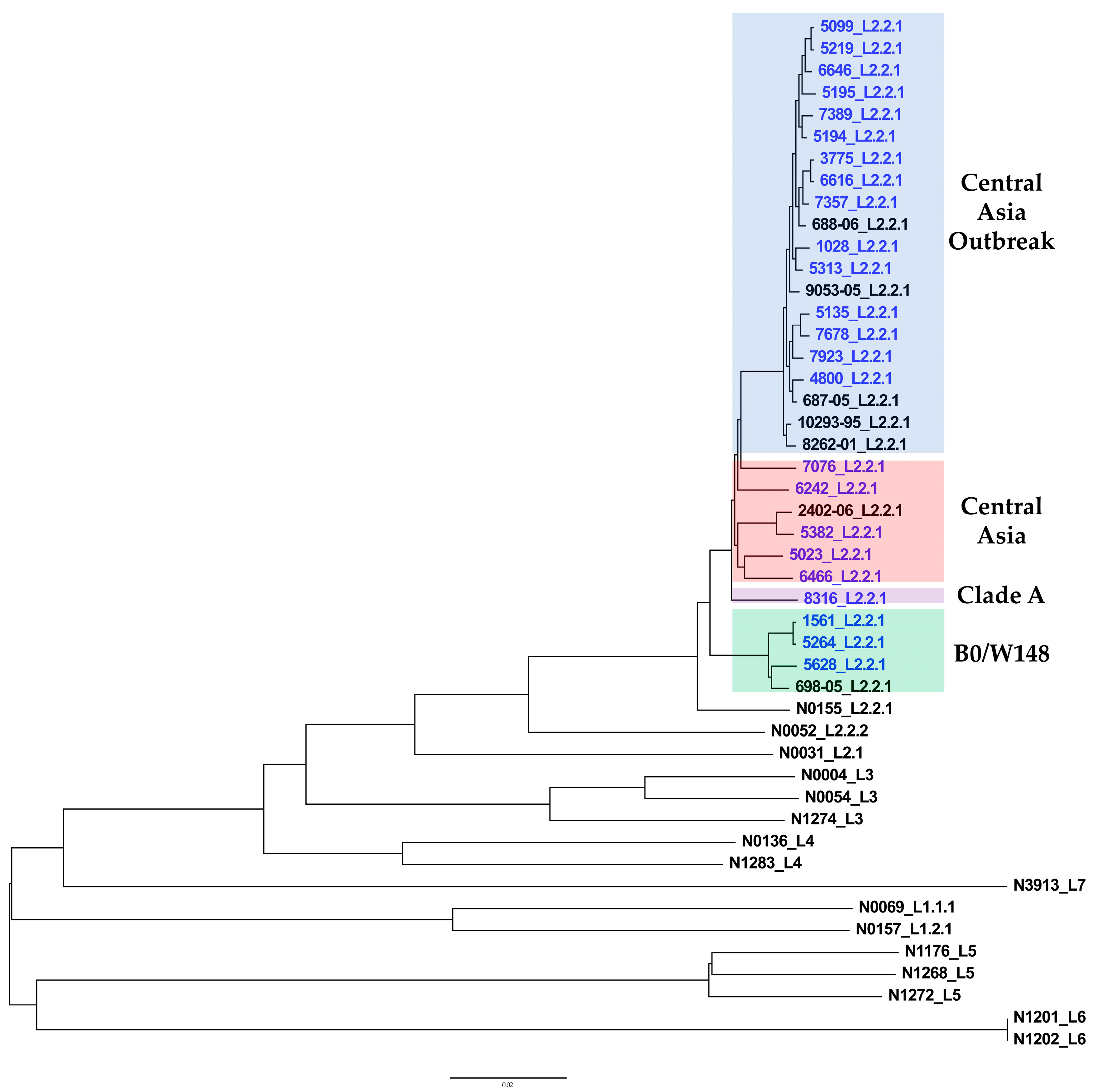
2.5. Phylogenetic Analysis
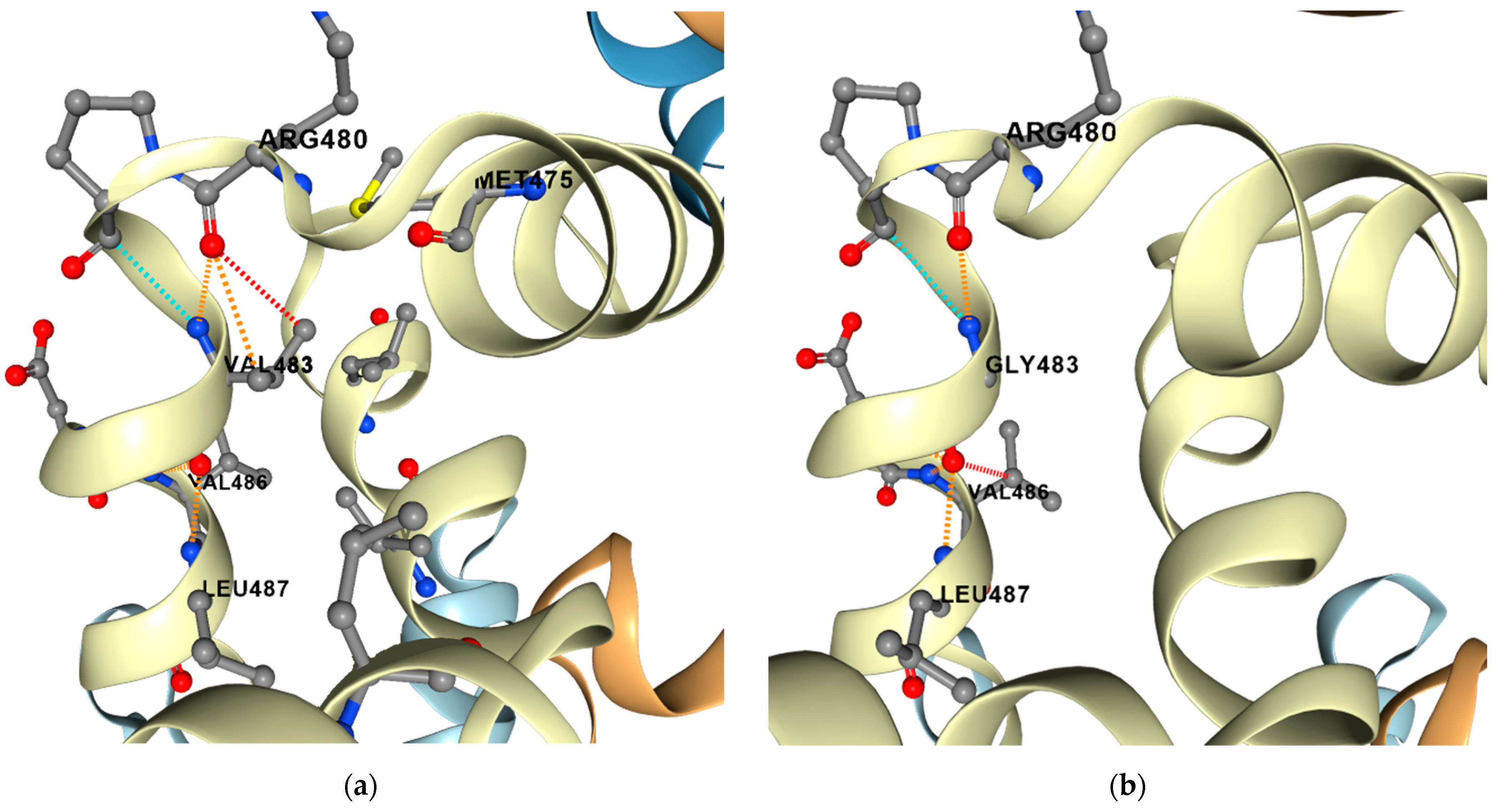
2.6. Compensatory Mutations
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Study Population and Drug Susceptibility Testing
5.2. DNA Isolation and Genotyping
5.3. Whole-Genome Sequencing
5.4. Prediction of Protein Stability and Dynamics
5.5. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. Global Tuberculosis Report 2022. 2022. Available online: https://www.who.int/publications/i/item/9789240061729 (accessed on 18 May 2023).
- Pai, M.; Kasaeva, T.; Swaminathan, S. COVID-19’s Devastating Effect on Tuberculosis Care—A Path to Recovery. N. Engl. J. Med. 2022, 386, 1490–1493. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.M.G.J.; Dodd, P.J. The Global Burden of Latent Tuberculosis Infection: A Re-Estimation Using Mathematical Modelling. PLoS Med. 2016, 13, e1002152. [Google Scholar] [CrossRef] [PubMed]
- Latent tuberculosis infection: Updated and consolidated guidelines for programmatic management. In WHO Guidelines Approved by the Guidelines Review Committee; World Health Organization: Geneva, Switzerland, 2018.
- Abeldenov, S.; Talhaoui, I.; Zharkov, D.O.; Ishchenko, A.A.; Ramanculov, E.; Saparbaev, M.; Khassenov, B. Characterization of DNA substrate specificities of apurinic/apyrimidinic endonucleases from Mycobacterium tuberculosis. DNA Repair. 2015, 33, 1–16. [Google Scholar]
- World Health Organisation. Tuberculosis Profile: Kazakhstan. Available online: https://worldhealthorg.shinyapps.io/tb_profiles/?_inputs_&entity_type=%22country%22&lan=%22EN%22&iso2=%22KZ%22 (accessed on 18 May 2023).
- Meehan, C.J.; Goig, G.A.; Kohl, T.A.; Verboven, L.; Dippenaar, A.; Ezewudo, M.; Farhat, M.R.; Guthrie, J.L.; Laukens, K.; Miotto, P.; et al. Whole genome sequencing of Mycobacterium tuberculosis: Current standards and open issues. Nat. Rev. Microbiol. 2019, 17, 533–545. [Google Scholar] [CrossRef]
- Tarlykov, P.; Atavliyeva, S.; Alenova, A.; Ramankulov, Y. Genomic analysis of Latin American-Mediterranean family of Mycobacterium tuberculosis clinical strains from Kazakhstan. Mem. Inst. Oswaldo Cruz 2020, 115, e200215. [Google Scholar] [CrossRef] [PubMed]
- Tarlykov, P.; Atavliyeva, S.; Alenova, A.; Ramankulov, Y. Draft Genome Sequence of an Extensively Drug-Resistant Mycobacterium tuberculosis Clinical Isolate, 3485_MTB, from Nur-Sultan, Kazakhstan. Microbiol. Resour. Announc. 2020, 9, e00025-20. [Google Scholar] [CrossRef] [PubMed]
- Kairov, U.; Kozhamkulov, U.; Molkenov, A.; Rakhimova, S.; Askapuli, A.; Zhabagin, M.; Akhmetova, A.; Yerezhepov, D.; Abilova, Z.; Abilmazhinova, A.; et al. Draft Genome Sequences of Two Clinical Isolates of Mycobacterium tuberculosis from Sputum of Kazakh Patients. Genome Announc. 2015, 3, e00466-15. [Google Scholar] [CrossRef] [PubMed]
- Mokrousov, I.; Shitikov, E.; Skiba, Y.; Kolchenko, S.; Chernyaeva, E.; Vyazovaya, A. Emerging peak on the phylogeographic landscape of Mycobacterium tuberculosis in West Asia: Definitely smoke, likely fire. Mol. Phylogenet. Evol. 2017, 116, 202–212. [Google Scholar] [CrossRef]
- Daniyarov, A.; Molkenov, A.; Rakhimova, S.; Akhmetova, A.; Yerezhepov, D.; Chingissova, L.; Bismilda, V.; Toksanbayeva, B.; Rakisheva, A.; Akilzhanova, A.; et al. Genomic Analysis of Multidrug-Resistant Mycobacterium tuberculosis Strains From Patients in Kazakhstan. Front. Genet. 2021, 12, 683515. [Google Scholar] [CrossRef]
- Mokrousov, I.; Chernyaeva, E.; Vyazovaya, A.; Skiba, Y.; Solovieva, N.; Valcheva, V.; Levina, K.; Malakhova, N.; Jiao, W.W.; Gomes, L.L.; et al. Rapid Assay for Detection of the Epidemiologically Important Central Asian/Russian Strain of the Mycobacterium tuberculosis Beijing Genotype. J. Clin. Microbiol. 2018, 56, e01551-17. [Google Scholar] [CrossRef]
- Dookie, N.; Rambaran, S.; Padayatchi, N.; Mahomed, S.; Naidoo, K. Evolution of drug resistance in Mycobacterium tuberculosis: A review on the molecular determinants of resistance and implications for personalized care. J. Antimicrob. Chemother. 2018, 73, 1138–1151. [Google Scholar] [CrossRef] [PubMed]
- Khawbung, J.L.; Nath, D.; Chakraborty, S. Drug resistant Tuberculosis: A review. Comp. Immunol. Microbiol. Infect. Dis. 2021, 74, 101574. [Google Scholar] [CrossRef]
- Telenti, A. Genetics and pulmonary medicine. 5. Genetics of drug resistant tuberculosis. Thorax 1998, 53, 793–797. [Google Scholar] [CrossRef][Green Version]
- Alame Emane, A.K.; Guo, X.; Takiff, H.E.; Liu, S. Drug resistance, fitness and compensatory mutations in Mycobacterium tuberculosis. Tuberculosis 2021, 129, 102091. [Google Scholar] [CrossRef] [PubMed]
- Mokrousov, I.; Vyazovaya, A.; Zhuravlev, V.; Otten, T.; Millet, J.; Jiao, W.W.; Shen, A.D.; Rastogi, N.; Vishnevsky, B.; Narvskaya, O. Real-time PCR assay for rapid detection of epidemiologically and clinically significant Mycobacterium tuberculosis Beijing genotype isolates. J. Clin. Microbiol. 2014, 52, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Xia, E.; Teo, Y.-Y.; Ong, R.T.-H. SpoTyping: Fast and accurate in silico Mycobacterium spoligotyping from sequence reads. Genome Med. 2016, 8, 19. [Google Scholar] [CrossRef]
- Coll, F.; McNerney, R.; Preston, M.D.; Guerra-Assunção, J.A.; Warry, A.; Hill-Cawthorne, G.; Mallard, K.; Nair, M.; Miranda, A.; Alves, A.; et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015, 7, 51. [Google Scholar] [CrossRef]
- Coll, F.; McNerney, R.; Guerra-Assuncao, J.A.; Glynn, J.R.; Perdigao, J.; Viveiros, M.; Portugal, I.; Pain, A.; Martin, N.; Clark, T.G. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 2014, 5, 4812. [Google Scholar] [CrossRef]
- Merker, M.; Blin, C.; Mona, S.; Duforet-Frebourg, N.; Lecher, S.; Willery, E.; Blum, M.G.; Rusch-Gerdes, S.; Mokrousov, I.; Aleksic, E.; et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat. Genet. 2015, 47, 242–249. [Google Scholar] [CrossRef]
- Thawornwattana, Y.; Mahasirimongkol, S.; Yanai, H.; Maung, H.M.W.; Cui, Z.; Chongsuvivatwong, V.; Palittapongarnpim, P. Revised nomenclature and SNP barcode for Mycobacterium tuberculosis lineage 2. Microb. Genom. 2021, 7, 000697. [Google Scholar] [CrossRef]
- Sonnenkalb, L.; Carter, J.J.; Spitaleri, A.; Iqbal, Z.; Hunt, M.; Malone, K.M.; Utpatel, C.; Cirillo, D.M.; Rodrigues, C.; Nilgiriwala, K.S.; et al. Bedaquiline and clofazimine resistance in Mycobacterium tuberculosis: An in-vitro and in-silico data analysis. Lancet Microbe 2023, 4, e358–e368. [Google Scholar] [CrossRef] [PubMed]
- Merker, M.; Barbier, M.; Cox, H.; Rasigade, J.P.; Feuerriegel, S.; Kohl, T.A.; Diel, R.; Borrell, S.; Gagneux, S.; Nikolayevskyy, V.; et al. Compensatory evolution drives multidrug-resistant tuberculosis in Central Asia. eLife 2018, 7, e38200. [Google Scholar] [CrossRef] [PubMed]
- Borrell, S.; Trauner, A.; Brites, D.; Rigouts, L.; Loiseau, C.; Coscolla, M.; Niemann, S.; De Jong, B.; Yeboah-Manu, D.; Kato-Maeda, M.; et al. Reference set of Mycobacterium tuberculosis clinical strains: A tool for research and product development. PLoS ONE 2019, 14, e0214088. [Google Scholar] [CrossRef]
- Walker, T.M.; Ip, C.L.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.W.; Wilson, D.J.; Hawkey, P.M.; Crook, D.W.; et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Daniyarov, A.; Akhmetova, A.; Rakhimova, S.; Abilova, Z.; Yerezhepov, D.; Chingissova, L.; Bismilda, V.; Takenov, N.; Akilzhanova, A.; Kairov, U.; et al. Whole-Genome Sequence-Based Characterization of Pre-XDR M. tuberculosis Clinical Isolates Collected in Kazakhstan. Diagnostics 2023, 13, 2005. [Google Scholar] [CrossRef]
- Skiba, Y.; Mokrousov, I.; Ismagulova, G.; Maltseva, E.; Yurkevich, N.; Bismilda, V.; Chingissova, L.; Abildaev, T.; Aitkhozhina, N. Molecular snapshot of Mycobacterium tuberculosis population in Kazakhstan: A country-wide study. Tuberculosis 2015, 95, 538–546. [Google Scholar] [CrossRef]
- Allix-Béguec, C.; Arandjelovic, I.; Bi, L.; Beckert, P.; Bonnet, M.; Bradley, P.; Cabibbe, A.M.; Cancino-Muñoz, I.; Caulfield, M.J.; Chaiprasert, A.; et al. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N. Engl. J. Med. 2018, 379, 1403–1415. [Google Scholar] [CrossRef]
- Jajou, R.; Kohl, T.A.; Walker, T.; Norman, A.; Cirillo, D.M.; Tagliani, E.; Niemann, S.; de Neeling, A.; Lillebaek, T.; Anthony, R.M.; et al. Towards standardisation: Comparison of five whole genome sequencing (WGS) analysis pipelines for detection of epidemiologically linked tuberculosis cases. Euro Surveill. Bull. Eur. Sur Les. Mal. Transm. 2019, 24, 1900130. [Google Scholar] [CrossRef]
- Klotoe, B.J.; Kacimi, S.; Costa-Conceicao, E.; Gomes, H.M.; Barcellos, R.B.; Panaiotov, S.; Haj Slimene, D.; Sikhayeva, N.; Sengstake, S.; Schuitema, A.R.; et al. Genomic characterization of MDR/XDR-TB in Kazakhstan by a combination of high-throughput methods predominantly shows the ongoing transmission of L2/Beijing 94-32 central Asian/Russian clusters. BMC Infect. Dis. 2019, 19, 553. [Google Scholar] [CrossRef]
- World Health, O. Catalogue of Mutations in Mycobacterium tuberculosis Complex and Their Association with Drug Resistance: Supplementary Document; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Snobre, J.; Villellas, M.C.; Coeck, N.; Mulders, W.; Tzfadia, O.; de Jong, B.C.; Andries, K.; Rigouts, L. Bedaquiline- and clofazimine- selected Mycobacterium tuberculosis mutants: Further insights on resistance driven largely by Rv0678. Sci. Rep. 2023, 13, 10444. [Google Scholar] [CrossRef]
- Vargas, R., Jr.; Freschi, L.; Spitaleri, A.; Tahseen, S.; Barilar, I.; Niemann, S.; Miotto, P.; Cirillo, D.M.; Köser, C.U.; Farhat, M.R. Role of Epistasis in Amikacin, Kanamycin, Bedaquiline, and Clofazimine Resistance in Mycobacterium tuberculosis Complex. Antimicrob. Agents Chemother. 2021, 65, e0116421. [Google Scholar] [CrossRef]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis: Module 4: Treatment: Drug-Resistant Tuberculosis Treatment; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- World Health Organization. Meeting Report of the WHO Expert Consultation on the Definition of Extensively Drug-Resistant Tuberculosis, 27–29 October 2020; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Gagneux, S. Fitness cost of drug resistance in Mycobacterium tuberculosis. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2009, 15 (Suppl. S1), 66–68. [Google Scholar] [CrossRef]
- Maisnier-Patin, S.; Andersson, D.I. Adaptation to the deleterious effects of antimicrobial drug resistance mutations by compensatory evolution. Res. Microbiol. 2004, 155, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Bhatter, P.; Chatterjee, A.; D’Souza, D.; Tolani, M.; Mistry, N. Estimating fitness by competition assays between drug susceptible and resistant Mycobacterium tuberculosis of predominant lineages in Mumbai, India. PLoS ONE 2012, 7, e33507. [Google Scholar] [CrossRef]
- Kodio, O.; Georges Togo, A.C.; Sadio Sarro, Y.D.; Fane, B.; Diallo, F.; Somboro, A.; Degoga, B.; Kone, M.; Coulibaly, G.; Tolofoudje, M.; et al. Competitive fitness of Mycobacterium tuberculosis in vitro. Int. J. Mycobacteriol 2019, 8, 287–291. [Google Scholar] [CrossRef]
- Song, T.; Park, Y.; Shamputa, I.C.; Seo, S.; Lee, S.Y.; Jeon, H.-S.; Choi, H.; Lee, M.; Glynne, R.J.; Barnes, S.W.; et al. Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the β′ subunit of RNA polymerase. Mol. Microbiol. 2014, 91, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Bainomugisa, A.; Lavu, E.; Hiashiri, S.; Majumdar, S.; Honjepari, A.; Moke, R.; Dakulala, P.; Hill-Cawthorne, G.A.; Pandey, S.; Marais, B.J.; et al. Multi-clonal evolution of multi-drug-resistant/extensively drug-resistant Mycobacterium tuberculosis in a high-prevalence setting of Papua New Guinea for over three decades. Microb. Genom. 2018, 4, e000147. [Google Scholar] [CrossRef]
- Comas, I.; Borrell, S.; Roetzer, A.; Rose, G.; Malla, B.; Kato-Maeda, M.; Galagan, J.; Niemann, S.; Gagneux, S. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 2011, 44, 106–110. [Google Scholar] [CrossRef]
- Li, Q.J.; Jiao, W.W.; Yin, Q.Q.; Xu, F.; Li, J.Q.; Sun, L.; Xiao, J.; Li, Y.J.; Mokrousov, I.; Huang, H.R.; et al. Compensatory Mutations of Rifampin Resistance Are Associated with Transmission of Multidrug-Resistant Mycobacterium tuberculosis Beijing Genotype Strains in China. Antimicrob. Agents Chemother. 2016, 60, 2807–2812. [Google Scholar] [CrossRef]
- Liu, Q.; Zuo, T.; Xu, P.; Jiang, Q.; Wu, J.; Gan, M.; Yang, C.; Prakash, R.; Zhu, G.; Takiff, H.E.; et al. Have compensatory mutations facilitated the current epidemic of multidrug-resistant tuberculosis? Emerg. Microbes Infect. 2018, 7, 98. [Google Scholar] [CrossRef]
- Wollenberg, K.R.; Desjardins, C.A.; Zalutskaya, A.; Slodovnikova, V.; Oler, A.J.; Quiñones, M.; Abeel, T.; Chapman, S.B.; Tartakovsky, M.; Gabrielian, A.; et al. Whole-Genome Sequencing of Mycobacterium tuberculosis Provides Insight into the Evolution and Genetic Composition of Drug-Resistant Tuberculosis in Belarus. J. Clin. Microbiol. 2017, 55, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Casali, N.; Nikolayevskyy, V.; Balabanova, Y.; Harris, S.R.; Ignatyeva, O.; Kontsevaya, I.; Corander, J.; Bryant, J.; Parkhill, J.; Nejentsev, S.; et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat. Genet. 2014, 46, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Brandis, G.; Hughes, D. Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found in clinical isolates. J. Antimicrob. Chemother. 2013, 68, 2493–2497. [Google Scholar] [CrossRef] [PubMed]
- Brandis, G.; Hughes, D. Mechanisms of fitness cost reduction for rifampicin-resistant strains with deletion or duplication mutations in rpoB. Sci. Rep. 2018, 8, 17488. [Google Scholar] [CrossRef]
- de Vos, M.; Müller, B.; Borrell, S.; Black, P.A.; van Helden, P.D.; Warren, R.M.; Gagneux, S.; Victor, T.C. Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob. Agents Chemother. 2013, 57, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, F.B.; Guthrie, J.L.; Neemuchwala, A.; Lastovetska, O.; Melano, R.G.; Mehaffy, C. Profiling of rpoB mutations and MICs for rifampin and rifabutin in Mycobacterium tuberculosis. J. Clin. Microbiol. 2014, 52, 2157–2162. [Google Scholar] [CrossRef]
- Meftahi, N.; Namouchi, A.; Mhenni, B.; Brandis, G.; Hughes, D.; Mardassi, H. Evidence for the critical role of a secondary site rpoB mutation in the compensatory evolution and successful transmission of an MDR tuberculosis outbreak strain. J. Antimicrob. Chemother. 2016, 71, 324–332. [Google Scholar] [CrossRef]
- San, L.L.; Aye, K.S.; Oo, N.A.T.; Shwe, M.M.; Fukushima, Y.; Gordon, S.V.; Suzuki, Y.; Nakajima, C. Insight into multidrug-resistant Beijing genotype Mycobacterium tuberculosis isolates in Myanmar. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2018, 76, 109–119. [Google Scholar] [CrossRef]
- Sarina, N.; Abeldenov, S.; Turgimbayeva, A.; Zhylkibayev, A.; Ramankulov, Y.; Khassenov, B.; Eskendirova, S. Obtaining and characterization of monoclonal antibodies against recombinant extracellular domain of human epidermal growth factor receptor 2. Hum. Antibodies 2018, 26, 103–111. [Google Scholar] [CrossRef]
- Favorov, M.; Belilovsky, E.; Aitmagambetova, I.; Ismailov, S.; White, M.E.; Chorba, T. Tuberculosis deaths averted by implementation of the DOTS strategy in Kazakhstan. Int. J. Tuberc. Lung Dis. 2010, 14, 1582–1588. [Google Scholar]
- Ibrayeva, A.; Kozhamkulov, U.; Raiymbek, D.; Alenova, A.; Igilikova, S.; Zholdybayeva, E.; Abildaev, T.; Momynaliev, K. Molecular epidemiology of Mycobacterium tuberculosis strains circulating in the penitentiary system of Kazakhstan [Short communication]. Int. J. Tuberc. Lung Dis. 2014, 18, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Di Gennaro, F.; Pizzol, D.; Cebola, B.; Stubbs, B.; Monno, L.; Saracino, A.; Luchini, C.; Solmi, M.; Segafredo, G.; Putoto, G.; et al. Social determinants of therapy failure and multi drug resistance among people with tuberculosis: A review. Tuberculosis 2017, 103, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Bumburidi, E.; Ajeilat, S.; Dadu, A.; Aitmagambetova, I.; Ershova, J.; Fagan, R.; Favorov, M.O. Progress toward tuberculosis control and determinants of treatment outcomes—Kazakhstan, 2000–2002. MMWR Suppl. 2006, 55, 11–15. [Google Scholar] [PubMed]
- van Soolingen, D.; Hermans, P.W.; de Haas, P.E.; Soll, D.R.; van Embden, J.D. Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: Evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J. Clin. Microbiol. 1991, 29, 2578–2586. [Google Scholar] [CrossRef] [PubMed]
- Kohl, T.A.; Utpatel, C.; Schleusener, V.; De Filippo, M.R.; Beckert, P.; Cirillo, D.M.; Niemann, S. MTBseq: A comprehensive pipeline for whole genome sequence analysis of Mycobacterium tuberculosis complex isolates. PeerJ 2018, 6, e5895. [Google Scholar] [CrossRef]
- Feuerriegel, S.; Schleusener, V.; Beckert, P.; Kohl, T.A.; Miotto, P.; Cirillo, D.M.; Cabibbe, A.M.; Niemann, S.; Fellenberg, K. PhyResSE: A Web Tool Delineating Mycobacterium tuberculosis Antibiotic Resistance and Lineage from Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 1908. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A. FigTree, version 1.4. 0; Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2012. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Isolate | Gender | Age | DR Type | Diagnosis (TB) | Case |
|---|---|---|---|---|---|
| 1028 | Female | 35 | Pre-XDR | Infiltrative pulmonary | New |
| 1561 | Male | 42 | Pre-XDR | Infiltrative pulmonary | Relapse |
| 3775 | Male | 44 | Pre-XDR | Miliary | New |
| 4800 | Male | 40 | Pre-XDR | Infiltrative pulmonary | Retreatment |
| 5023 | Female | 34 | Sens | Infiltrative pulmonary | New |
| 5099 | Female | 60 | Pre-XDR | Infiltrative pulmonary | Relapse |
| 5135 | Male | 29 | Pre-XDR | Infiltrative pulmonary | New |
| 5194 | Female | 30 | MDR | Infiltrative pulmonary | New |
| 5195 | Male | 25 | Pre-XDR | Infiltrative pulmonary | New |
| 5219 | Female | 60 | Pre-XDR | Infiltrative pulmonary | Relapse |
| 5264 | Male | 41 | Pre-XDR | Fibrocystic cavernous | Relapse |
| 5313 | Male | 31 | MDR | Infiltrative pulmonary | New |
| 5382 | Female | 61 | MDR | Infiltrative pulmonary | New |
| 5628 | Male | 47 | Pre-XDR | Infiltrative pulmonary | Relapse |
| 6242 | Female | 44 | Other (S) | Infiltrative pulmonary | Relapse |
| 6466 | Female | 52 | MDR | Infiltrative pulmonary | Relapse |
| 6616 | Male | 44 | Pre-XDR | Miliary | New |
| 6646 | Male | 50 | Pre-XDR | Infiltrative pulmonary | Retreatment |
| 7076 | Male | 39 | Sens | Infiltrative pulmonary | New |
| 7357 | Male | 54 | MDR | Fibrocystic cavernous | Relapse |
| 7389 | Male | 54 | Pre-XDR | Infiltrative pulmonary | Relapse |
| 7678 | Male | 37 | Pre-XDR | Infiltrative pulmonary | New |
| 7923 | Female | 39 | XDR | Infiltrative pulmonary | New |
| 8316 | Female | 49 | Pre-XDR | Infiltrative pulmonary | New |
| Isolate | Spoligotype | RD | Sublineage [21] | Sublineage [22] | Sublineage [23] |
|---|---|---|---|---|---|
| 1028 | 000000000003771 | RD105, RD207, RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 1561 | 000000000003771 | RD105, RD207, RD181 | 2.2.1 | Europe/Russian W148 outbreak | L2.2.M4.5 |
| 3775 | 000000000003771 | RD105, RD207, RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 4800 | 000000000003771 | RD105, RD207, RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 5023 | 000000000003771 | RD105, RD207, RD181 | 2.2.1 | Central Asia | L2.2.M4.9 |
| 5099 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 5135 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 5194 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 5195 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 5219 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 5264 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Europe/Russian W148 outbreak | L2.2.M4.5 |
| 5313 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 5382 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia | L2.2.M4.9 |
| 5628 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Europe/Russian W148 outbreak | L2.2.M4.5 |
| 6242 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia | L2.2.M4.9 |
| 6466 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia | L2.2.M4.9 |
| 6616 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 6646 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 7076 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia | L2.2.M4.9 |
| 7357 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 7389 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 7678 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 7923 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Central Asia outbreak | L2.2.M4.9.1 |
| 8316 | 000000000003771 | RD105; RD207; RD181 | 2.2.1 | Clade A | L2.2.M4.9.2 |
| Drug | INH | RIF | EMB | STM | PZA | OFX | KAN | AMK | CAP | ETO | BDQ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Isolate | |||||||||||
| 1028 | R | R | R | R | S | R | S | S | S | S | S |
| 1561 | R | R | R | R | R | R | S | S | S | S | R |
| 3775 | R | R | R | R | S | R | S | S | S | S | S |
| 4800 | R | R | R | R | R | R | R | R | R | S | S |
| 5023 | S | S | S | S | S | S | S | S | S | S | S |
| 5099 | R | R | R | R | S | R | S | S | S | S | S |
| 5135 | R | R | R | R | S | R | S | S | S | S | S |
| 5194 | R | R | R | R | R | S | S | S | S | S | S |
| 5195 | R | R | R | R | R | R | S | S | S | S | S |
| 5219 | R | R | R | R | S | R | S | S | S | S | S |
| 5264 | R | R | R | R | R | R | S | S | S | S | R |
| 5313 | R | R | R | R | S | S | S | S | S | S | S |
| 5382 | R | R | R | R | R | S | R | R | R | S | S |
| 5628 | R | R | R | R | R | R | R | S | S | S | S |
| 6242 | S | S | S | R | S | S | S | S | S | S | S |
| 6466 | R | R | R | R | S | S | S | S | S | R | S |
| 6616 | R | R | R | R | S | R | S | S | S | S | S |
| 6646 | R | R | R | R | S | R | S | S | R | S | S |
| 7076 | S | S | S | S | S | S | S | S | S | S | S |
| 7357 | R | R | R | R | R | S | S | S | S | S | S |
| 7389 | R | R | R | R | R | R | S | S | S | S | R |
| 7678 | R | R | R | R | S | R | S | S | S | S | S |
| 7923 | R | R | R | R | S | R | S | S | S | S | R |
| 8316 | R | R | R | S | S | R | R | S | S | R | S |
| Gene | Compensatory Mutation | Frequency | P/NP | HP/HB | Energy (ΔΔG, kcal/mol) | Flexibility (ΔΔSVib, kcal/mol) |
|---|---|---|---|---|---|---|
| rpoC | V1039A | 4 | NP-NP | HB-HB | 1.266 | −3.570 |
| rpoC | D485N | 2 | P-P | HP-HP | 0.917 | −4.029 |
| rpoC | G519S | 2 | NP-P | HB-HP | 1.024 | −3.801 |
| rpoC | K717Q | 2 | P-P | HP-HP | 1.028 | −3.533 |
| rpoA | T187A | 2 | P-NP | HP-HB | 0.143 | 0.104 |
| rpoC | V431M | 1 | NP-NP | HB-HB | 1.064 | −4.096 |
| rpoC | V483G | 1 | NP-NP | HB-HB | −0.793 | −3.064 |
| rpoC | V517L | 1 | NP-NP | HB-HB | 1.785 | −4.121 |
| rpoB | Y564H | 1 | P-P | HP-HP | 1.399 | −3.783 |
| rpoB | T585S | 1 | P-P | HP-HP | 0.025 | −4.287 |
| rpoB | E761D | 1 | P-P | HP-HP | 1.463 | −4.275 |
| rpoA | D190A | 1 | P-NP | HP-HB | 0.025 | 0.595 |
| rpoA | G31S | 1 | NP-P | HB-HP | 0.083 | −0.448 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auganova, D.; Atavliyeva, S.; Amirgazin, A.; Akisheva, A.; Tsepke, A.; Tarlykov, P. Genomic Characterization of Drug-Resistant Mycobacterium tuberculosis L2/Beijing Isolates from Astana, Kazakhstan. Antibiotics 2023, 12, 1523. https://doi.org/10.3390/antibiotics12101523
Auganova D, Atavliyeva S, Amirgazin A, Akisheva A, Tsepke A, Tarlykov P. Genomic Characterization of Drug-Resistant Mycobacterium tuberculosis L2/Beijing Isolates from Astana, Kazakhstan. Antibiotics. 2023; 12(10):1523. https://doi.org/10.3390/antibiotics12101523
Chicago/Turabian StyleAuganova, Dana, Sabina Atavliyeva, Asylulan Amirgazin, Akmaral Akisheva, Anna Tsepke, and Pavel Tarlykov. 2023. "Genomic Characterization of Drug-Resistant Mycobacterium tuberculosis L2/Beijing Isolates from Astana, Kazakhstan" Antibiotics 12, no. 10: 1523. https://doi.org/10.3390/antibiotics12101523
APA StyleAuganova, D., Atavliyeva, S., Amirgazin, A., Akisheva, A., Tsepke, A., & Tarlykov, P. (2023). Genomic Characterization of Drug-Resistant Mycobacterium tuberculosis L2/Beijing Isolates from Astana, Kazakhstan. Antibiotics, 12(10), 1523. https://doi.org/10.3390/antibiotics12101523