Abstract
Salmonella enterica subspecies enterica is one of the most important foodborne pathogens and the causative agent of salmonellosis, which affects both humans and animals producing numerous infections every year. The study and understanding of its epidemiology are key to monitoring and controlling these bacteria. With the development of whole-genome sequencing (WGS) technologies, surveillance based on traditional serotyping and phenotypic tests of resistance is being replaced by genomic surveillance. To introduce WGS as a routine methodology for the surveillance of food-borne Salmonella in the region, we applied this technology to analyze a set of 141 S. enterica isolates obtained from various food sources between 2010 and 2017 in the Comunitat Valenciana (Spain). For this, we performed an evaluation of the most relevant Salmonella typing methods, serotyping and sequence typing, using both traditional and in silico approaches. We extended the use of WGS to detect antimicrobial resistance determinants and predicted minimum inhibitory concentrations (MICs). Finally, to understand possible contaminant sources in this region and their relationship to antimicrobial resistance (AMR), we performed cluster detection combining single-nucleotide polymorphism (SNP) pairwise distances and phylogenetic and epidemiological data. The results of in silico serotyping with WGS data were highly congruent with those of serological analyses (98.5% concordance). Multi-locus sequence typing (MLST) profiles obtained with WGS information were also highly congruent with the sequence type (ST) assignment based on Sanger sequencing (91.9% coincidence). In silico identification of antimicrobial resistance determinants and minimum inhibitory concentrations revealed a high number of resistance genes and possible resistant isolates. A combined phylogenetic and epidemiological analysis with complete genome sequences revealed relationships among isolates indicative of possible common sources for isolates with separate sampling in time and space that had not been detected from epidemiological information. As a result, we demonstrate the usefulness of WGS and in silico methods to obtain an improved characterization of S. enterica enterica isolates, allowing better surveillance of the pathogen in food products and in potential environmental and clinical samples of related interest.
1. Introduction
The genus Salmonella is classified into different serovars or serotypes on the basis of surface antigens according to the White–Kauffmann–Le Minor scheme [1]. This scheme has been a useful and efficient way to subtype Salmonella, facilitating international communication and comparison. Nevertheless, this typing scheme does not always correctly reflect the evolutionary relationships among the different strains of S. enterica subsp. enterica. On the one hand, most serotypes are polyphyletic, deriving from multiple independent ancestors [2]. On the other hand, homologous recombination and chromosomal mutations have resulted in genotypically and phenotypically diverse subtypes evolved from the same serovars [3]. Furthermore, serotyping has been traditionally performed by serological tests. This is a laborious technique, and the recognition of the wide range of serovars that abound in food products requires a wide panel of antigens that is not available in many laboratories. Moreover, it may lead to imprecise results [4].
Serotyping by serology has been progressively complemented or replaced by molecular typing methods, such as multi-locus sequence-based typing (MLST) [5,6,7,8]. However, these methods do not have enough discriminatory power to differentiate among closely related strains [8,9]. This limitation renders them insufficient for many epidemiological studies.
High-throughput sequencing (HTS) technologies have allowed rapid and affordable analyses of complete genomes [10]. Whole-genome sequences (WGS) allow an unprecedented level of discrimination among genetically related isolates, which can be applied to many interesting questions, such as accurate phylogenetic and phylogenomic analyses [11]. Moreover, WGS also facilitates the examination of serotype- or subtype-determining genes [12]. This in silico typing approach is being accepted and used for source attribution and epidemiological surveillance [13]. Several analytical approaches have been developed towards this goal [14,15,16], and various studies have shown the potential of WGS in epidemiological investigations [17,18].
The increasing use, often abuse, of antibiotics, especially for human health and for farm animals, has contributed to an increase in the prevalence of antimicrobial resistance. AMR isolates are found in many different environments, from the clinic to wastewaters and farms, which has prompted actions at many different levels and targets [19]. A reduction in the overuse of antibiotics in human health and farming and aquaculture is one of these, and a much closer monitoring of the genetic determinants of resistance is another example. Antimicrobial resistance is recognized as one of the major threats for human health for international organizations, such as WHO, FAO, and OECD [20]. This has led to the implementation of surveillance of antimicrobial resistance in human pathogens using WGS, especially for bacteria in the ESKAPE group (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.) and other major pathogens, such as Mycobacterium tuberculosis or highly pathogenic strains of Escherichia coli. The ECDC [21] and the WHO [22] have recommended the use of WGS as the gold standard methodology for surveillance of bacterial pathogens.
Access to the WGS of bacterial isolates also allows performing additional analyses that are not considered in routine surveillance. Among these, evaluating the presence of genetic determinants of antimicrobial resistance is one of the most relevant, especially under the One Health paradigm [23]. This is becoming the de facto main framework for the surveillance and control of antimicrobial resistance. Food-related pathogens are key players under this framework [24,25], and using genome information to identify new resistance determinants and monitor the spread of those already known is a major improvement over previous methodologies [26,27]. However, its implementation must consider not only the technological issues but also the subsequent analyses and the comparison with the regular methods used in each laboratory [28].
Here, we derived whole genome sequences of 141 Salmonella spp. isolates derived from public health surveillance studies of Salmonella in food products at the Comunitat Valenciana (Spain) from 2010 until 2017. We have obtained the sequence type and serotype of these isolates by traditional methods (Sanger sequencing and serology) and in silico. For in silico serotyping, we used two different programs, SISTR and SeqSero2, which are considered the most reliable open-source tools for in silico serovar prediction of Salmonella. In addition, we used the genomic information to determine the presence of antimicrobial resistance genes and to evaluate the minimum inhibitory concentrations (MICs) of a panel of antibiotics. Finally, we performed a detailed study of the relationships among them in order to detect possible clusters and transmissions.
2. Results
2.1. Quality of Sequencing Reads, Genome Assembly, and Annotation
We obtained complete genome sequences from 141 isolates (Table S1, Supplementary Material). After filtering, the average number of reads per isolate decreased from 777,649.2 to 675,013.9, and their average size increased from 139.7 to 143.8 bp (Table S2, Supplementary Material). The assemblies resulted in an average of 116.6 contigs per isolate and a genome size of 4,868,712 bp, with an average N50 value of 216,087.2 (Table S3, Supplementary Material).
From the 141 initial isolates, we removed 6 from further analysis, because they were not identified as Salmonella enterica by Kraken (n = 3) or because they did not attain the minimum values of assembly quality (n = 3). The 135 genomes retained for further analyses had an average of 71.3 contigs (range 29–249).
2.2. Comparison between Phenotypic and In Silico Typing
The 135 isolates retained were tested by both serology and in silico serotyping with two different widely used programs, SISTR and SeqSero2. Serology resulted in 23 different serotypes, with serotype Typhimurium being the most abundant (36.6%). We could not determine the serotype in four isolates (Table S4, Supplementary Material).
Both in silico serotyping programs resulted in 25 different serotypes and assigned the same serotype in 132 of the 135 isolates (97.8%) (Table S4, Supplementary Material). The remaining three isolates (Se_V_139, Se_V_141 and Se_V_142) could not be classified by SISTR because it could not identify the O antigen (Table 1).

Table 1.
Serology results and in silico determination of the antigens by SeqSero2 and SISTR for the isolates with discordant results between serology and in silico or between in silico serotyping programs. ND: Not Determined.
For these discordant cases, serology results did not agree with those obtained in silico (Se_V_139) or did not provide results (Se_V_141 and Se_V_142). Moreover, for Se_V_141, SeqSero2 and SISTR identified different antigenic factors for the fljB gene, which encodes the H2 antigen. In this sample, this gene showed many differences with the corresponding antigenic factor in both databases, which led us to suspect that a recombination event involving the fljB gene might have occurred.
The serology results were not coincident with the in silico serotyping tools for only two isolates (1.48%): Se_V_139, discussed above, and Se_V_3. Both in silico tools identified the latter as Paratyphi B, while it was assigned to serotype Schleissheim by serology. Both serotypes share the same antigenic factor for the fliC gene, but the Schleissheim serotype is monophasic, while Paratyphi B is biphasic and, therefore, has an additional fljB gene. The fljB gene was detected by both in silico tools, with a maximum score of 100.
The most common ST in our sampling was ST34 (38 isolates, 28.1%), followed by ST198 (14, 10.4%). Of the 30 STs obtained, 15 were represented by only 1 isolate. All the STs detected, except ST5689, were associated with an eBURST Group (eBG) (Table S4, Supplementary Material). The STs obtained by Sanger sequencing and MLST analysis of WGS sequences agreed in 124 isolates (91.9%). Six of the eleven isolates in which discrepancies were found belonged to the Typhimurium serotype, and the different STs obtained by each method differed in a single allele (ST19 and ST34, 1 SNP). For the remaining five isolates, an ST could be assigned only by WGS.
MLST and serology definitions of the population lineages were highly similar. Each ST was normally associated with a serotype, except for three serotypes (Cerro, Derby, and Typhimurium) that presented more than one ST (Table 2) and a polyphyletic distribution.
Table 2.
Number of isolates and STs of each serotype identified in the dataset (135 isolates from CV and 235 from the RefSeq database). Undetermined STs (n = 3) are not included.
2.3. Core Genome and Phylogenetic Analysis
In order to study possible transmission clusters and/or common sources among the isolates, we performed a phylogenetic study. In addition to the 135 isolates sequenced in this work, we included 308 additional S. enterica assemblies from RefSeq belonging to 19 of the serotypes identified in our dataset. However, some of the less common serotypes (n = 9) were left without representatives.
The pangenome of the dataset resulted in 11,024 different genes, and we obtained a core genome of 2225 genes. The resulting multiple alignment contained 2,078,455 nt and 136,511 variable sites. A maximum likelihood tree was reconstructed using the evolutionary model GTR + F + R3. The phylogenetic tree revealed that the different STs and serotypes found in this study are well-defined by long branches and high bootstrap support values (>90%) (Figure 1).
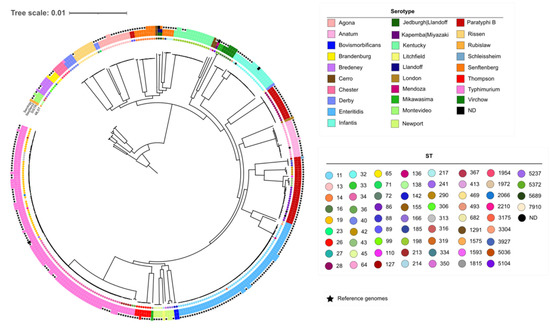
Figure 1.
Maximum likelihood phylogenetic tree from the strict core alignment of the 135 studied isolates and 235 reference genomes. All the internal branches have bootstrap values larger than 90%. Inner dots indicate the ST. The circles around the tree show the serotypes inferred by SISTR (inner circle), SeqSero2 (middle circle), and the serology (outer circle). (Accessible at https://itol.embl.de/tree/1957716215329871633951913).
2.4. Antimicrobial Resistance Analysis
Antimicrobial resistance was studied by two approaches, the identification of known genetic determinants using STARAMR and predicting the MIC with machine learning. The two approaches yielded very similar results.
According to the predictions obtained by STARAMR (Table 3 and Table S5 Supplementary Material), 29 of the 135 isolates (21.48%) did not carry any resistance determinant (genes or point mutations) to any antibiotic family. For the remainder, we found resistance to at least one antibiotic. In total, 39 different resistance genes were detected. Resistance to tetracycline was the most prevalent (73 isolates), followed by sulfisoxazole (70 isolates) and streptomycin (70 isolates). In addition, the analysis revealed 4 different point mutations in the gene gyrA (S83F, S83Y, D87N, D87Y) in 38 isolates. These mutations confer resistance to ciprofloxacin and nalidixic acid. The predicted MIC values revealed a high proportion of resistance against streptomycin (112 isolates), tetracycline (72 isolates), and ampicillin (46 isolates) (Table 3 and Table S5 Supplementary Material). Only 18 isolates (13.3%) showed susceptible predicted MICs to any antibiotic.
Table 3.
Number of isolates predicted as resistant by in silico MIC determination and by STARAMR. Genes involved in resistance detected by STARAMR are also shown.
Resistance determinants were not distributed evenly among serotypes (Figure 2). Serotype Chester is characterized by six resistance genes common to all its isolates, which are not present in other close serotypes. For the Rissen serotype, inferences of antibiotic sensitivity from the presence of ARGs or MIC values reflect important variation among the samples. Isolates from the Infantis serotype show two resistance patterns: while five isolates do not have any resistance gene, and seven isolates share the ant(3″)- Ia, sul1 and tet(A) genes. This also occurs in the Derby serotype, with the resistance genes aadA2, dfrA12, sul3, and tet(A). Moreover, the monophasic variant of S. typhimurium is related with 4 resistance genes and several isolates to predicted phenotypic resistance to sulfisoxazole (8 isolates) and ampicillin (15 isolates). For Senftenberg, Agona, Enteritidis, Newport, and most isolates of the Typhimurium serotype, no resistance genes were detected. Remarkably, the only S. mikawasima isolate of this study was predicted to be resistant to ceftriaxone, a first-choice antibiotic used to treat human salmonellosis.
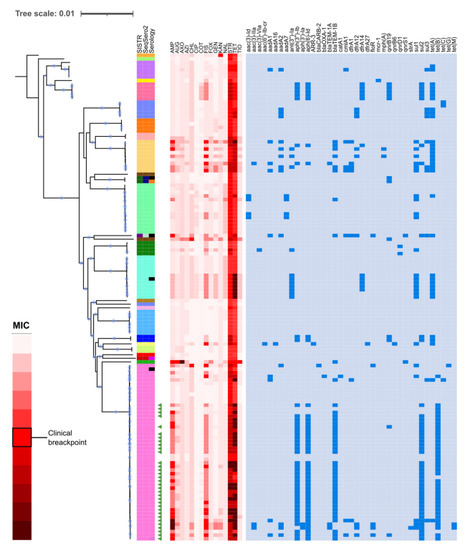
Figure 2.
Relationships between the phenotypic and genotypic resistance determinants in the different serotypes. The red heat-map indicates the MIC predicted for each isolate. In the blue heat-map, the resistance genes found in each serotype are indicated. AMP: Ampicillin, AUG: Augmentin, AXO: Ceftriaxone, AZI: Azithromycin, CHL: Chloramphenicol, CIP: Ciprofloxacin, COT: Co-trimoxazole, FIS: Sulfisoxazole, FOX: Cefoxitin, GEN: Gentamicin, KAN: Kanamycin, NAL: Nalidixic acid, STR: Streptomycin, TET: Tetracycline, and TIO: Ceftiofur. Accessible at: https://itol.embl.de/tree/1957716215415541614592390.
Interestingly, we detected the mcr-1 gene, conferring resistance to colistin in the only ST334 and Brandenburg serotype isolate analyzed. The gene was located in an IncX4 conjugative plasmid with no additional resistance genes. We found that the full plasmid was assembled in one single contig. This plasmid had a 99% identity and 100% coverage with the S. enterica strain CFSAN064033 plasmid pGMI17-001_2 (accession NZ_CP028174.1). A putative relaxase ORF1_3 was found at position 21,365–22,594, with an identity of 38.39%. Figure S1 shows the sequence and structural comparison between pGMI17-001_2 and the contig.
2.5. Cluster Definitions and Epidemiological Investigations
Using the core genome alignment, we clustered the isolates in high-similarity clusters (HSC) of 10, 5, or 3 pairwise SNPs distances. Of the 135 samples, 82 were included in one of the HSC identified. We identified 26, 26, and 24 clusters with two or more isolates for thresholds of 10 (Clusters C1–C26), 5 (70 isolates, C27–C81), and 3 SNPs (62 isolates, C65–C88), respectively (Table S5, Supplementary Material). All the samples grouped in the same cluster belong to the same ST and serotype (Figure 3).
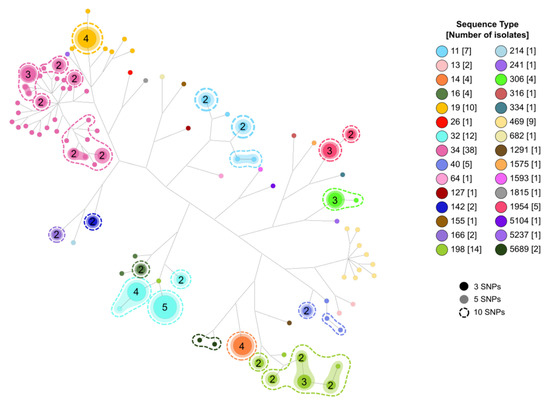
Figure 3.
Network analysis showing the relationships among isolates and STs and the clusters obtained according to the different threshold levels. Numbers in circles represent identical isolates.
As expected, most isolates that grouped in HSC also shared consistent epidemiological information (Table S5, Supplementary Material). However, we found some cases of isolates included in the same HSC cluster with reported different epidemiological data (Table 4). Most discrepancies correspond to different types of samples in the same or close locality and year. However, we also detected one case in which four highly similar isolates were sampled through 3 years in different localities, although all the samples were related to poultry products.
Table 4.
Detected clusters for which epidemiological data are not consistent with genetic similarity.
In most isolates in HSCs, we detected AMR determinants (66/82, 80.5%) which resulted in predicted resistance to one or more antibiotics. Similarly, the inferred MIC values for these isolates led to predicted resistance to at least one antibiotic for the same total number of isolates, but there were several cases of discrepancy between the two methods. In fact, sensitivity to all tested antibiotics was inferred in only eight isolates.
A more detailed analysis of the inferred sensitivity profiles within HSC revealed additional discrepancies. Most of these were detected at the highest clustering level (10 SNPs), with 8 cases in which at least 1 of the isolates differed in the inferred resistance profile from ARGs (n = 5) and/or CMIs (n = 6). Some of these discrepancies were also observed at the 5 SNPs (n = 6) and even the 3 SNPs (n = 6) clustering levels (Supplementary Table S5).
3. Discussion
Applying the One Health approach to the surveillance and control of antimicrobial resistance requires the implementation of methodologies that can provide comparable results in different settings and for samples of different sources [29]. Traditionally, surveillance has been organism-driven, focusing on genotypic and/or phenotypic features that are more relevant for each pathogen. This led to the establishment of many different methodologies with different problems of reproducibility, transferability, standardization, and scalability across laboratories and pathogens. Some of these problems were minimized through strict protocols and regular quality and harmonization controls, apart from the use of common controls and platforms for sharing results. PulseNet was the best example of how much this strategy could achieve [30].
However, the introduction of high-throughput sequencing and the possibility of obtaining complete or near-complete genome sequences of bacterial isolates at reasonable costs and time has led to a dramatic change in how to achieve surveillance goals and solve most of the aforementioned problems. This has not been fulfilled completely yet, but the path seems clear, and most agencies are shifting to genome-based surveillance. Among the many advantages that this methodology offers, those that allow connecting the results or surveillance from different levels, sites, and species are especially relevant for the One Health approach.
In this work, we obtained a high level of agreement (98.52%) between in silico determined serotypes and those determined by serology. This level of agreement is higher than that obtained previously with a larger sample size [11]. In one of the cases of disagreement, the discordance between the Paratyphi B and Schleissheim serotypes is likely due to a lack of detection of agglutination for the H2 antigen, as suggested by the phylogenetic tree that places this isolate in a clade formed by the serotype Paratyphi B. In the other case, a completely different antigenic factor of the fliC gene was identified phenotypically and in silico.
For in silico serotyping, we employed two of the most reliable tools currently available, SeqSero2 and SISTR [31]. They reported a coincident result in 97.8% of the isolates (132/135). However, in two cases, SISTR could not determine one of the antigenic factors, while SeqSero2 unambiguously identified them. The serotype reported by SeqSero2 usually corresponds to the ST assigned to these isolates. However, due to the low number of representative genomes of the serotype assigned in the RefSeq database, SeqSero2 prediction could not be confirmed by core genome analysis.
In most isolates, the results of the MLST analysis performed by the two methods were consistent. However, the allele combination obtained by Sanger-based MLST did not correspond to known STs in five of the isolates, while by complete genome-based MLST, we could assign an ST to all of them. It is likely that the STs currently determined in four of them were not established in the database when these isolates were typed (between 2014 and 2017). The other discordant isolate presented STs with only one different allele. Their location in the phylogenetic tree suggests that the ST obtained from complete genomes is correct.
Although the results of the resistance genes analysis show that the isolates have a high content of resistance genes to several antibiotic families, this does not necessarily correspond to clinical resistance, as shown by the results of the MIC predictor (Figure 1). In addition, for several of the predicted resistant isolates, especially those resistant to streptomycin, no resistance determinants were identified. Similarly, we found an isolate resistant to ceftriaxone, but we have not detected any resistance gene or mutation that could originate it. This fact can be explained by the presence of not-detected genes related to resistance to these antibiotics. It is necessary to have more samples to investigate the genetic determinant that confers resistance to this antibiotic.
For the remaining isolates, the predicted phenotypes do not involve antibiotics currently used in the treatment of salmonellosis, so our isolates are not clinically relevant. However, we found the mcr-1 gene in the only study isolate classified as Brandenburg serotype and ST334. This gene is responsible for the colistin resistance. It has been identified worldwide in Salmonella isolates obtained from humans, animals, the environment, and food, but as far as we know, it has not been reported in the Brandenburg serotype. The IncX4 plasmid that carries the gene mcr-1 has been detected in Salmonella food isolates belonging to other serotypes in different countries [32,33,34]. In Spain, this plasmid has been found in Escherichia coli isolates [35,36], but to our knowledge, it has not been reported in Salmonella.
Colistin has been widely used in animal production in several countries for prophylaxis and growth promotion [37]. In 2015, the use of colistin in swine production in Spain was estimated to be 50 mg/PCU (population correction units), but in the past decade, the spread of carbapenemase-producing Enterobacteriaceae led to re-implementing the use of colistin as a last therapeutic option for the treatment of human infections. For this reason, a reduction target of 5 mg/PCU was set for the next years [38]. Now, colistin is classified by The World Health Organization as an antibiotic of critical importance in human clinical settings [39]. Genetic analysis and epidemiological surveillance are crucial for minimizing the spread of colistin resistance, and early identification of colistin-resistant isolates in any setting is one of the most useful outcomes of WGS-based surveillance.
The phylogenetic reconstruction highlights the genetic heterogeneity of several serotypes [40,41,42]. In relation to our study isolates, each clade of the polyphyletic serotypes corresponds to a specific ST, except in the case of S. typhimurium and its monophasic variant. Although sequence typing generally allows very efficient lineage discrimination, it can group isolates as different from these (Figure 2). Furthermore, STs do not always correspond to ancestral relationships, as in the case of ST19, which has a paraphyletic distribution. On the other hand, MLST schemes lack the necessary resolution for adequate discrimination of outbreaks. However, epidemiological information in combination with genomic information and phylogenetic analysis can provide this higher level of discriminatory power, allowing the identification of closely related samples.
Using this information, we have been able to detect closely related isolates that may be associated with the same transmission source. High-similarity clusters were generally consistent with epidemiological data. However, for some of these clusters, the isolates were obtained from apparently unrelated products (Clusters C7, C18, and C65) and/or at distant dates (Cluster C6) (Table S5, Supplementary Material). For all these cases, it would be convenient to study in more detail the production chain of the products and the context in which the samples were obtained.
The most obvious limitation of this work is the lack of information on sensitivity profiles to antibiotics, as these tests were not included in the routine procedures at the Public Health Laboratory. Lack of this information undermines our in silico evaluation of antimicrobial sensitivity profiles, but it also highlights one of the potential benefits of using WGS for surveillance of food-related products because this allows the early identification of pathogens with potential resistance to various antibiotics. In addition, the use of machine learning approaches to infer AMR profiles and MICs, such as PATRIC [43], is becoming popular, as these methods are revealing high accuracy and reliability for different pathogenic bacteria [44,45,46]. In any case, these predictions should be tested more extensively to rule out and/or establish limits to machine learning inferences before they can be used to adopt appropriate actions and implement recommendations for human and animal public health.
Another limitation is that we used a core genome approach to define HSCs, while several of these reveal differences in antimicrobial susceptibility and epidemiological features. It is possible that some of these HSCs will be further subdivided into separate groups should their accessory genomes be considered in the analysis, thus generating more coherent and congruent subgroups. Nevertheless, this problem is much more easily tackled with data from WGS analysis than with results from serological or direct multi-locus sequence typing, where resolving these discrepancies is not possible.
4. Materials and Methods
4.1. Sample Selection
We used a set of 258 Salmonella spp. isolates collected by the Public Health Laboratory of the Comunitat Valenciana (CV, Spain) between 2010 and 2017. This collection contains information for each isolate on serotype, sequence type (ST), and epidemiological data, such as sampling source, date, and location.
Serotyping was performed by slide agglutination with O and H antigen-specific sera according to the White–Kauffmann–Le Minor scheme [1], while STs were obtained following the Warwick University protocols (available at https://enterobase.readthedocs.io/en/latest/mlst/mlst-legacy-info.html).
We used epidemiological and serological data to select samples from the collection that were likely to be associated with possible transmission chains or serotype misidentification or belonged to infrequent serotypes. We selected 141 isolates following these criteria (Table S1, Supplementary Material).
4.2. Whole-Genome Sequencing and Gene Annotation
DNA was extracted using EasyMag (Biomérieux, Lyon, France). Isolates were sequenced using Illumina NextSeq technology. Sequencing libraries were prepared with the Nextera XT DNA library preparation kit according to the manufacturer’s protocol. As a result, we obtained paired-end reads of 2 × 150 bp. Raw sequence data generated in this study were deposited in the European Nucleotide Archive (ENA) under project PRJEB49974.
FastQC and MultiQC were used to assess the quality of the raw reads. Short reads (<100 bp) or bad quality reads (mean quality < 25) were removed from ensuing analyses using PrinSEQ [47]. To discard contaminated or misidentified isolates, we examined the taxonomic content of each sample using Kraken [48]. Cleaned and filtered sequence reads were assembled de novo using SPAdes [49]. Assembly quality was evaluated using QUAST [50]. Assemblies with more than 600 contigs or a GC content lower than 50% or higher than 52% were discarded from subsequent analyses. Gene annotation was performed using PROKKA [51].
4.3. In Silico Isolate Characterization: Serotypes, STs, and AMR
We used seq2MLST v. 1.0.1 [52] to identify the STs of each sample. For in silico serotyping, we compared the results of SeqSero2 [15] and SISTR [16].
Antibiotic resistance genes, mutations, and plasmids were identified using STARAMR 0.7.2 (https://github.com/phac-nml/staramr), which uses the ResFinder [53], PointFinder [54], and PlasmidFinder [55] databases. This tool provides a drug resistance prediction based on gene identification, but it does not imply clinical resistance. For this reason, we used a method based on machine learning developed by the PathoSystems Resource Integration Center (PATRIC) [43] to predict the minimum inhibitory concentration (MIC) for 15 clinically important antibiotics. Table 5 shows the breakpoints used to consider resistance.
Table 5.
Breakpoints used for antibiotic resistance testing. Breakpoints established by CLSI were used when available. There are no CLSI breakpoints for streptomycin or ceftiofur, so NARMS-established breakpoints (https://www.cdc.gov/narms/antibiotics-tested.html) were used instead.
4.4. Core Genome and Evolutionary Analysis
All publicly available complete S. enterica subsp. enterica genome assemblies were retrieved from RefSeq on 29 June 2020. We used SISTR and SeqSero2 to determine the serotype of each genome. We only kept those genomes of serotypes represented in our sample collection. In addition, we removed genomes with a discrepancy between the serotype indicated at the NCBI and the one obtained using SISTR and SeqSero2.
To analyze the relationships among the different isolates, we studied their pangenome. Shared orthologous genes were determined with Proteinortho5 [56]. Public genomes that did not contain most of the genes present in the study samples were discarded. In addition, duplicate genes were removed from the pangenome. For each isolate, genes in the strict core genome were concatenated. We used AMAS [57] to remove genes with an abnormally large number of undetermined characters or a high proportion of variable sites.
To reconstruct the global phylogeny of the isolates, we used IQTREE [58] employing the ultrafast bootstrap option [59] with 1000 replicates. Finally, MEGAX [60] was used to obtain the distance matrix among the analyzed genomes. To represent the different trees and include the serotyping results, we used iTOL (https://itol.embl.de).
4.5. Cluster Definition and Epidemiological Investigation
With the aim of identifying clusters that were more likely to be associated with the same contamination source, we used the core genome alignment to group the isolates into clusters of increasing levels of similarity [61]. For this, we performed single-linkage clustering of the SNP distance matrix at three threshold levels (10, 5, and 3 SNPs), i.e., the maximum length separating the sequences in the same cluster. Clustering was performed using the R library DECIPHER [62]. Moreover, we related these clusters with epidemiological information (source/product, geographic location, and collection date) and predicted antimicrobial sensitivity (see above) for each isolate.
5. Conclusions
This study shows the potential of complete genome sequencing in epidemiological surveillance of S. enterica and highlights the need to incorporate WGS and bioinformatic techniques into routine surveillance analyses. As we have shown, an in silico approach is advantageous for the identification of these serotypes. However, the addition of complete genomes sequences of the less prevalent serotypes in Salmonella databases is essential for improving the quality of in silico methods for typing Salmonella isolates and resolve ambiguities and misassignments derived from serology.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/antibiotics12050883/s1, Table S1: Sampling information of the Salmonella enterica isolates included in this study. Table S2: Summary of sequence read quality. Table S3: Summary of assembly results. Table S4: Serotypes and STs. Table S5: Isolates included in HSC and their features. Figure S1: Sequence and structure of the contig containing the mcr-1 gene with the Incx4 plasmid.
Author Contributions
Conceptualization: N.G.-G. and F.G.-C.; data curation: A.S.-S.; formal analysis: A.S.-S., S.O.-M., M.L.-L. and N.G.-G.; funding acquisition: F.G.-C.; investigation: A.S.-S., L.M., S.O.-M. and M.L.-L.; methodology: A.S.-S. and N.G.-G.; project administration: M.L.C. and F.G.-C.; resources: L.M., M.L.C., S.O.-M. and M.L.-L.; visualization: A.S.-S. and N.G.-G.; writing—original draft: A.S.-S. and N.G.-G.; writing—review and editing: N.G.-G. and F.G.-C.; supervision: N.G.-G. and F.G.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by projects BFU2017-89594R from MICIN (Spanish Government), CIPROM2021-053 from Conselleria de Educació, Ciència i Universitats (Generalitat Valenciana), and Conselleria de Sanitat Universal i Salut Publica (Generalitat Valenciana).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grimont, P.A.D.; Weill, F.-X. Others Antigenic Formulae of the Salmonella Serovars. WHO Collab. Cent. Ref. Res. Salmonella 2007, 9, 1–166. [Google Scholar]
- Alikhan, N.-F.; Zhou, Z.; Sergeant, M.J.; Achtman, M. A Genomic Overview of the Population Structure of Salmonella. PLoS Genet. 2018, 14, e1007261. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Paul, S.; Kisiela, D.I.; Linardopoulou, E.V.; Sokurenko, E.V. Convergent Molecular Evolution of Genomic Cores in Salmonella enterica and Escherichia coli. J. Bacteriol. 2012, 194, 5002–5011. [Google Scholar] [CrossRef]
- Ibrahim, G.M.; Morin, P.M. Salmonella Serotyping Using Whole Genome Sequencing. Front. Microbiol. 2018, 9, 2993. [Google Scholar] [CrossRef] [PubMed]
- Wattiau, P.; Boland, C.; Bertrand, S. Methodologies for Salmonella enterica Subsp. enterica Subtyping: Gold Standards and Alternatives. Appl. Environ. Microbiol. 2011, 77, 7877–7885. [Google Scholar] [CrossRef]
- Achtman, M.; Wain, J.; Weill, F.-X.; Nair, S.; Zhou, Z.; Sangal, V.; Krauland, M.G.; Hale, J.L.; Harbottle, H.; Uesbeck, A.; et al. Multilocus Sequence Typing as a Replacement for Serotyping in Salmonella enterica. PLoS Pathog. 2012, 8, e1002776. [Google Scholar] [CrossRef]
- Mather, A.E.; Reid, S.W.J.; Maskell, D.J.; Parkhill, J.; Fookes, M.C.; Harris, S.R.; Brown, D.J.; Coia, J.E.; Mulvey, M.R.; Gilmour, M.W.; et al. Distinguishable Epidemics of Multidrug-Resistant Salmonella Typhimurium DT104 in Different Hosts. Science 2013, 341, 1514–1517. [Google Scholar] [CrossRef]
- Scaltriti, E.; Sassera, D.; Comandatore, F.; Morganti, M.; Mandalari, C.; Gaiarsa, S.; Bandi, C.; Zehender, G.; Bolzoni, L.; Casadei, G.; et al. Differential Single Nucleotide Polymorphism-Based Analysis of an Outbreak Caused by Salmonella enterica Serovar Manhattan Reveals Epidemiological Details Missed by Standard Pulsed-Field Gel Electrophoresis. J. Clin. Microbiol. 2015, 53, 1227–1238. [Google Scholar] [CrossRef]
- Taylor, A.J.; Lappi, V.; Wolfgang, W.J.; Lapierre, P.; Palumbo, M.J.; Medus, C.; Boxrud, D. Characterization of Foodborne Outbreaks of Salmonella enterica Serovar Enteritidis with Whole-Genome Sequencing Single Nucleotide Polymorphism-Based Analysis for Surveillance and Outbreak Detection. J. Clin. Microbiol. 2015, 53, 3334–3340. [Google Scholar] [CrossRef]
- Imanian, B.; Donaghy, J.; Jackson, T.; Gummalla, S.; Ganesan, B.; Baker, R.C.; Henderson, M.; Butler, E.K.; Hong, Y.; Ring, B.; et al. The Power, Potential, Benefits, and Challenges of Implementing High-Throughput Sequencing in Food Safety Systems. NPJ Sci. Food 2022, 6, 35. [Google Scholar] [CrossRef]
- Yachison, C.A.; Yoshida, C.; Robertson, J.; Nash, J.H.E.; Kruczkiewicz, P.; Taboada, E.N.; Walker, M.; Reimer, A.; Christianson, S.; Nichani, A.; et al. The Validation and Implications of Using Whole Genome Sequencing as a Replacement for Traditional Serotyping for a National Salmonella Reference Laboratory. Front. Microbiol. 2017, 8, 1044. [Google Scholar] [CrossRef] [PubMed]
- Banerji, S.; Simon, S.; Tille, A.; Fruth, A.; Flieger, A. Genome-Based Salmonella Serotyping as the New Gold Standard. Sci. Rep. 2020, 10, 4333. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Biological Hazards (EFSA BIOHAZ Panel); Koutsoumanis, K.; Allende, A.; Alvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Hilbert, F.; et al. Whole Genome Sequencing and Metagenomics for Outbreak Investigation, Source Attribution and Risk Assessment of Food-Borne Microorganisms. EFSA J. 2019, 17, e05898. [Google Scholar] [PubMed]
- Tewolde, R.; Dallman, T.; Schaefer, U.; Sheppard, C.L.; Ashton, P.; Pichon, B.; Ellington, M.; Swift, C.; Green, J.; Underwood, A. MOST: A Modified MLST Typing Tool Based on Short Read Sequencing. PeerJ 2016, 4, e2308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; den Bakker, H.C.; Li, S.; Chen, J.; Dinsmore, B.A.; Lane, C.; Lauer, A.C.; Fields, P.I.; Deng, X. SeqSero2: Rapid and Improved Salmonella Serotype Determination Using Whole-Genome Sequencing Data. Appl. Environ. Microbiol. 2019, 85, e01746-19. [Google Scholar] [CrossRef]
- Yoshida, C.E.; Kruczkiewicz, P.; Laing, C.R.; Lingohr, E.J.; Gannon, V.P.J.; Nash, J.H.E.; Taboada, E.N. The Salmonella in Silico Typing Resource (SISTR): An Open Web-Accessible Tool for Rapidly Typing and Subtyping Draft Salmonella Genome Assemblies. PLoS ONE 2016, 11, e0147101. [Google Scholar] [CrossRef] [PubMed]
- Rounds, J.M.; Taylor, A.J.; Eikmeier, D.; Nichols, M.M.; Lappi, V.; Wirth, S.E.; Boxrud, D.J.; Smith, K.E.; Medus, C. Prospective Salmonella Enteritidis Surveillance and Outbreak Detection Using Whole Genome Sequencing, Minnesota 2015–2017. Epidemiol. Infect. 2020, 148, e254. [Google Scholar] [CrossRef]
- Ford, L.; Carter, G.P.; Wang, Q.; Seemann, T.; Sintchenko, V.; Glass, K.; Williamson, D.A.; Howard, P.; Valcanis, M.; Castillo, C.F.S.; et al. Incorporating Whole-Genome Sequencing into Public Health Surveillance: Lessons from Prospective Sequencing of Salmonella typhimurium in Australia. Foodborne Pathog. Dis. 2018, 15, 161–167. [Google Scholar] [CrossRef]
- Available online: https://apps.who.int/gb/ebwha/pdf_files/WHA68/A68_R7-en.pdf (accessed on 6 April 2023).
- Foodborne Antimicrobial Resistance; FAO: Rome, Italy; WHO: Geneva, Switzerland, 2022; ISBN 978925135734.
- Available online: https://www.ecdc.europa.eu/sites/default/files/documents/framework-for-genomic-surveillance.pdf (accessed on 6 April 2023).
- World Health Organization. GLASS Whole-Genome Sequencing for Surveillance of Antimicrobial Resistance. Available online: https://apps.who.int/iris/bitstream/handle/10665/334354/9789240011007-eng.pdf (accessed on 6 April 2023).
- Mancilla-Becerra, L.M.; Lías-Macías, T.; Ramírez-Jiménez, C.L.; Barba León, J. Multidrug-Resistant Bacterial Foodborne Pathogens: Impact on Human Health and Economy. In Pathogenic Bacteria; IntechOpen: London, UK, 2020; ISBN 9781789859874. [Google Scholar]
- Doyle, M.E. Multidrug-Resistant Pathogens in the Food Supply. Foodborne Pathog. Dis. 2015, 12, 261–279. [Google Scholar] [CrossRef]
- Caniça, M.; Manageiro, V.; Abriouel, H.; Moran-Gilad, J.; Franz, C.M.A.P. Antibiotic Resistance in Foodborne Bacteria. Trends Food Sci. Technol. 2019, 84, 41–44. [Google Scholar] [CrossRef]
- Pightling, A.W.; Pettengill, J.B.; Luo, Y.; Baugher, J.D.; Rand, H.; Strain, E. Interpreting Whole-Genome Sequence Analyses of Foodborne Bacteria for Regulatory Applications and Outbreak Investigations. Front. Microbiol. 2018, 9, 1482. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Dwivedi, V.D.; Kirtipal, N. Application of Whole Genome Sequencing (WGS) Approach against Identification of Foodborne Bacteria. In Microbial Genomics in Sustainable Agroecosystems; Tripathi, V., Kumar, P., Tripathi, P., Kishore, A., Eds.; Springer: Singapore, 2019; Volume 1, pp. 131–148. ISBN 9789811387395. [Google Scholar]
- Collineau, L.; Boerlin, P.; Carson, C.A.; Chapman, B.; Fazil, A.; Hetman, B.; McEwen, S.A.; Parmley, E.J.; Reid-Smith, R.J.; Taboada, E.N.; et al. Integrating Whole-Genome Sequencing Data into Quantitative Risk Assessment of Foodborne Antimicrobial Resistance: A Review of Opportunities and Challenges. Front. Microbiol. 2019, 10, 1107. [Google Scholar] [CrossRef] [PubMed]
- Gerner-Smidt, P.; Besser, J.; Concepción-Acevedo, J.; Folster, J.P.; Huffman, J.; Joseph, L.A.; Kucerova, Z.; Nichols, M.C.; Schwensohn, C.A.; Tolar, B. Whole Genome Sequencing: Bridging One-Health Surveillance of Foodborne Diseases. Front Public Health 2019, 7, 172. [Google Scholar] [CrossRef] [PubMed]
- Nadon, C.; Van Walle, I.; Gerner-Smidt, P.; Campos, J.; Chinen, I.; Concepcion-Acevedo, J.; Gilpin, B.; Smith, A.M.; Man Kam, K.; Perez, E.; et al. PulseNet International: Vision for the Implementation of Whole Genome Sequencing (WGS) for Global Food-Borne Disease Surveillance. Eurosurveillance 2017, 22, 30544. [Google Scholar] [CrossRef]
- Uelze, L.; Borowiak, M.; Deneke, C.; Szabó, I.; Fischer, J.; Tausch, S.H.; Malorny, B. Performance and Accuracy of Four Open-Source Tools for In Silico Serotyping of Salmonella Spp. Based on Whole-Genome Short-Read Sequencing Data. Appl. Environ. Microbiol. 2020, 86, e02265-19. [Google Scholar] [CrossRef]
- Rau, R.B.; de Lima-Morales, D.; Wink, P.L.; Ribeiro, A.R.; Barth, A.L. Salmonella enterica mcr-1 Positive from Food in Brazil: Detection and Characterization. Foodborne Pathog. Dis. 2020, 17, 202–208. [Google Scholar] [CrossRef]
- Moreno, L.Z.; Gomes, V.T.M.; Moreira, J.; de Oliveira, C.H.; Peres, B.P.; Silva, A.P.S.; Thakur, S.; La Ragione, R.M.; Moreno, A.M. First Report of Mcr-1-Harboring Salmonella enterica Serovar Schwarzengrund Isolated from Poultry Meat in Brazil. Diagn. Microbiol. Infect. Dis. 2019, 93, 376–379. [Google Scholar] [CrossRef]
- Garcia-Graells, C.; De Keersmaecker, S.C.J.; Vanneste, K.; Pochet, B.; Vermeersch, K.; Roosens, N.; Dierick, K.; Botteldoorn, N. Detection of Plasmid-Mediated Colistin Resistance, Mcr-1 and Mcr-2 Genes, in Salmonella Spp. Isolated from Food at Retail in Belgium from 2012 to 2015. Foodborne Pathog. Dis. 2018, 15, 114–117. [Google Scholar] [CrossRef]
- Migura-Garcia, L.; González-López, J.J.; Martinez-Urtaza, J.; Aguirre Sánchez, J.R.; Moreno-Mingorance, A.; Perez de Rozas, A.; Höfle, U.; Ramiro, Y.; Gonzalez-Escalona, N. Mcr-Colistin Resistance Genes Mobilized by IncX4, IncHI2, and IncI2 Plasmids in Escherichia coli of Pigs and White Stork in Spain. Front. Microbiol. 2019, 10, 3072. [Google Scholar] [CrossRef]
- Hernández, M.; Iglesias, M.R.; Rodríguez-Lázaro, D.; Gallardo, A.; Quijada, N.; Miguela-Villoldo, P.; Campos, M.J.; Píriz, S.; López-Orozco, G.; de Frutos, C.; et al. Co-Occurrence of Colistin-Resistance Genes mcr-1 and mcr-3 among Multidrug-Resistant Escherichia coli Isolated from Cattle, Spain, September 2015. Eurosurveillance 2017, 22, 30586. [Google Scholar] [CrossRef]
- Lima, T.; Domingues, S.; Da Silva, G.J. Plasmid-Mediated Colistin Resistance in Salmonella enterica: A Review. Microorganisms 2019, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/updated-advice-use-colistin-products-animals-within-european-union-development-resistance-possible_en-0.pdf (accessed on 6 April 2023).
- World Health Organization. Critically Important Antimicrobials for Human Medicine, 6th revision; World Health Organization: Geneva, Switzerland, 2019; ISBN 978-92-4-151552-8. [Google Scholar]
- Worley, J.; Meng, J.; Allard, M.W.; Brown, E.W.; Timme, R.E. Salmonella enterica Phylogeny Based on Whole-Genome Sequencing Reveals Two New Clades and Novel Patterns of Horizontally Acquired Genetic Elements. mBio 2018, 9, 2303. [Google Scholar] [CrossRef] [PubMed]
- Sangal, V.; Harbottle, H.; Mazzoni, C.J.; Helmuth, R.; Guerra, B.; Didelot, X.; Paglietti, B.; Rabsch, W.; Brisse, S.; Weill, F.-X.; et al. Evolution and Population Structure of Salmonella enterica Serovar Newport. J. Bacteriol. 2010, 192, 6465–6476. [Google Scholar] [CrossRef] [PubMed]
- Timme, R.E.; Pettengill, J.B.; Allard, M.W.; Strain, E.; Barrangou, R.; Wehnes, C.; Van Kessel, J.S.; Karns, J.S.; Musser, S.M.; Brown, E.W. Phylogenetic Diversity of the Enteric Pathogen Salmonella enterica Subsp. enterica Inferred from Genome-Wide Reference-Free SNP Characters. Genome Biol. Evol. 2013, 5, 2109–2123. [Google Scholar] [CrossRef]
- Nguyen, M.; Long, S.W.; McDermott, P.F.; Olsen, R.J.; Olson, R.; Stevens, R.L.; Tyson, G.H.; Zhao, S.; Davis, J.J. Using Machine Learning to Predict Antimicrobial MICs and Associated Genomic Features for Nontyphoidal Salmonella. J. Clin. Microbiol. 2019, 57, e01260-18. [Google Scholar] [CrossRef]
- Anahtar, M.N.; Yang, J.H.; Kanjilal, S. Applications of Machine Learning to the Problem of Antimicrobial Resistance: An Emerging Model for Translational Research. J. Clin. Microbiol. 2021, 59, e0126020. [Google Scholar] [CrossRef]
- Khaledi, A.; Weimann, A.; Schniederjans, M.; Asgari, E.; Kuo, T.-H.; Oliver, A.; Cabot, G.; Kola, A.; Gastmeier, P.; Hogardt, M.; et al. Predicting Antimicrobial Resistance in Pseudomonas aeruginosa with Machine Learning-Enabled Molecular Diagnostics. EMBO Mol. Med. 2020, 12, e10264. [Google Scholar] [CrossRef]
- Eyre, D.W.; De Silva, D.; Cole, K.; Peters, J.; Cole, M.J.; Grad, Y.H.; Demczuk, W.; Martin, I.; Mulvey, M.R.; Crook, D.W.; et al. WGS to Predict Antibiotic MICs for Neisseria gonorrhoeae. J. Antimicrob. Chemother. 2017, 72, 1937–1947. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality Control and Preprocessing of Metagenomic Datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 2014, 15, 1–12. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Carroll, L.M. seq2mlst: In Silico Multi-Locus Sequence Typing 2017. Available online: https://github.com/lmc297/seq2mlst (accessed on 23 April 2023).
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A Novel Web Tool for WGS-Based Detection of Antimicrobial Resistance Associated with Chromosomal Point Mutations in Bacterial Pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Möller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-) orthologs in Large-Scale Analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef]
- Borowiec, M.L. AMAS: A Fast Tool for Alignment Manipulation and Computing of Summary Statistics. PeerJ 2016, 4, e1660. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Waldram, A.; Dolan, G.; Ashton, P.M.; Jenkins, C.; Dallman, T.J. Epidemiological Analysis of Salmonella Clusters Identified by Whole Genome Sequencing, England and Wales 2014. Food Microbiol. 2018, 71, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.S. Using DECIPHER v2. 0 to Analyze Big Biological Sequence Data in R. R J. 2016, 8, 352. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).