

Synthetic and Semisynthetic Compounds as Antibacterials Targeting Virulence Traits in Resistant Strains: A Narrative Updated Review

 , , and
, , and
Abstract
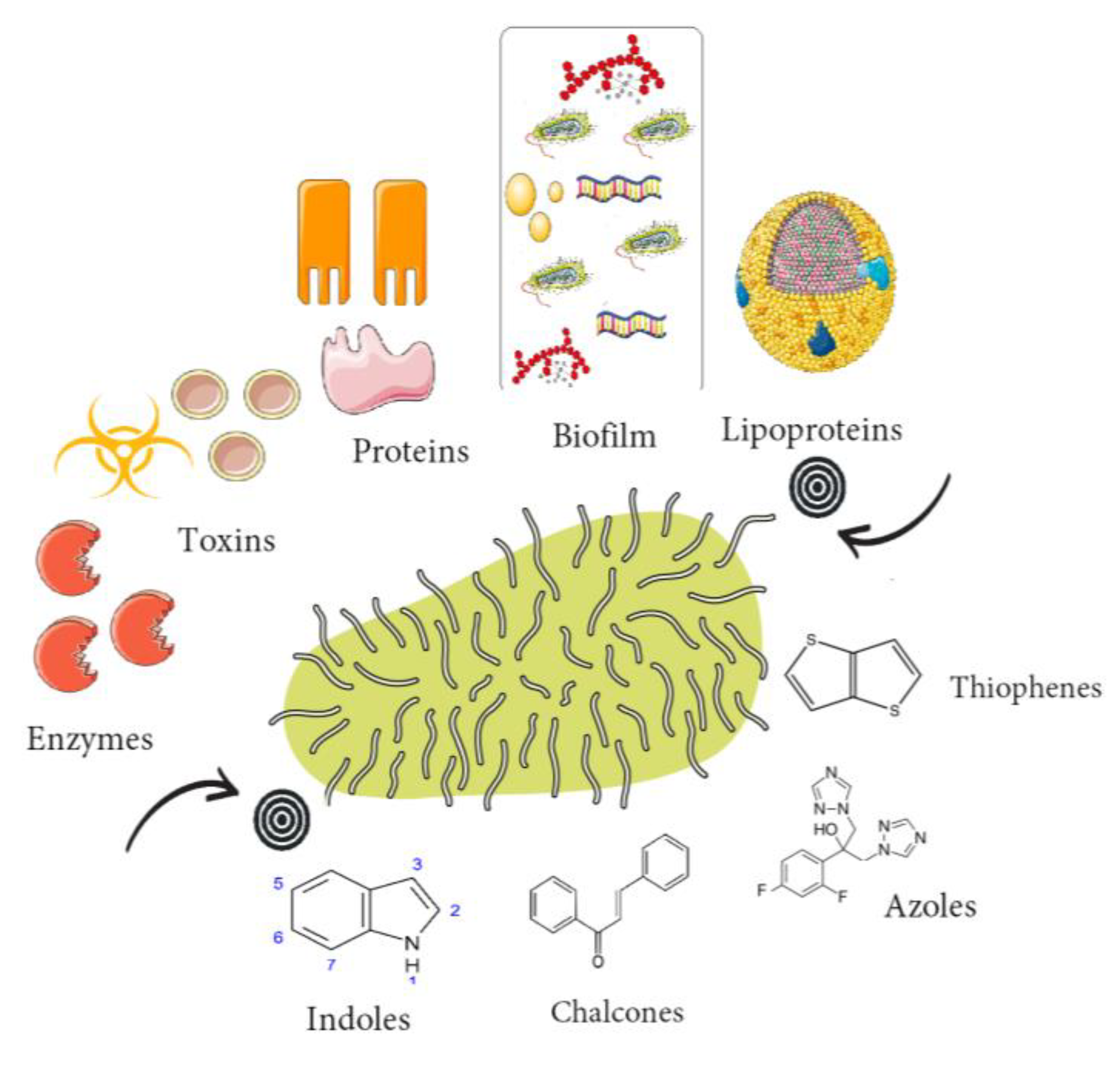
1. Introduction
2. Indoles
3. Azoles
4. Thiophenes
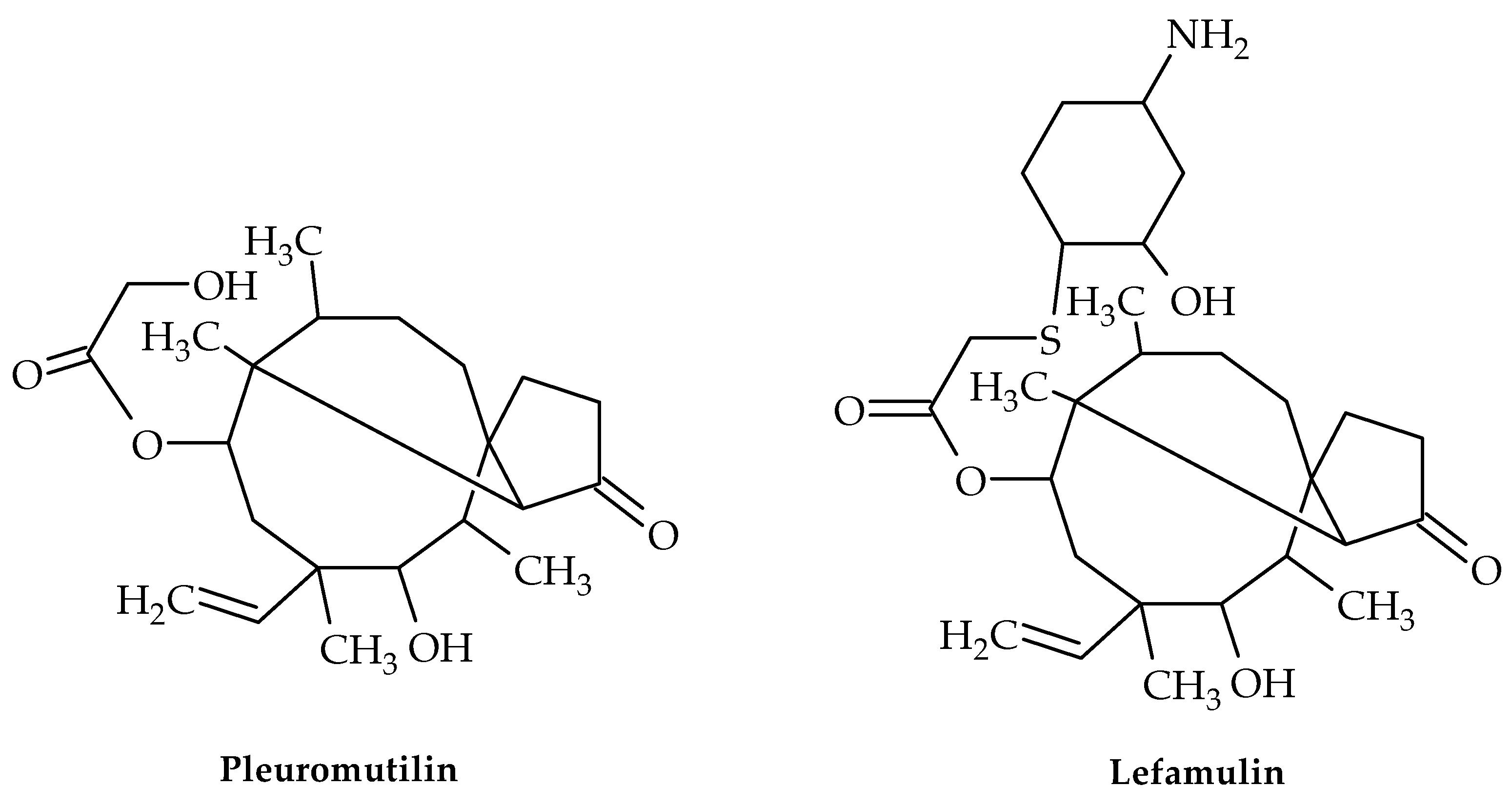
5. Pleuromutilin Derivatives
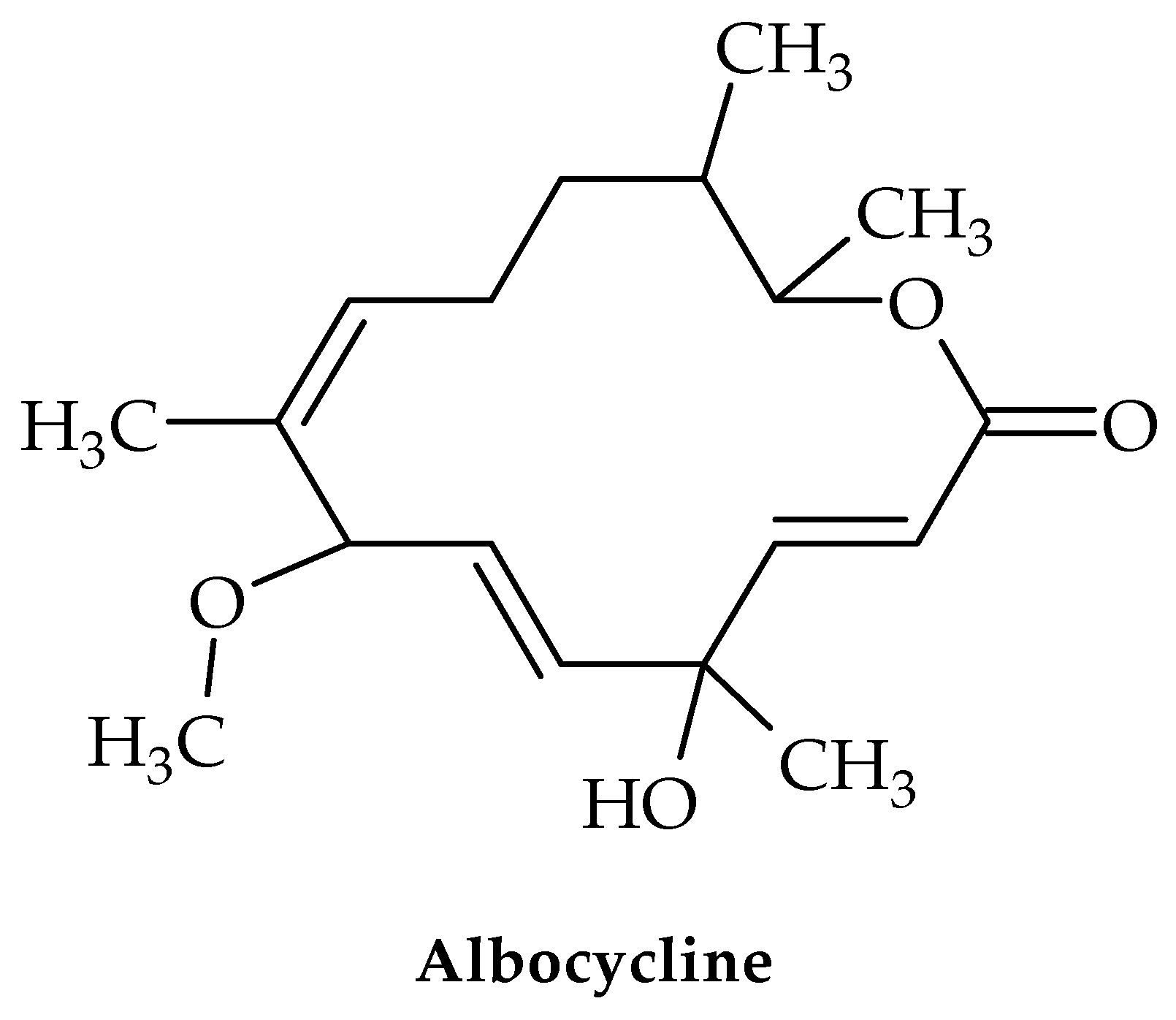
6. Albocyclin and General Lactone Derivatives
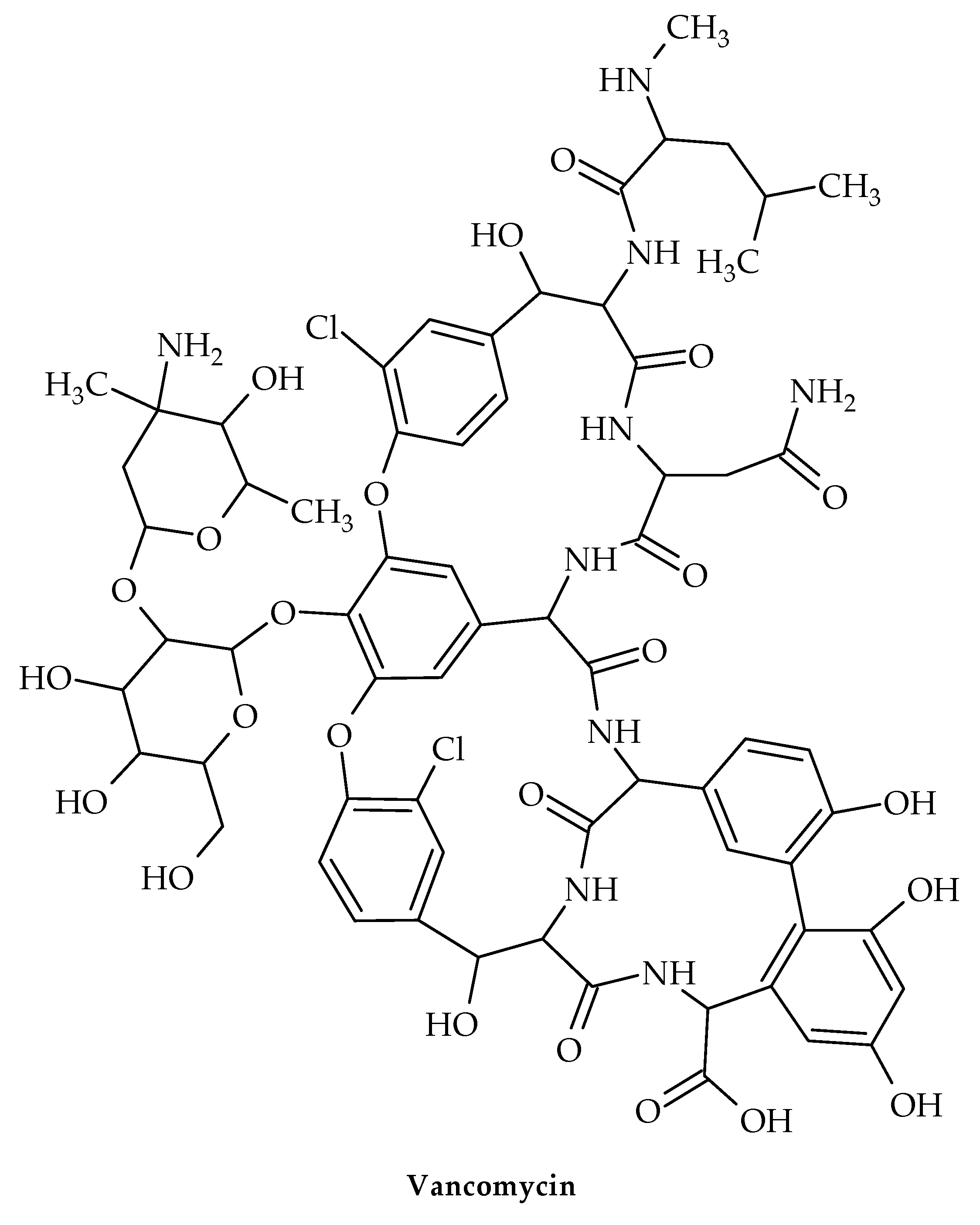
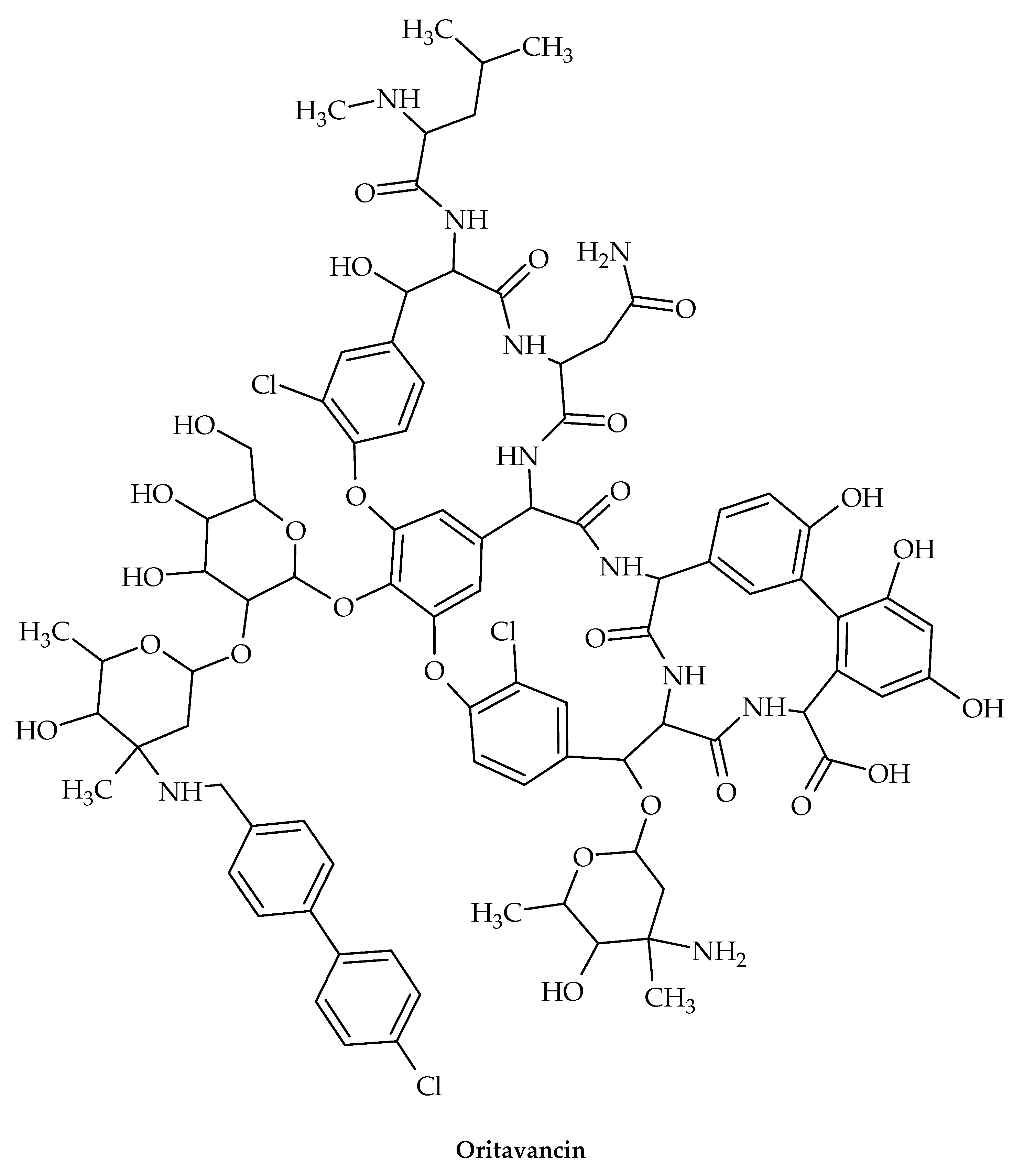
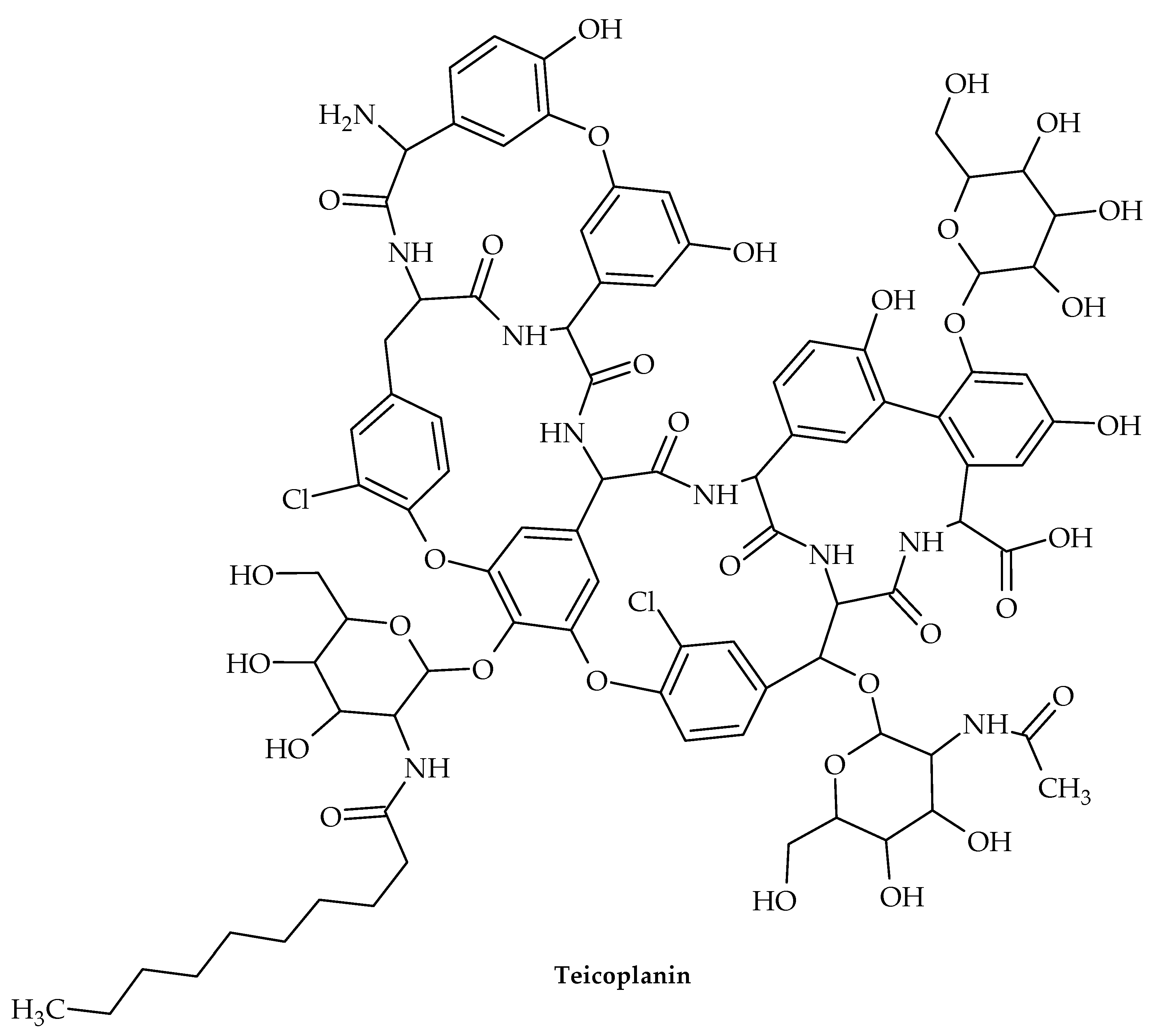
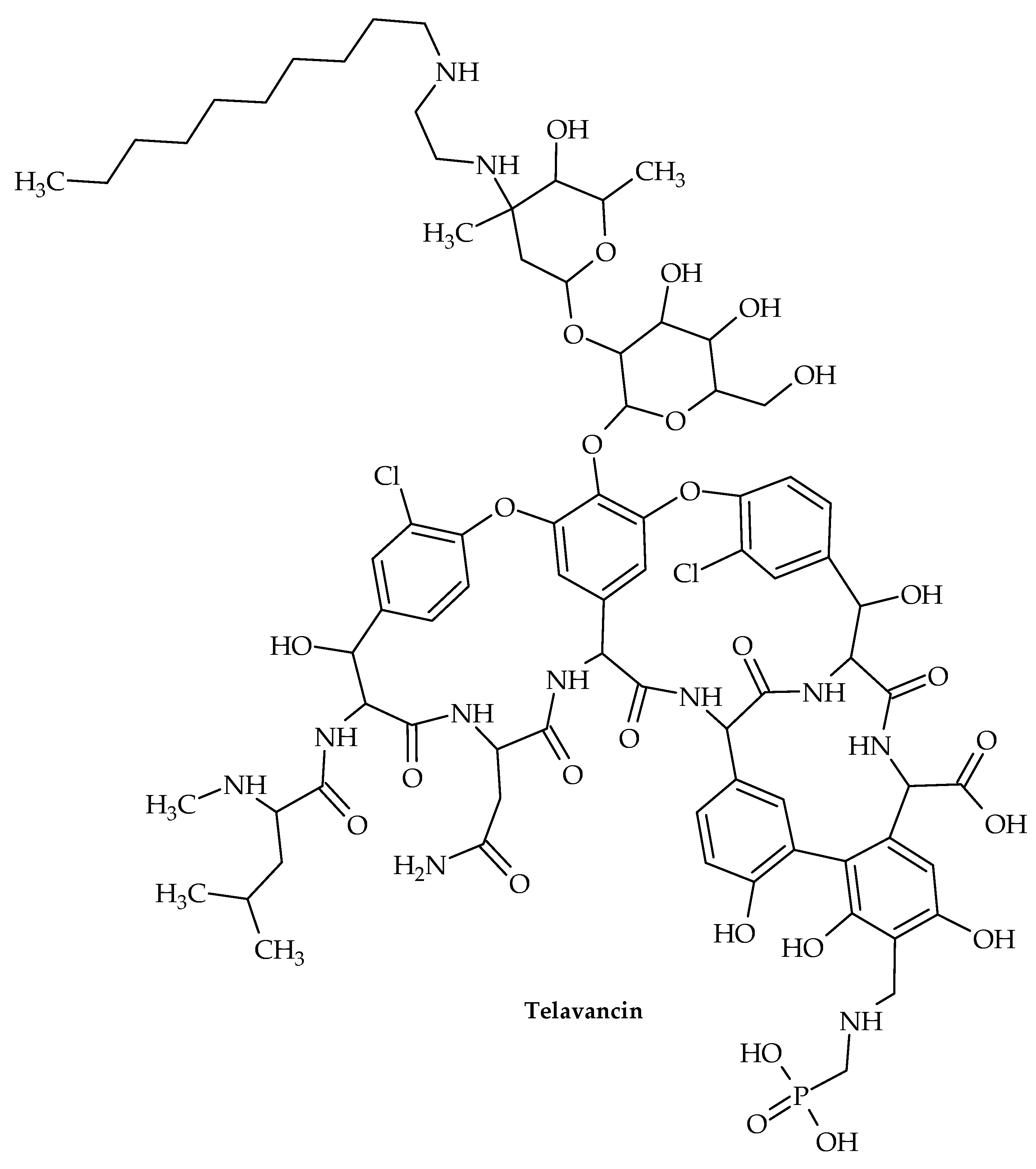
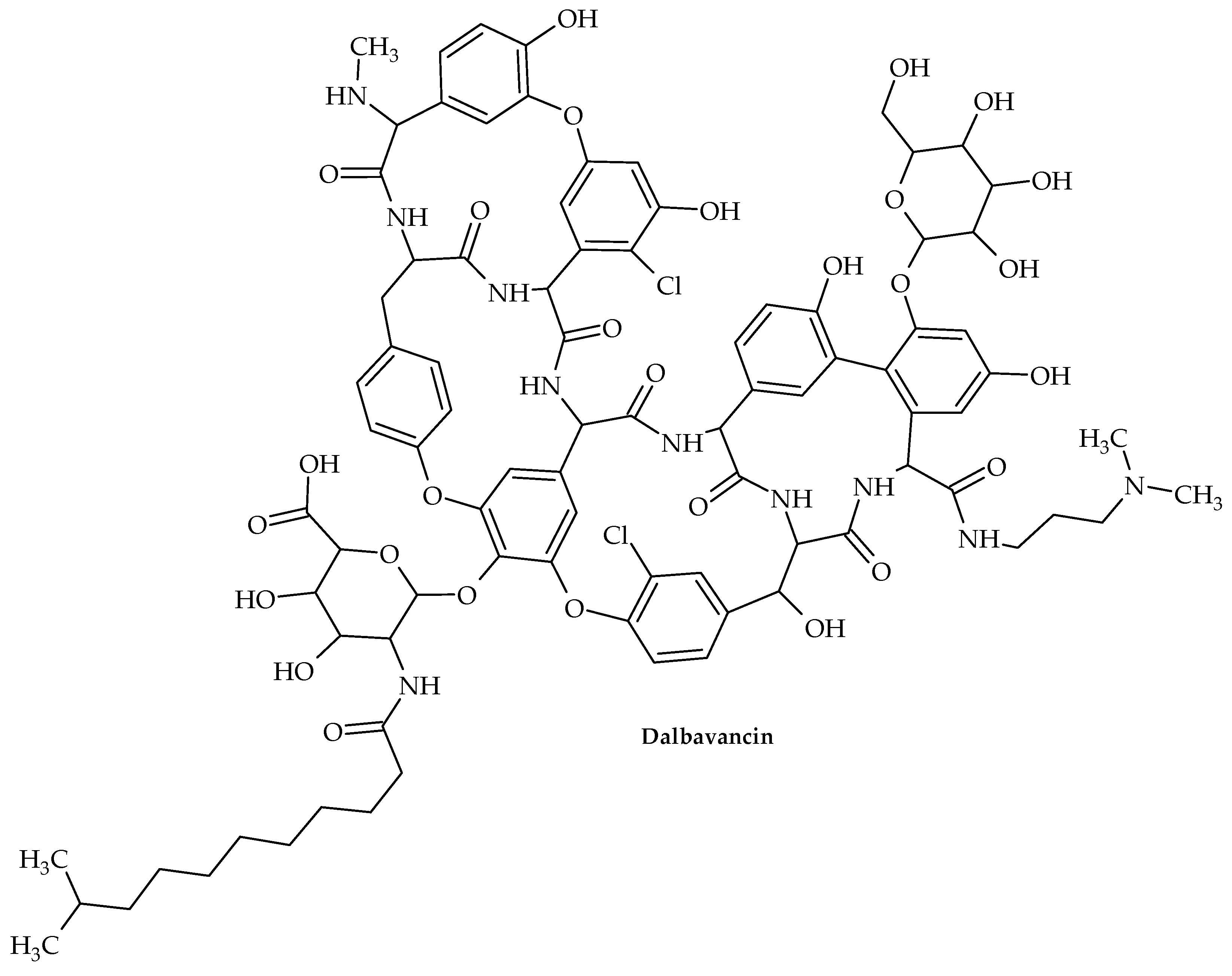
7. Glycopeptides
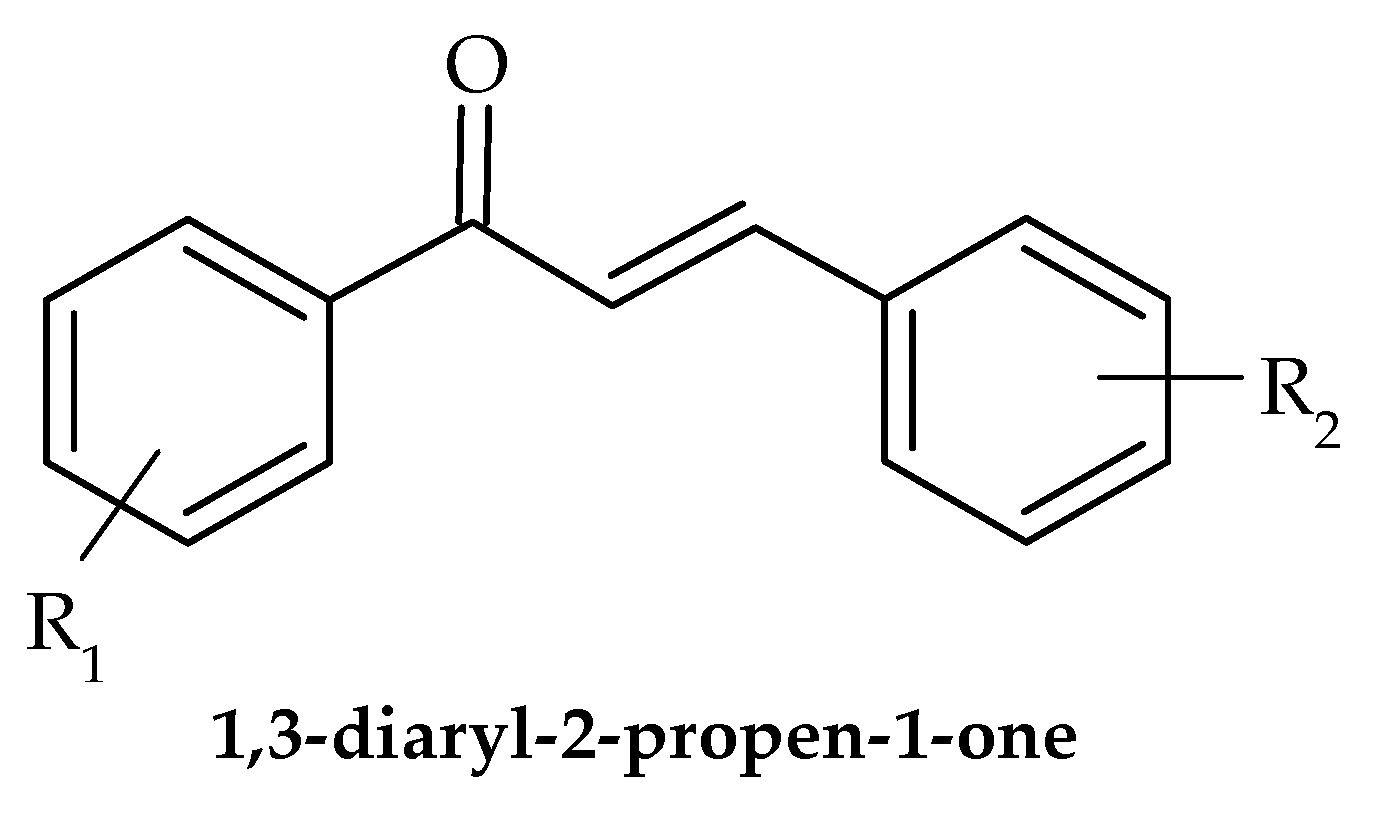
8. Chalcones
9. Future Perspectives
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Piddock, L.J. Reflecting on the final report of the O’Neill review on antimicrobial resistance. Lancet Infect. Dis. 2016, 16, 767–768. [Google Scholar] [CrossRef]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- Zhang, F.; Cheng, W. The Mechanism of Bacterial Resistance and Potential Bacteriostatic Strategies. Antibiotics 2022, 11, 1215. [Google Scholar] [CrossRef]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, G. Strategies to Overcome Antimicrobial Resistance (AMR) Making Use of Non-Essential Target Inhibitors: A Review. Int. J. Mol. Sci. 2019, 20, 5844. [Google Scholar] [CrossRef]
- Dörr, T.; Lewis, K.; Vulic, M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 2009, 5, e1000760. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Kalia, V.C. Quorum sensing inhibitors: An overview. Biotechnol. Adv. 2013, 31, 224–245. [Google Scholar] [CrossRef]
- Bhat, A.; Ahmad, I. Anti-virulence strategies: Stalling the micromachines of bacterial pathogens. Front. Microbiol. 2018, 9, 1456. [Google Scholar] [CrossRef]
- Jeong, J.W.; Lee, K.Y.; Kim, J.S. Natural products as sources of new fungicides. J. Microbiol. Biotechnol. 2009, 19, 1263–1270. [Google Scholar]
- Gutiérrez-Barranquero, J.A.; Reen, F.J.; McCarthy, R.R.; O’Gara, F. Deciphering the role of coumarin as a novel quorum sensing inhibitor suppressing virulence phenotypes in bacterial pathogens. Appl. Microbiol. Biotechnol. 2015, 99, 3303–3316. [Google Scholar] [CrossRef] [PubMed]
- Frederich, M.; Tits, M.; Angenot, L. Potential antimalarial activity of indole alkaloids. Trans R. Soc. Trop. Med. Hyg. 2008, 102, 11–19. [Google Scholar] [CrossRef]
- Singh, R.; Dubey, A.K. Lactone derivatives as potential antibacterial agents. Med. Chem. Res. 2018, 27, 1589–1611. [Google Scholar]
- Okano, A.; Isley, N.A.; Boger, D.L. Peripheral modifications of [9]dodecanylglycylgramicidin A: Discovery of novel antimicrobial agents with high potency and selectivity against methicillin-resistant Staphylococcus aureus (MRSA). J. Am. Chem. Soc. 2017, 139, 944–955. [Google Scholar]
- El-Messery, S.M.; Habib, E.E.; Al-Rashood, S.T.A.; Hassan, G.S. Synthesis, antimicrobial, anti-biofilm evaluation, and molecular modelling study of new chalcone linked amines derivatives. J. Enzym. Inhib. Med. Chem. 2018, 33, 818–832. [Google Scholar] [CrossRef]
- Peng, X.M.; Cai, G.X.; Zhou, C.H. Recent developments in azole compounds as antibacterial and antifungal agents. Curr. Top. Med. Chem. 2013, 13, 1963–2010. [Google Scholar] [CrossRef]
- Deryabin, D.; Inchagova, K.; Rusakova, E.; Duskaev, G. Coumarin’s Anti-Quorum Sensing Activity Can Be Enhanced When Combined with Other Plant-Derived Small Molecules. Molecules 2021, 26, 208. [Google Scholar] [CrossRef]
- Sethupathy, S.; Sathiyamoorthi, E.; Kim, Y.G.; Lee, J.H.; Lee, J. Antibiofilm and Antivirulence Properties of Indoles Against Serratia marcescens. Front. Microbiol. 2020, 11, 584812. [Google Scholar] [CrossRef]
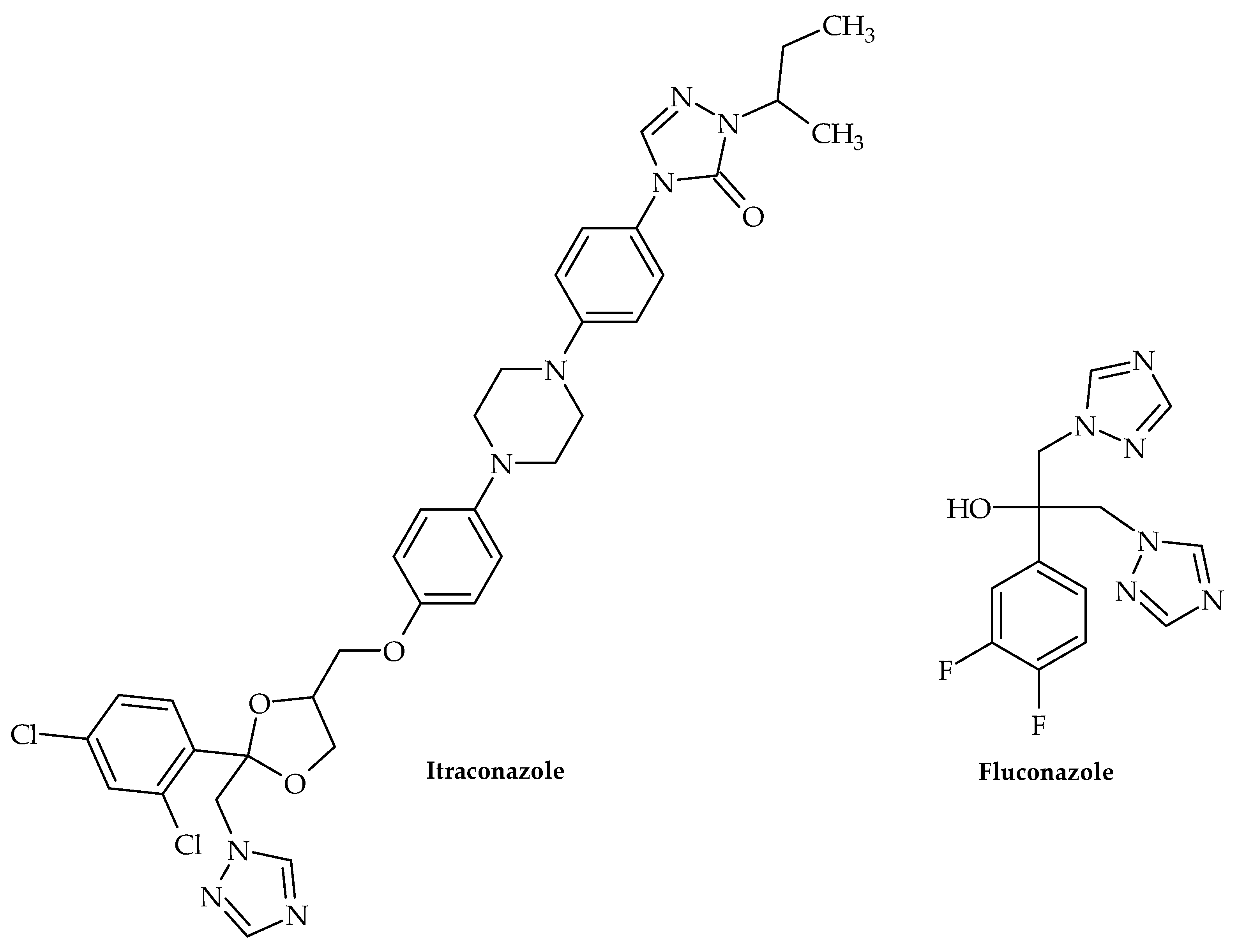
- Hooper, D.C.; Jacoby, G.A. Topoisomerase Inhibitors: Fluoroquinolone Mechanisms of Action and Resistance. Cold Spring Harb. Perspect.Med. 2016, 6(9), a025320. [Google Scholar] [CrossRef]
- Cappiello, F.; Loffredo, M.R.; Del Plato, C.; Cammarone, S.; Casciaro, B.; Quaglio, D.; Mangoni, M.L.; Botta, B.; Ghirga, F. The Revaluation of Plant-Derived Terpenes to Fight Antibiotic-Resistant Infections. Antibiotics 2020, 9, 325. [Google Scholar] [CrossRef]
- van Groesen, E.; Innocenti, P.; Martin, N.I. Recent Advances in the Development of Semisynthetic Glycopeptide Antibiotics: 2014–2022. ACS Infect. Dis. 2022, 8, 1381–1407. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.; Wang, S.; Xu, X.; Yi, Y.; Guo, W.; Liu, Y.; Liang, J. Chemical synthesis and biological activities of novel pleuromutilin derivatives with substituted amino moiety. PLoS ONE 2013, 8, e82595. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Design and Synthesis of Novel Antimicrobial Agents. Antibiotics 2023, 12, 628. [Google Scholar] [CrossRef]
- Balcerek, M.; Szmigiel-Bakalarz, K.; Lewańska, M.; Günther, D.; Oeckler, O.; Malik, M.; Morzyk-Ociepa, B. Experimental and computational study on dimers of 5-halo-1H-indole-2-carboxylic acids and their microbiological activity. J. Mol. Struct. 2023, 1274, 134492. [Google Scholar] [CrossRef]
- Salem, M.A.; Ragab, A.; El-Khalafawy, A.; Makhlouf, A.H.; Askar, A.A.; Ammar, Y.A. Design, synthesis, in vitro antimicrobial evaluation and molecular docking studies of indol-2-one tagged with morpholinosulfonyl moiety as DNA gyrase inhibitors. Bioorg. Chem. 2020, 96, 103619. [Google Scholar] [CrossRef]
- Alzahrani, Y.A.; Ammar, M.; Abu-Elghait, M.A.; Salem, M.; Assiri, T.E.; Ali, R. Development of novel indolin-2-one derivative incorporating thiazole moiety as DHFR and quorum sensing inhibitors: Synthesis, antimicrobial, and antibiofilm activities with molecular modelling study. Bioorg. Chem. 2022, 119, 105571. [Google Scholar] [CrossRef]
- Srikanth, D.; Joshi, S.V.; Shaik, M.G.; Pawar, G.; Bujji, S.; Kanchupalli, V.; Chopra, S.; Nanduri, S. A comprehensive review on potential therapeutic inhibitors of nosocomial Acinetobacter baumannii superbugs. Bioorg. Chem. 2022, 124, 105849. [Google Scholar] [CrossRef]
- Raorane, C.J.; Lee, J.H.; Lee, J. Rapid Killing and Biofilm Inhibition of Multidrug-Resistant Acinetobacter baumannii Strains and Other Microbes by Iodoindoles. Biomolecules 2020, 10, 1186. [Google Scholar] [CrossRef]
- Kim, Y.G.; Lee, J.H.; Park, S.; Lee, J. The Anticancer Agent 3,3′-Diindolylmethane Inhibits Multispecies Biofilm Formation by Acne-Causing Bacteria and Candida albicans. Microbiol. Spectr. 2022, 10, e0205621. [Google Scholar] [CrossRef]
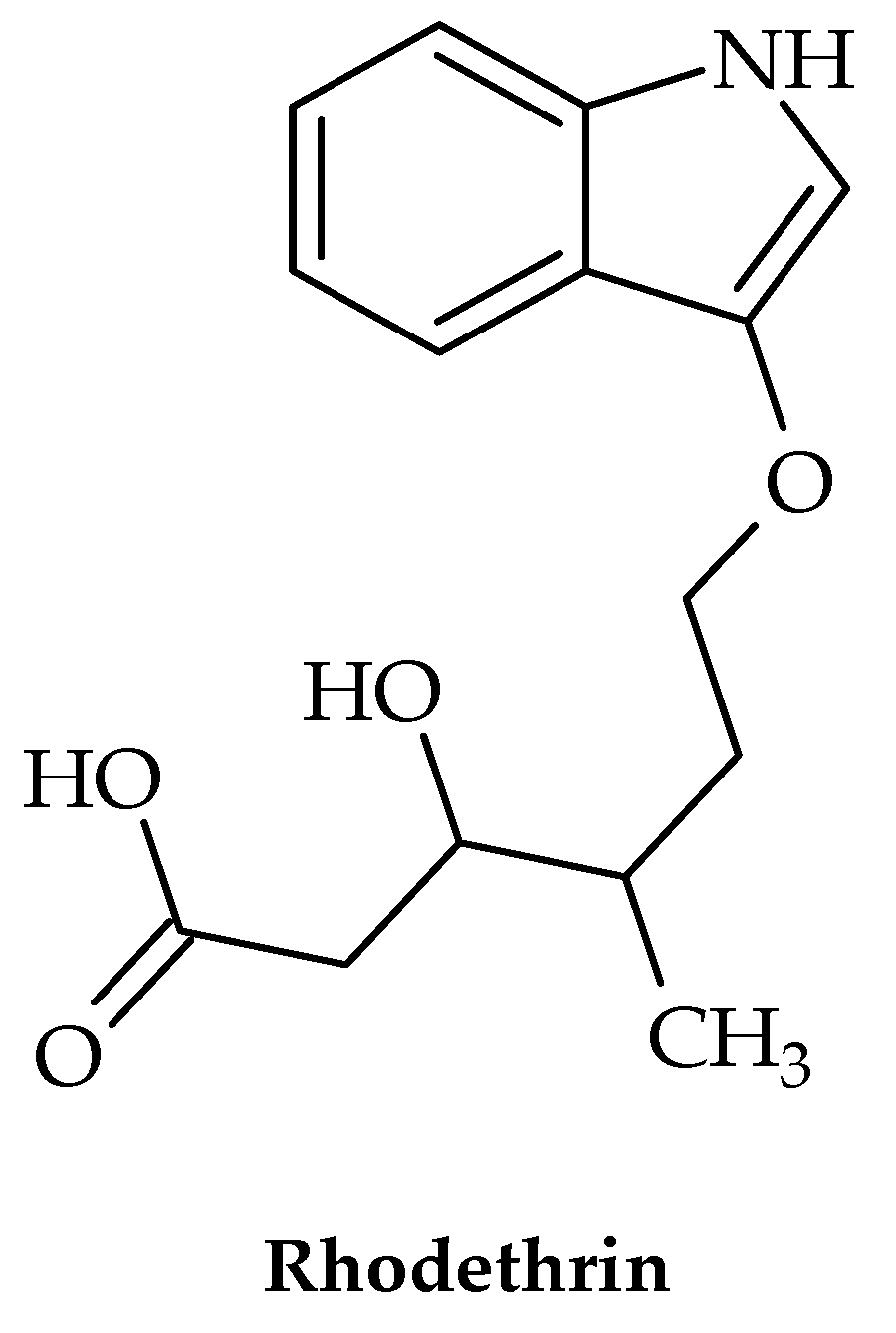
- Tatta, E.R.; Kumavath, R. Attenuation of Enterococcus faecalis biofilm formation by Rhodethrin: A combinatorial study with an antibiotic. Microb. Pathog. 2022, 163, 105401. [Google Scholar] [CrossRef]
- Amer, M.A.; Ramadan, M.A.; Attia, A.S.; Wasfi, R. Silicone Foley catheter simpregnated with microbial indole derivatives inhibit crystalline biofilm formation by Proteus mirabilis. Front. Cell. Infect. Microbiol. 2022, 12, 1010625. [Google Scholar] [CrossRef] [PubMed]
- Boya, B.R.; Lee, J.H.; Lee, J. Antibiofilm and Antimicrobial Activities of Chloroindoles Against Uropathogenic Escherichia coli. Front. Microbiol. 2022, 13, 872943. [Google Scholar] [CrossRef]
- Cernicchi, G.; Felicetti, T.; Sabatini, S. Microbial Efflux Pump Inhibitors: A Journey around Quinoline and Indole Derivatives. Molecules 2021, 26, 6996. [Google Scholar] [CrossRef]
- Ahmed, B.; Jailani, A.; Lee, J.H.; Lee, J. Effect of halogenated indoles on biofilm formation, virulence, and root surface colonization by Agrobacterium tumefaciens. Chemosphere 2022, 293, 133603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, Q.; Fu, S.; Janssen, C.; Eggermont, M.; Defoirdt, T. Indole decreases the virulence of the bivalve model pathogens Vibrio tasmaniensis LGP32 and Vibrio crassostreae J2-9. Sci. Rep. 2022, 12, 5749. [Google Scholar] [CrossRef]
- Sathiyamoorthi, E.; Faleye, O.S.; Lee, J.H.; Raj, V.; Lee, J. Antibacterial and Antibiofilm Activities of Chloroindoles Against Vibrio parahaemolyticus. Front. Microbiol. 2021, 12, 714371. [Google Scholar] [CrossRef]
- Li, Z.; Sun, F.; Fu, X.; Chen, Y. 5-Methylindole kills various bacterial pathogens and potentiates aminoglycoside against methicillin-resistant Staphylococcus aureus. PeerJ 2022, 10, e14010. [Google Scholar] [CrossRef]
- Nobre, L.S.; Todorovic, S.; Tavares, A.F.; Oldfield, E.; Hildebrandt, P.; Teixeira, M.; Saraiva, L.M. Binding of azole antibiotics to Staphylococcus aureus flavohemoglobin increases intracellular oxidative stress. J. Bacteriol. 2010, 192, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Olaifa, K.; Ajunwa, O.; Marsili, E. Electroanalytic evaluation of antagonistic effect of azole fungicides on Acinetobacter baumannii biofilms. Electrochim. Acta 2022, 405, 139837. [Google Scholar] [CrossRef]
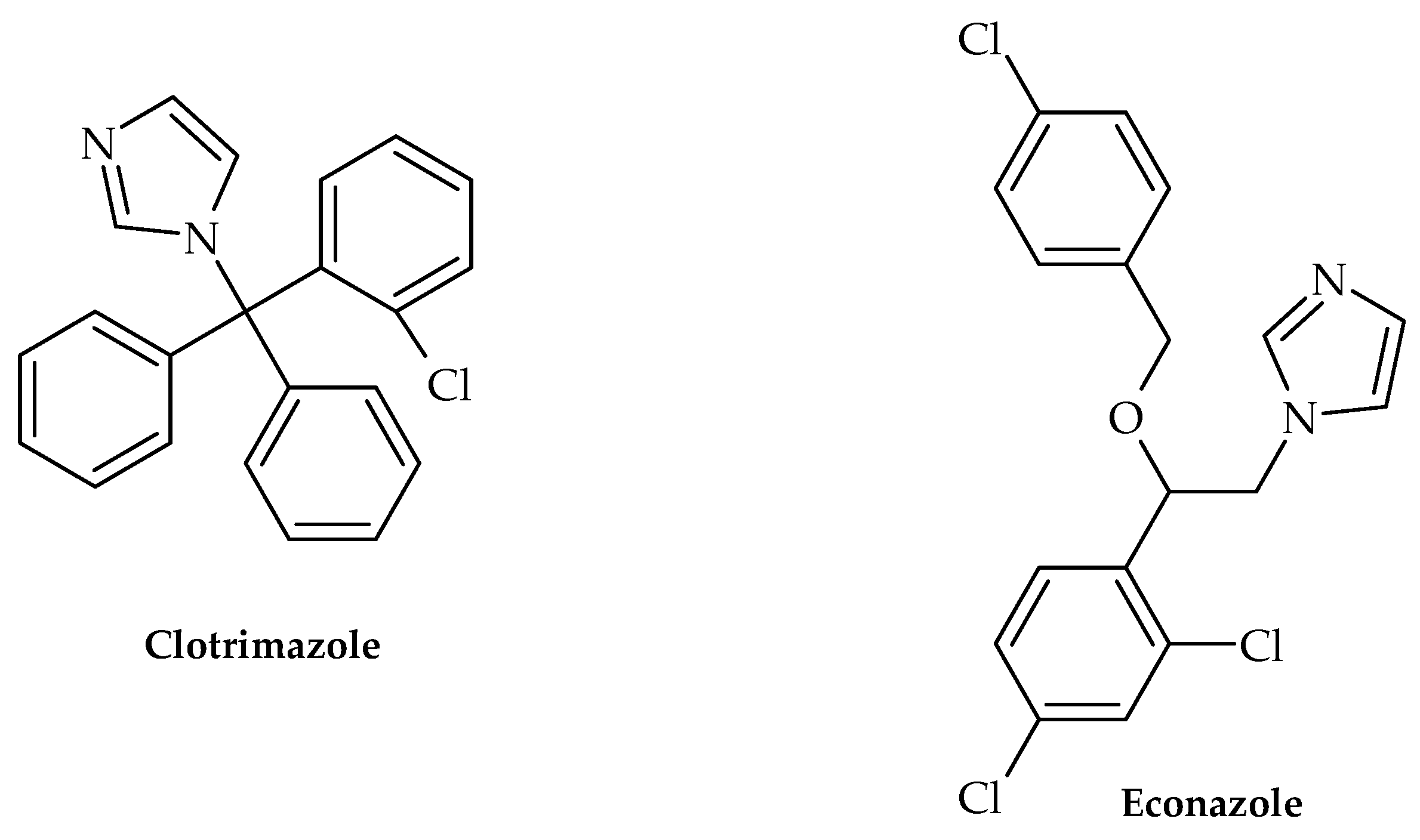
- Qiu, W.; Ren, B.; Dai, H.; Zhang, L.; Zhang, Q.; Zhou, X.; Li, Y. Clotrimazole and econazole inhibit Streptococcus mutans biofilm and virulence in vitro. Arch. Oral. Biol. 2017, 73, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Nasr, M.; Bondock, S.; Eid, S. Design, synthesis, antimicrobial evaluation and molecular docking studies of some new thiophene, pyrazole and pyridone derivatives bearing sulfisoxazole moiety. Eur. J. Med. Chem. 2014, 84, 491–504. [Google Scholar] [CrossRef]
- Gomes, M.P.; Correia, E.M.; Gomes, M.W.L.; Santos, C.C.C.; Barros, C.S.; Abreu, F.V.; de Antunes, L.S.; Ferreira, V.F.; Gonçalves, M.C.; Pinto, C.E.C.; et al. Antibacterial profile in vitro and in vivo of new 1,4-naphthoquinones tethered to 1,2,3-1h-triazoles against the planktonic growth of Streptococcus mutans. J. Braz. Chem. Soc. 2022, 33, 1028–1040. [Google Scholar] [CrossRef]
- Sapijanskaite-Banevic, B.; Palskys, V.; Vaickelioniene, R.; Šiugždaite, J.; Kavaliauskas, P.; Grybaite, B.; Mickevicius, V. Synthesis and Antibacterial Activity of New Azole, Diazole and Triazole Derivatives Based on p-Aminobenzoic Acid. Molecules 2021, 26, 2597. [Google Scholar] [CrossRef]
- Nechaeva, O.V.; Tikhomirova, E.I.; Zayarsky, D.A.; Bespalova, N.V.; Glinskaya, E.V.; Shurshalova, N.F.; Al Bayati, B.M.; Babailova, A.I. AntiBiofilm Activity of Polyazolidinammonium Modified with Iodine Hydrate Ions against Microbial Biofilms of Uropathogenic Coliform Bacteria. Bull. Exp. Biol. Med. 2017, 162, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Dawoud, N.T.A.; El-Fakharany, E.M.; Abdallah, A.E.; El-Gendi, H.; Lotfy, D.R. Synthesis, and docking studies of novel heterocycles incorporating the indazolyl thiazole moiety as antimicrobial and anticancer agents. Sci. Rep. 2022, 12, 3424. [Google Scholar] [CrossRef]
- Rando, D.G.; Doriguetto, A.C.; Tomich de Paula da Silva, C.H.; Ellena, J.; Sato, D.N.; Leite, C.Q.; Varanda, E.A.; Ferreira, E.I. A duplicated nitrotienyl derivative with antimycobacterial activity: Synthesis, X-ray crystallography, biological and mutagenic activity tests. Eur. J. Med. Chem. 2006, 41, 1196–1200. [Google Scholar] [CrossRef]
- Scotti, L.; Oliveira Lima, E.; da Silva, M.S.; Ishiki, H.; Oliveira Lima, I.; Oliveira Pereira, F.; Mendonça Junior, F.J.; Scotti, M.T. Docking and PLS studies on a set of thiophenes RNA polymerase inhibitors against Staphylococcus aureus. Curr. Top. Med. Chem. 2014, 14, 64–80. [Google Scholar] [CrossRef]
- Ramalingam, A.; Sarvanan, J. Synthesis, Docking and Antimicrobial Activity Studies of Some Novel Fused Thiophenes of Biological Interest. J. Young Pharm. 2020, 12, 118–124. [Google Scholar] [CrossRef]
- Metwally, H.M.; Khalaf, N.A.; Abdel-Latif, E. Synthesis, DFT investigations, antioxidant, antibacterial activity and SAR-study of novel thiophene-2-carboxamide derivatives. BMC Chem. 2023, 17, 6. [Google Scholar] [CrossRef]
- Kavanagh, F.; Hervey, A.; Robbins, W.J. Antibiotic substances from basidiomycetes. Proc. Natl. Acad. Sci. USA 1951, 37, 570–574. [Google Scholar] [CrossRef]
- Li, B.; Zhang, Z.; Zhang, J.F.; Liu, J.; Zuo, X.Y.; Chen, F.; Zhang, G.Y.; Fang, H.Q.; Jin, Z.; Tang, Y.Z. Design, synthesis and biological evaluation of pleuromutilin-Schiff base hybrids as potent anti-MRSA agents in vitro and in vivo. Eur. J. Med. Chem. 2021, 223, 113624. [Google Scholar] [CrossRef] [PubMed]
- Hogenauer, G. The mode of action of pleuromutilin derivatives: Location and properties of pleuromutilin binding site on Escherichia coli ribosomes. Eur. J. Biochem. 1975, 52, 93–98. [Google Scholar] [CrossRef]
- Paukner, S.; Riedl, R. Pleuromutilins: Potent drugs for resistant bugs-mode of action and resistance. Cold Spring Harb. Perspect. Med. 2017, 7, a027110. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, S.M.; Karlsson, M.; Johansson, L.B.; Vester, B. The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Mol. Microbiol. 2001, 41, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Hodgin, L.A.; Högenauer, G. The mode of action of pleuromutilin derivatives. Effect on cell-free polypeptide synthesis. Eur. J. Biochem. 1974, 47, 527–533. [Google Scholar] [CrossRef]
- Gürel, G.; Blaha, G.; Moore, P.B.; Steitz, T.A. U2504 determines the species specificity of the A-site cleft antibiotics: The structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J. Mol. Biol. 2009, 389, 146–156. [Google Scholar] [CrossRef]
- Adhikary, S.; Duggal, M.K.; Nagendran, S.; Chintamaneni, M.; Tuli, H.S.; Kaur, G. Lefamulin: A new hope in the field of community-acquired bacterial pneumonia. Cur. Pharm. Rep. 2022, 8, 418–426. [Google Scholar] [CrossRef]
- Veve, M.P.; Wagner, J.L. Lefamulin: Review of a promising novel pleuromutilin antibiotic. Pharmacotherapy 2018, 38, 935–946. [Google Scholar] [CrossRef]
- Yi, Y.; Zhang, J.; Zuo, J.; Zhang, M.; Yang, S.; Huang, Z.; Li, G.; Shang, R.; Lin, S. Novel pyridinium cationic pleuromutilin analogues overcoming bacterial multidrug resistance. Eur. J. Med. Chem. 2023, 251, 115269. [Google Scholar] [CrossRef]
- Deng, Y.; Tang, D.; Wang, Q.R.; Huang, S.; Fu, L.Z.; Li, C.H. Semi-synthesis, antibacterial activity, and molecular docking study of novel pleuromutilin derivatives bearing cinnamic acids moieties. Arch. Pharm. 2019, 352, e1800266. [Google Scholar] [CrossRef]
- Farney, E.P.; Feng, S.S.; Schafers, F.; Reisman, S.E. Total synthesis of (+)-pleuromutilin. J. Am. Chem. Soc. 2018, 140, 1267–1270. [Google Scholar] [CrossRef]
- Alberti, F.; Khairudin, K.; Venegas, E.R.; Davies, J.A.; Hayes, P.M.; Willis, C.L.; Bailey, A.M.; Foster, G.D. Heterologous expression reveals the biosynthesis of the antibiotic pleuromutilin and generates bioactive semi-synthetic derivatives. Nat. Commun. 2017, 8, 1831. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Yang, S.; Liu, Y.; Yin, B.; Zhao, Z.; Li, G.; Huang, Z.; Chen, L.; Liu, F.; Shang, R.; et al. Antibiotic resistance and drug modification: Synthesis, characterization and bioactivity of newly modified potent pleuromutilin derivatives with a substituted piperazine moiety. Bioorg. Chem. 2023, 132, 106353. [Google Scholar] [CrossRef]
- Novak, R. Are pleuromutilin antibiotics finally fit for human use? Ann. N. Y. Acad. Sci. 2011, 1241, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Waites, K.B.; Crabb, D.M.; Duffy, L.B.; Jensen, J.S.; Liu, Y.; Paukner, S. In vitro activities of lefamulin and other antimicrobial agents against macrolidesusceptible and macrolide-resistant Mycoplasma pneumoniae from the United States, Europe, and China. Antimicrob. Agents Chemother. 2017, 61, e02008-16. [Google Scholar] [CrossRef]
- Sader, H.S.; Biedenbach, D.J.; Paukner, S.; Ivezic-Schoenfeld, Z.; Jones, R.N. Antimicrobial activity of the investigational pleuromutilin compound BC3781 tested against Gram-positive organisms commonly associated with acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 2012, 56, 1619–1623. [Google Scholar] [CrossRef]
- Afshari, A.; Taheri, S.; Hashemi, M.; Norouzy, A.; Nematy, M.; Mohamadi, S. Methicillin- and Vancomycin-Resistant Staphylococcus aureus and Vancomycin-Resistant Enterococci Isolated from Hospital Foods: Prevalence and Antimicrobial Resistance Patterns. Curr. Microbiol. 2022, 79, 326. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Z.; Liu, Y.H.; Chen, J.X. Pleuromutilin and its derivatives-the lead compounds for novel antibiotics. Mini Rev. Med. Chem. 2012, 12, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Killeavy, E.E.; Jogl, G.; Gregory, S.T. Tiamulin-Resistant Mutants of the Thermophilic Bacterium Thermus thermophilus. Antibiotics 2020, 9, 313. [Google Scholar] [CrossRef]
- Klitgaard, R.N.; Ntokou, E.; Norgaard, K.; Biltoft, D.; Hansen, L.H.; Traedholm, N.M.; Kongsted, J.; Vester, B. Mutations in the bacterial ribosomal protein l3 and their association with antibiotic resistance. Antimicrob. Agents Chemother. 2015, 59, 3518–3528. [Google Scholar] [CrossRef]
- Patel, A.B.; Lighter, J.; Fulmer, Y.; Copin, R.; Ratner, A.J.; Shopsin, B. Retapamulin Activity Against Pediatric Strains of Mupirocin-resistant Methicillin-resistant Staphylococcus aureus. Pediatr. Infect. Dis. J. 2021, 40, 637–638. [Google Scholar] [CrossRef]
- Singh, R.; Gombosev, A.; Dutciuc, T.; Evans, K.; Portillo, L.M.; Hayden, M.K.; Gillen, D.; Peterson, E.; Tjoa, T.; Cao, C.; et al. Randomized Double-Blinded Placebo-Controlled Trial to Assess the Effect of Retapamulin for Nasal Decolonization of Mupirocin-Resistant Methicillin-Resistant Staphylococcus aureus Nasal Carriers. Open Forum. Infect. Dis. 2016, 3 (Suppl. S1), 301. [Google Scholar] [CrossRef]
- Surowiak, A.K.; Balcerzak, L.; Lochyński, S.; Strub, D.J. Biological Activity of Selected Natural and Synthetic Terpenoid Lactones. Int. J. Mol. Sci. 2021, 22, 5036. [Google Scholar] [CrossRef] [PubMed]
- Sartori, S.K.; Diaz, M.A.N.; Diaz-Muñoz, G. Lactones: Classification, Synthesis, Biological Activities, and Industrial Applications. Tetrahedron 2021, 84, 132001. [Google Scholar] [CrossRef]
- Fan, B.Z.; Hiasa, H.; Lv, W.; Brody, S.; Yang, Z.Y.; Aldrich, C.; Cushman, M.; Liang, J.H. Design, Synthesis and Structure-Activity Relationships of Novel 15-Membered Macrolides: Quinolone/Quinoline-Containing Sidechains Tethered to the C-6 Position of Azithromycin Acylides. Eur. J. Med. Chem. 2020, 193, 112222. [Google Scholar] [CrossRef]
- Mazur, M.; Masłowiec, D. Antimicrobial Activity of Lactones. Antibiotics 2022, 11, 1327. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, P.; Gawdzik, B.; Trzepizur, D.; Szymczak, M.; Skiba, G.; Raj, S.; Kramkowski, K.; Lizut, R.; Ostaszewski, R. δ-Lactones-A New Class of Compounds That Are Toxic to E. coli K12 and R2-R4 Strains. Materials 2021, 14, 2956. [Google Scholar] [CrossRef]
- Yokoe, H.; Yoshida, M.; Shishido, K. Total synthesis of (-)-Xanthatin. Tetrahedron Lett. 2008, 49, 3504–3506. [Google Scholar] [CrossRef]
- Matsuo, K.; Ohtsuki, K.; Yoshikawa, T.; Shisho, K.; Yokotani-Tomita, K.; Shinto, M. Total synthesis of xanthanolides. Tetrahedron 2010, 66, 8407–8419. [Google Scholar] [CrossRef]
- Nagahama, N.; Suzuki, M.; Awataguc, S.; Okuda, T. Studies on a new antibiotic, albocycline. I. isolation, purification and properties. J. Antibiot. 1967, 20, 261–266. [Google Scholar]
- Koyama, N.; Yotsumoto, M.; Onaka, H.; Tomoda, H. New structural scaffold 14-membered macrocyclic lactone ring for selective inhibitors of cell wall peptidoglycan biosynthesis in Staphylococcus aureus. J. Antibiot. 2013, 66, 303–304. [Google Scholar] [CrossRef]
- Daher, S.S.; Franklin, K.P.; Scherzi, T.; Dunman, P.M.; Andrade, R.B. Synthesis and biological evaluation of semi-synthetic albocycline analogs. Bioorg. Med. Chem. Lett. 2020, 30, 127509. [Google Scholar] [CrossRef]
- Wilson, D.N. The A-Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 393. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhou, G.; Ge, Y.; D’Ambrosio, E.A.; Eidem, T.M.; Blanchard, C.; Shehatou, C.; Chatare, V.K.; Dunman, P.M.; Valentine, A.M.; et al. Elucidating the inhibition of peptidoglycan biosynthesis in Staphylococcus aureus by albocycline, a macrolactone isolated from Streptomyces maizeus. Bioorg. Med. Chem. 2018, 26, 3453–3460. [Google Scholar] [CrossRef] [PubMed]
- Barna, J.C.J.; Williams, D.H. The structure and mode of action of glycopeptide antibiotics of the vancomycin group. Annu. Rev. Microbiol. 1984, 38, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, R. Glycopeptide antibiotics. In Drugs and the Pharmaceutical Sciences; Marcel Dekker: New York, NY, USA, 1994; Volume 63. [Google Scholar]
- Zeng, D.; Debabov, D.; Hartsell, T.L.; Cano, R.J.; Adams, S.; Schuyler, J.A.; McMillan, R.; Pace, J.L. Approved Glycopeptide Antibacterial Drugs: Mechanism of Action and Resistance. Cold Spring Harb. Perspect Med. 2016, 6, a026989. [Google Scholar] [CrossRef]
- Singh, M.; Chang, J.; Coffman, L.; Kim, S.J. Hidden mode of action of glycopeptide antibiotics: Inhibition of wall teichoic acid biosynthesis. J. Phys. Chem. B 2017, 121, 3925–3932. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Santa Maria, J.P.; Walker, S. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef]
- Cegelski, L.; Kim, S.J.; Hing, A.W.; Studelska, D.R.; O’Connor, R.D.; Mehta, A.K.; Schaefer, J. Rotational-echo double resonance characterization of the effects of vancomycin on cell wall synthesis in Staphylococcus aureus. Biochemistry 2002, 41, 13053–13058. [Google Scholar] [CrossRef]
- Muller, A.; Klockner, A.; Schneider, T. Targeting a cell wall biosynthesis hot spot. Nat. Prod. Rep. 2017, 34, 909–932. [Google Scholar] [CrossRef] [PubMed]
- Olademehin, O.P.; Shuford, K.L.; Kim, S.J. Molecular dynamics simulations of the secondary-binding site in disaccharide-modified glycopeptide antibiotics. Sci. Rep. 2022, 12, 7087. [Google Scholar] [CrossRef]
- Brotz, H.; Bierbaum, G.; Reynolds, P.E.; Sahl, H.G. The lantibiotic mersacidin inhibits peptidoglycan biosynthesis at the level of transglycosylation. Eur. J. Biochem. 1997, 246, 193–199. [Google Scholar] [CrossRef]
- Arthur, M.; Molinas, C.; Bugg, T.D.; Wright, G.D.; Walsh, C.T.; Courvalin, P. Evidence for in vivo incorporation of D-lactate into peptidoglycan precursors of vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 1992, 36, 867–869. [Google Scholar] [CrossRef]
- Nicas, T.I.; Mullen, D.L.; Flokowitsch, J.E.; Preston, D.A.; Snyder, N.J.; Zweifel, M.J.; Wilkie, S.C.; Rodriguez, M.J.; Thompson, R.C.; Cooper, R.D. Semisynthetic glycopeptide antibiotics derived from LY264826 active against vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 1996, 40, 2194–2199. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, D.; Stoneburner, A.; Shinabarger, D.L.; Arhin, F.F.; Belley, A.; Moeck, G.; Pillar, C.M. Comparative in vitro activity of oritavancin and other agents against vancomycin-susceptible and -resistant enterococci. J. Antimicrob. Chemother. 2016, 72, 622–624. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Tanaka, K.S.; Dietrich, E.; Far, A.R.; Schaefer, J. Locations of the hydrophobic side chains of lipoglycopeptides bound to the peptidoglycan of Staphylococcus aureus. Biochemistry 2013, 52, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
- Patti, G.J.; Kim, S.J.; Yu, T.Y.; Dietrich, E.; Tanaka, K.S.; Parr, T.R., Jr.; Far, A.R.; Schaefer, J. Vancomycin and oritavancin have different modes of action in Enterococcus faecium. J. Mol. Biol. 2009, 392, 1178–1191. [Google Scholar] [CrossRef]
- Kim, S.J.; Matsuoka, S.; Patti, G.J.; Schaefer, J. Vancomycin derivative with damaged D-Ala-D-Ala binding cleft binds to cross-linked peptidoglycan in the cell wall of Staphylococcus aureus. Biochemistry 2008, 47, 3822–3831. [Google Scholar] [CrossRef]
- Kim, S.J.; Cegelski, L.; Stueber, D.; Singh, M.; Dietrich, E.; Tanaka, K.S.; Parr, T.R.; Far, A.R.; Schaefer, J. Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis in Staphylococcus aureus. J. Mol. Biol. 2008, 377, 281–293. [Google Scholar] [CrossRef]
- Chun, T.; Pattem, J.; Gillis, R.B.; Dinu, V.T.; Yakubov, G.E.; Corfield, A.P.; Harding, S.E. Self-association of the glycopeptide antibiotic teicoplanin A2 in aqueous solution studied by molecular hydrodynamics. Sci. Rep. 2023, 13, 1969. [Google Scholar] [CrossRef] [PubMed]
- Parenti, F.; Beretta, G.; Berti, M.; Arioli, V. Teichomycins, new antibiotics from actinoplanes teichomyceticus nov. sp. J. Antibiot. 2006, 31, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.E. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1989, 8, 943–950. [Google Scholar] [CrossRef]
- Vimberg, V.; Gazak, R.; Szűcs, Z.; Borbás, A.; Herczegh, P.; Cavanagh, J.P.; Zieglerova, L.; Závora, J.; Adámková, V.; Novotna, G.B. Fluorescence assay to predict activity of the glycopeptide antibiotics. J. Antibiot. 2019, 72, 114–117. [Google Scholar] [CrossRef]
- Pintér, G.; Batta, G.; Kéki, S.; Mándi, A.; Komáromi, I.; Takács-Novák, K.; Sztaricskai, F.; Röth, E.; Ostorházi, E.; Rozgonyi, F.; et al. Diazo transfer–click reaction route to new, lipophilic teicoplanin and ristocetin aglycon derivatives with high antibacterial and anti-influenza virus activity: An aggregation and receptor binding study. J. Med. Chem. 2009, 52, 6053–6061. [Google Scholar] [CrossRef] [PubMed]
- Tollas, S.; Bereczki, I.; Sipos, A.; Rőth, E.; Batta, G.; Daróczi, L.; Kéki, S.; Ostorházi, E.; Rozgonyi, F.; Herczegh, P. Nano-sized clusters of a teicoplanin ψ-aglycon-fullerene conjugate. Synthesis, antibacterial activity and aggregation studies. Eur. J. Med. Chem. 2012, 54, 943–948. [Google Scholar] [CrossRef]
- Corno, G.; Coci, M.; Giardina, M.; Plechuk, S.; Campanile, F.; Stefani, S. Antibiotics promote aggregation within aquatic bacterial communities. Front. Microbiol. 2014, 5, 297. [Google Scholar] [CrossRef]
- Higgins, D.L.; Chang, R.; Debabov, D.V.; Leung, J.; Wu, T.; Krause, K.M.; Sandvik, E.; Hubbard, J.M.; Kaniga, K.; Schmidt, D.E., Jr.; et al. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2005, 49, 1127–1134. [Google Scholar] [CrossRef]
- Draghi, D.C.; Benton, B.M.; Krause, K.M.; Thornsberry, C.; Pillar, C.; Sahm, D.F. Comparative surveillance study of telavancin activity against recently collected gram-positive clinical isolates from across the United States. Antimicrob. Agents Chemother. 2008, 52, 2383–2388. [Google Scholar] [CrossRef]
- Food and Drug Administration. FDA Labelling Information. 2009. Available online: https://www.fda.gov/files/food/published/Food-Labeling-Guide-%28PDF%29.pdf (accessed on 15 November 2010).
- Das, B.; Sarkar, C.; Das, D.; Gupta, A.; Kalra, A.; Sahni, S. Telavancin: A novel semisynthetic lipoglycopeptide agent to counter the challenge of resistant Gram-positive pathogens. Ther. Adv. Infect. Dis. 2017, 2, 49–73. [Google Scholar] [CrossRef]
- Breukink, E.J.; Humphrey, P.P.A.; Benton, B.M.; Visscher, I. Evidence for a multivalent interaction between telavancin and membrane-bound lipid II. In Proceedings of the 46th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, USA, 27–30 September 2006. [Google Scholar]
- Benton, B.; Breukink, E.; Visscher, I.; Debabov, D.; Lunde, C.; Janc, J.; Humphrey, P. Telavancin inhibits peptidoglycan biosynthesis through preferential targeting of transglycosylation: Evidence for a multivalent interaction between telavancin and lipid II. Int. J. Antimicrob. Agents 2007, 29, 51–52. [Google Scholar] [CrossRef]
- Lunde, C.S.; Hartouni, S.R.; Janc, J.W.; Mammen, M.; Humphrey, P.P.; Benton, B.M. Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor lipid II. Antimicrob. Agents Chemother. 2009, 53, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Dunne, M.W.; Puttagunta, S.; Sprenger, C.R.; Rubino, C.; Van Wart, S.; Baldassarre, J. Extended –duration dosing and distribution of dalbavancin into bone and artic ular tissue. Antimicrob. Agents Chemother. 2015, 59, 1849–1855. [Google Scholar] [CrossRef]
- Fazili, T.; Bansal, E.; Garner, D.; Gomez, M.; Stornelli, N. Dalbavancin as sequential therapy for infective endocarditis due to Gram-positive organisms: A review. Int. J. Antimicrob. Agents 2023, 61, 106749. [Google Scholar] [CrossRef]
- Zhang, Y.; Sass, A.; Van Acker, H.; Wille, J.; Verhasselt, B.; Van Nieuwerburgh, F.; Kaever, V.; Crabbé, A.; Coenye, T. Coumarin Reduces Virulence and Biofilm Formation in Pseudomonas aeruginosa by Affecting Quorum Sensing, Type III Secretion and C-di-GMP Levels. Front. Microbiol. 2018, 9, 1952. [Google Scholar] [CrossRef] [PubMed]
- Ušjak, D.; Ivković, B.; Božić, D.D.; Bošković, L.; Milenković, M. Antimicrobial activity of novel chalcones and modulation of virulence factors in hospital strains of Acinetobacter baumannii and Pseudomonas aeruginosa. Microb. Pathog. 2019, 131, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Ušjak, D.; Dinić, M.; Novović, K.; Ivković, B.; Filipović, N.; Stevanović, M.; Milenković, M.T. Methoxy-Substituted Hydroxychalcone Reduces Biofilm Production, Adhesion and Surface Motility of Acinetobacter baumannii by Inhibiting ompA Gene Expression. Chem. Biodivers. 2021, 18, e2000786. [Google Scholar] [CrossRef]
- Qi, P.; Wang, N.; Zhang, T.; Feng, Y.; Zhou, X.; Zeng, D.; Meng, J.; Liu, L.; Jin, L.; Yang, S. Anti-Virulence Strategy of Novel Dehydroabietic Acid Derivatives: Design, Synthesis, and Antibacterial Evaluation. Int. J. Mol. Sci. 2023, 24, 2897. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.W.; Ruan, L.Y.; Chen, H.J.; Luo, H.Z.; Jiang, H.; Wang, J.S.; Jia, A.Q. Inhibition of Quorum Sensing and Virulence in Serratia marcescens by Hordenine. J. Agric. Food Chem. 2019, 67, 784–795. [Google Scholar] [CrossRef]
- Du, Y.; Sun, J.; Gong, Q.; Wang, Y.; Fu, P.; Zhu, W. New α-Pyridones with Quorum-Sensing Inhibitory Activity from Diversity-Enhanced Extracts of a Streptomyces sp. Derived from Marine Algae. J. Agric. Food Chem. 2018, 66, 1807–1812. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Compounds | Compound | Bacteria | Type of Activity | Reference |
|---|---|---|---|---|
| Indole | 5-halo-1H-indole-2-carboxylic acids | Listeria monocytogenes | Inhibits the growth of bacteria | [25] |
| Indole | indol-2-one with morpholinosulfonyl | Staphylococcusaureus | Inhibitor of DNA gyrase | [26] |
| Indole | thiazolo-indolin-2-one | S. aureus (ATCC 29213) P. aeruginosa (ATCC 9027) | Inhibits biofilm formation | [27] |
| Indole | d-pyrimido[4,5-b] indole | Acinetobacter baumanii | Inhibits the growth of bacteria | [28] |
| Indole | 3-amino indoles | Multi-drug resistant A. baumanii | Inhibits the growth of bacteria | [28] |
| Indole | 4-hydroxy-2-pyridone derivatives containing indolyl | Multi-drug resistant A. baumanii | Inhibits the growth of bacteria | [28] |
| Indole | 2-hydrazino2-imidazoline | Multi-drug resistant A. baumanii | Inhibits the growth of bacteria | [28] |
| Indole | bis-indolyl methane | Multi-drug resistant A. baumanii | Inhibits the growth of bacteria | [28] |
| Indole | 5-iodoindole | A. baumanii, Escherichia coli, S. aureus | Inhibits the growth of bacteria, decreases motility, disrupts biofilm formation | [29] |
| Indole | 3,3′-diindolylmethane | Cutibacterium acnes S. aureus | Inhibits the growth of bacteria | [30] |
| Indole | indole terpenoid compound rhodethrin | Enterococcus faecalis | Inhibits biofilm formation | [31] |
| Indole | indole extract from the supernatant of the rhizobacterium Enterobacter sp. Zch127 | Proteus mirabilis | Inhibits biofilm formation | [32] |
| Indole | 4-chloroindole, 5-chloroindole, 5-chloro 2-methyl indole | E. coli | Decreases bacterial motility, disrupts biofilm formation | [33] |
| Indole | 4-chloroindole, 6-iodoindole, 5-chloro-2-methyl indole | Agrobacterium tumefaciens | Decreases swimming motility, the production of exopolysaccharide and exoprotease, and cell surface hydrophobicity and biofilm formation | [35] |
| Indole | indole | V. tasmaniensis LGP32 and V. crassostreae J2-9 | Decreases swimming motility, inhibits biofilm formation | [36] |
| Indole | 4-chloroindole, 7-chloroindole, 4-iodoindole, and 7-iodoindole | V. parahaemolyticus | Inhibits biofilm formation | [37] |
| Azole | naphthalimide-containing nitroimidazoles | A. baumannii | Inhibits the growth of bacteria | [28] |
| Azole | itraconazole and fluconazole | A. baumannii | Inhibits biofilm formation | [40] |
| Azole | clotrimazole, econazole | Streptococcus mutans | Inhibits biofilm formation | [41] |
| Azole | pyrazole 30 | Pneumocystis vulgaris Klebsiella pneumoniae | Inhibits the growth of bacteria | [42] |
| Azole | 1,4-naphthoquinones linked to 1,2,3-1H-triazoles—compounds (9e, 9h, 9i, and 9j) | S. mutans | Inhibits the growth of bacteria | [43] |
| Azole | binaphthyl-1,2,3-triazole peptidomimetics | A. baumannii | Inhibits the growth of bacteria | [28] |
| Azole | heterocycle compounds with indazolylthiazole moiety (compounds 2, 3, 7, and 8) | S. mutans, P.aeruginosa | Inhibits biofilm production | [46] |
| Azole | N-(2-(1H-imidazol-4-yl)ethyl)-2-(2,3-dihydroxyphenyl)-N-hydroxy-5-methyloxazole-4-carboxamide | A. baumannii | Inhibits the growth of bacteria | [28] |
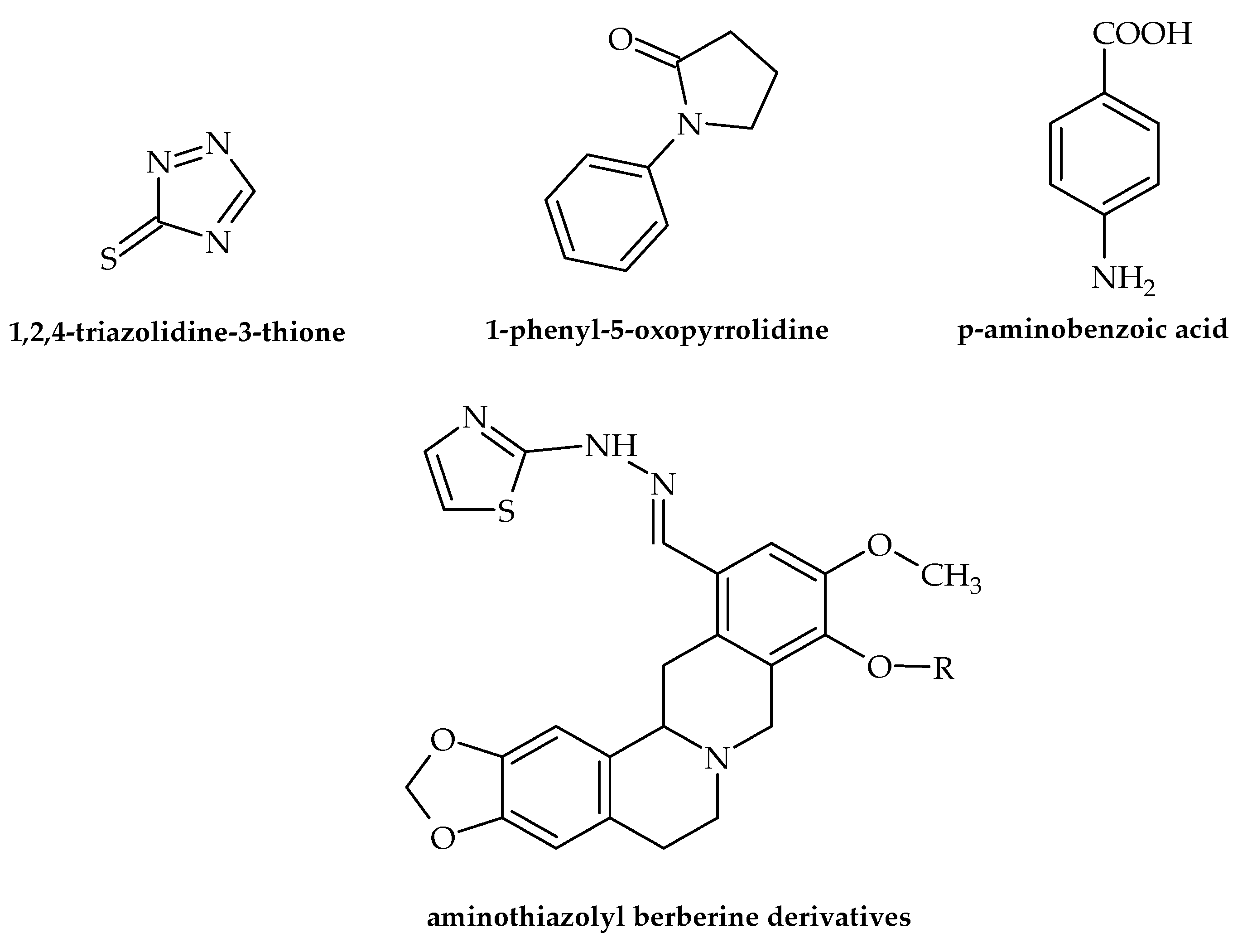
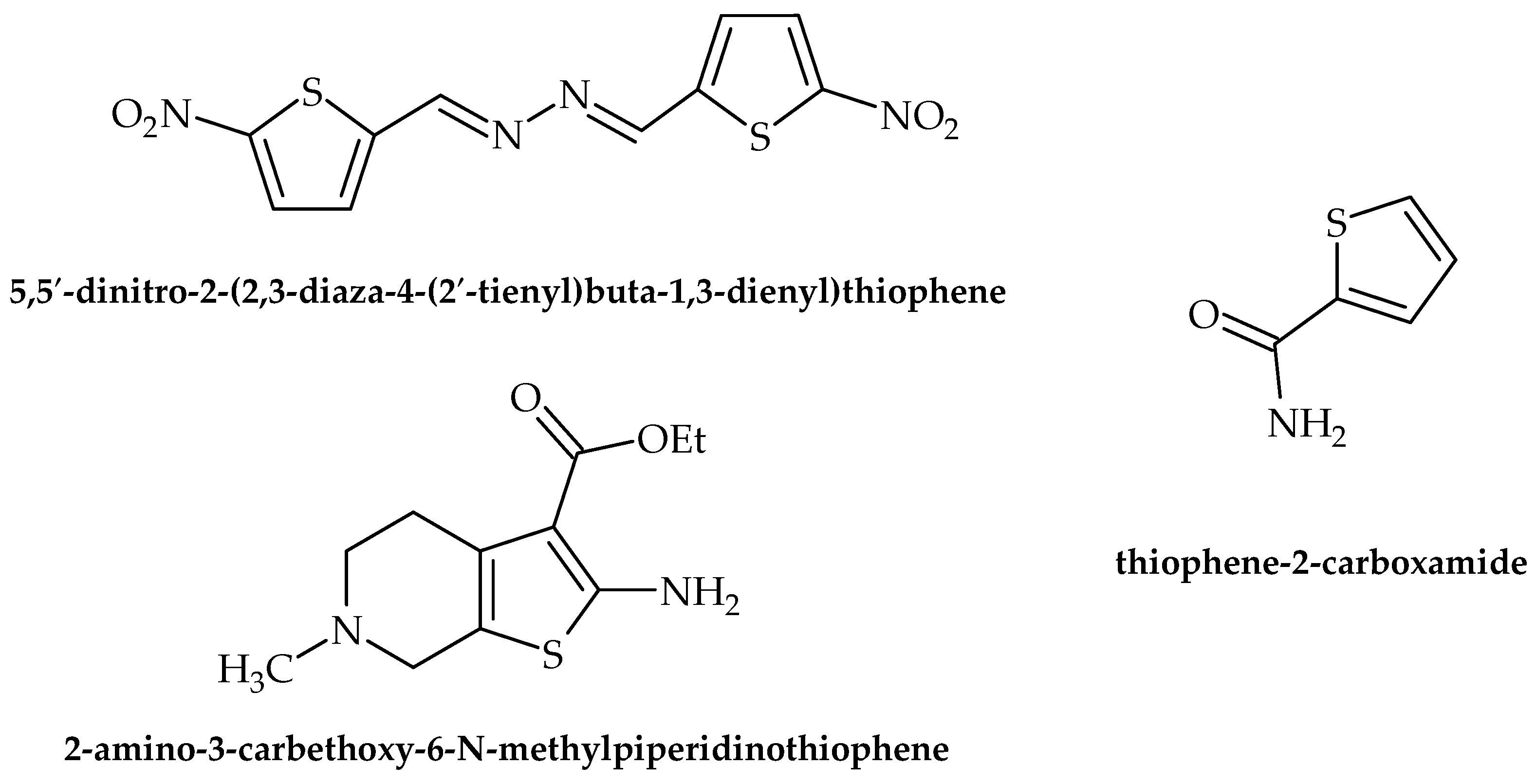
| Thiophene | 5,5′-dinitro-2-(2,3-diaza-4-(2′-tienyl)buta-1,3-dienyl)thiophene | Mycobacterium avium M. kansasei | Inhibits the growth of bacteria | [47] |
| Thiophene | 2 -amino-3-carbethoxy-6-N methyl piperidino thiophene | B. subtilis E. coli | Inhibits the growth of bacteria | [49] |
| Thiophene | thiophene-2-carboxamide | S. aureus, B. subtilis, E. coli, P. aeruginosa | Inhibits the growth of bacteria | [50] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stojković, D.; Petrović, J.; Carević, T.; Soković, M.; Liaras, K. Synthetic and Semisynthetic Compounds as Antibacterials Targeting Virulence Traits in Resistant Strains: A Narrative Updated Review. Antibiotics 2023, 12, 963. https://doi.org/10.3390/antibiotics12060963
Stojković D, Petrović J, Carević T, Soković M, Liaras K. Synthetic and Semisynthetic Compounds as Antibacterials Targeting Virulence Traits in Resistant Strains: A Narrative Updated Review. Antibiotics. 2023; 12(6):963. https://doi.org/10.3390/antibiotics12060963
Chicago/Turabian StyleStojković, Dejan, Jovana Petrović, Tamara Carević, Marina Soković, and Konstantinos Liaras. 2023. "Synthetic and Semisynthetic Compounds as Antibacterials Targeting Virulence Traits in Resistant Strains: A Narrative Updated Review" Antibiotics 12, no. 6: 963. https://doi.org/10.3390/antibiotics12060963
APA StyleStojković, D., Petrović, J., Carević, T., Soković, M., & Liaras, K. (2023). Synthetic and Semisynthetic Compounds as Antibacterials Targeting Virulence Traits in Resistant Strains: A Narrative Updated Review. Antibiotics, 12(6), 963. https://doi.org/10.3390/antibiotics12060963