

Complex Networks Analyses of Antibiofilm Peptides: An Emerging Tool for Next-Generation Antimicrobials’ Discovery

 ,
,  ,
,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
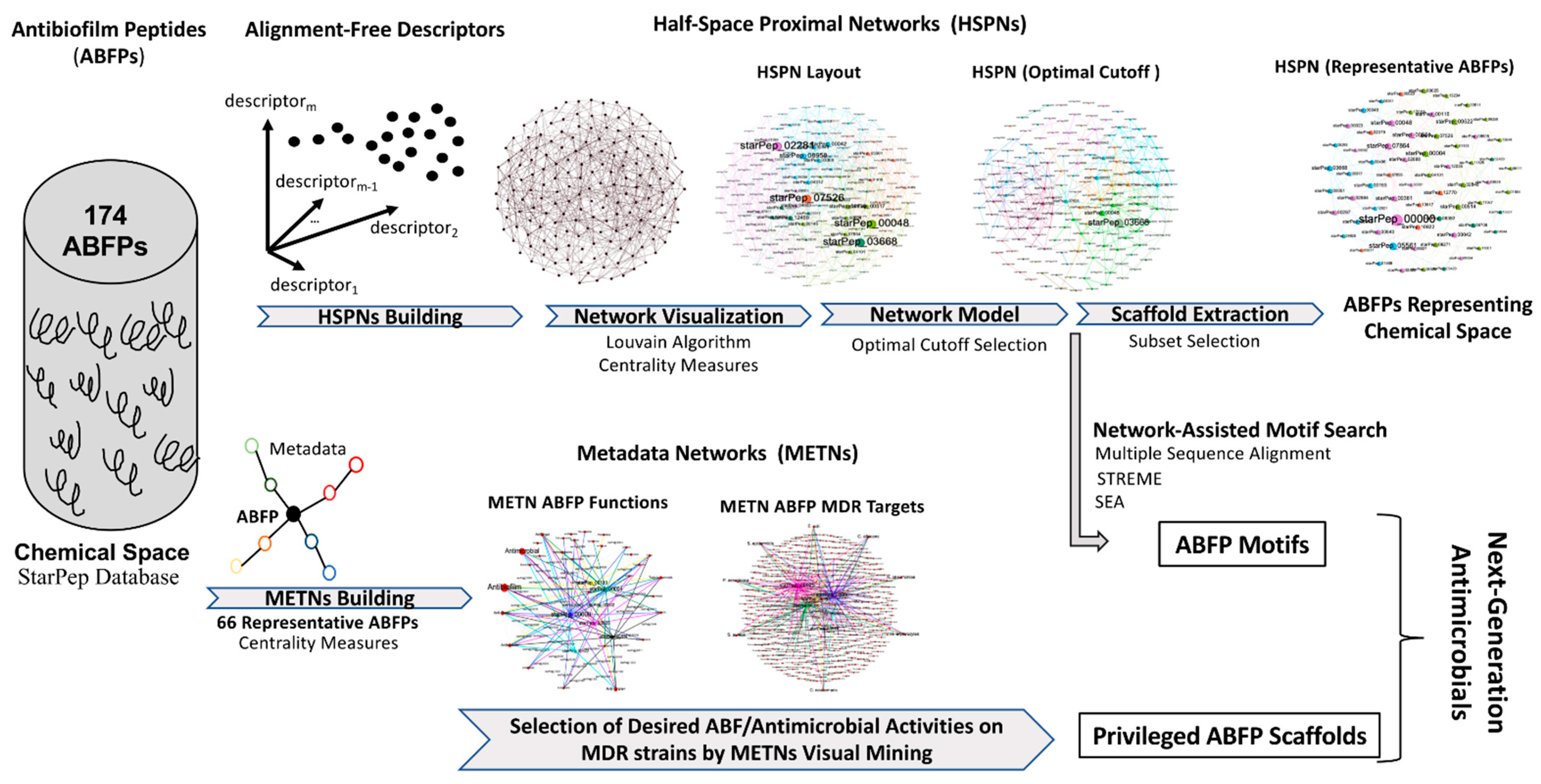
2.1. Half-Space Proximal Network Building
2.2. Metadata Networks
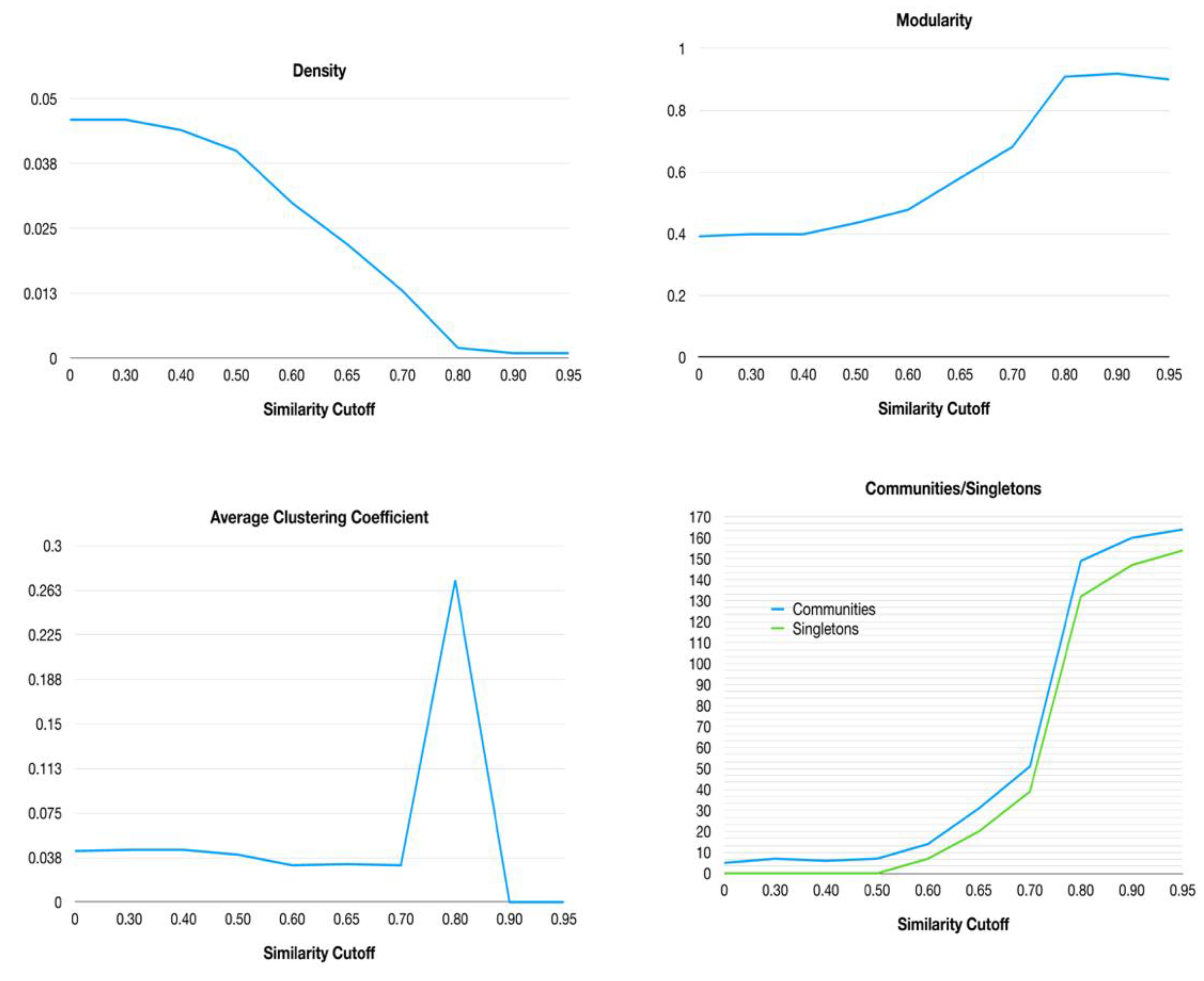
2.3. Networks Similarity Cutoff Analysis
2.4. Network Visualization
2.5. Scaffold Extraction by Centrality Measures
2.6. Selection of the Most Representative Extracted Subset
2.7. Motif Discovery
2.7.1. Multiple Sequence Alignments
- -
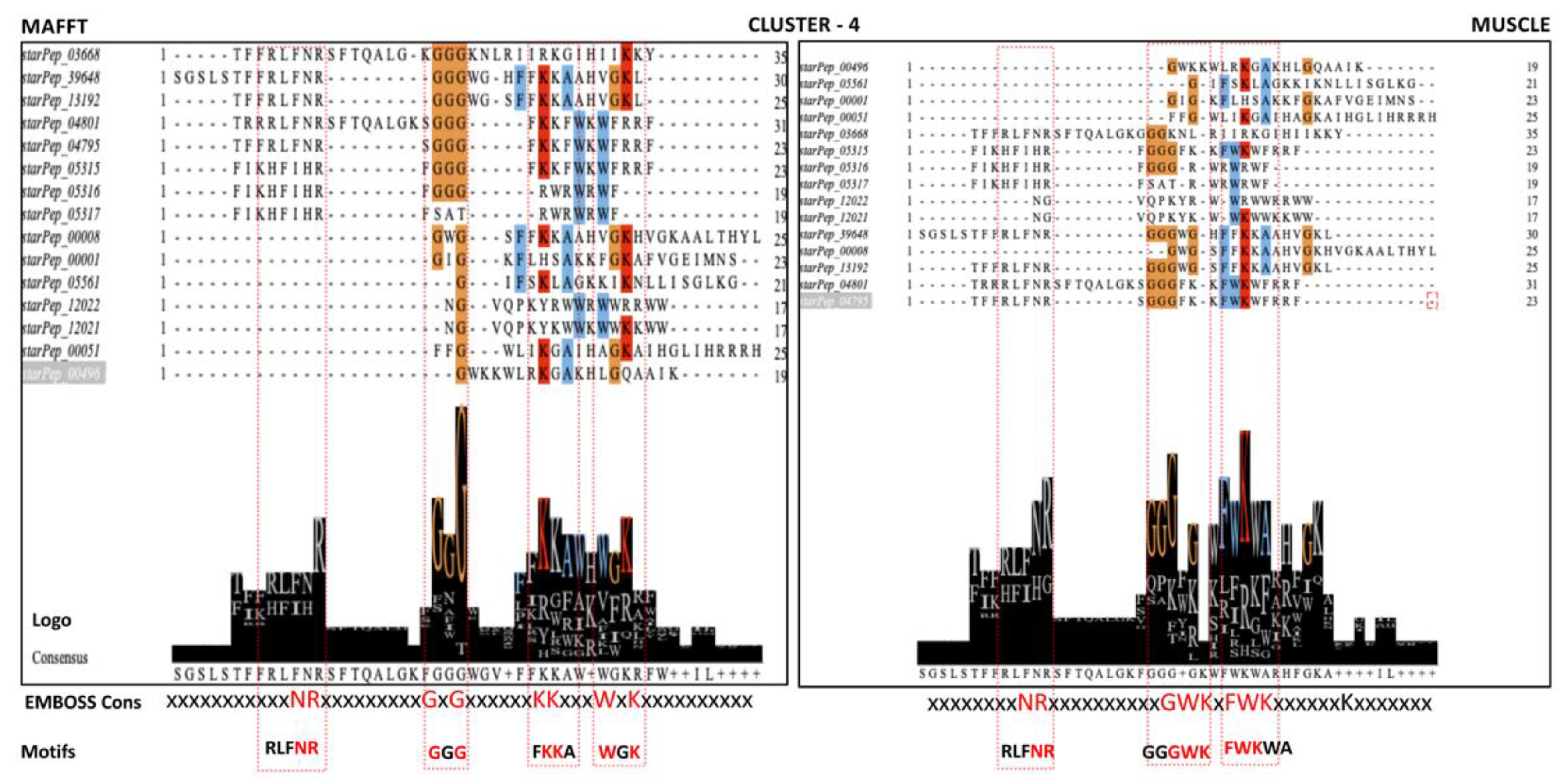
- Communities with more than 2 ABFPs, including the one containing 20 singletons, were aligned independently using multiple sequence alignment (MSA) algorithms. The algorithms of choice were MAFFT (Multiple Alignment using Fast Fourier Transform) v7.487 with the iterative refinement FFT-NS-i option [42] and MUSCLE (Multiple Sequence Comparison by Log- Expectation) v3.8 [43], publicly available at https://www.ebi.ac.uk/Tools/msa/, EMBL-EBI, Cambridgeshire, UK, accessed on 22 December 2022.
- -
- The conserved motifs were detected by jointly analyzing the consensus sequences and Seq2Logo, implemented in the Jalview v2.11.2.5 program [44] and EMBOSS Cons v6.6.0, available at https://www.ebi.ac.uk/Tools/msa/emboss_cons, EMBL-EBI, Cambridgeshire, UK, accessed on 22 December 2022).
- -
2.7.2. Alignment-Free (AF) Detection
2.7.3. Motif Enrichment Analysis
2.8. Overall Workflow Integrating Complex Networks to Next-Generation Antimicrobials Development
3. Results and Discussion
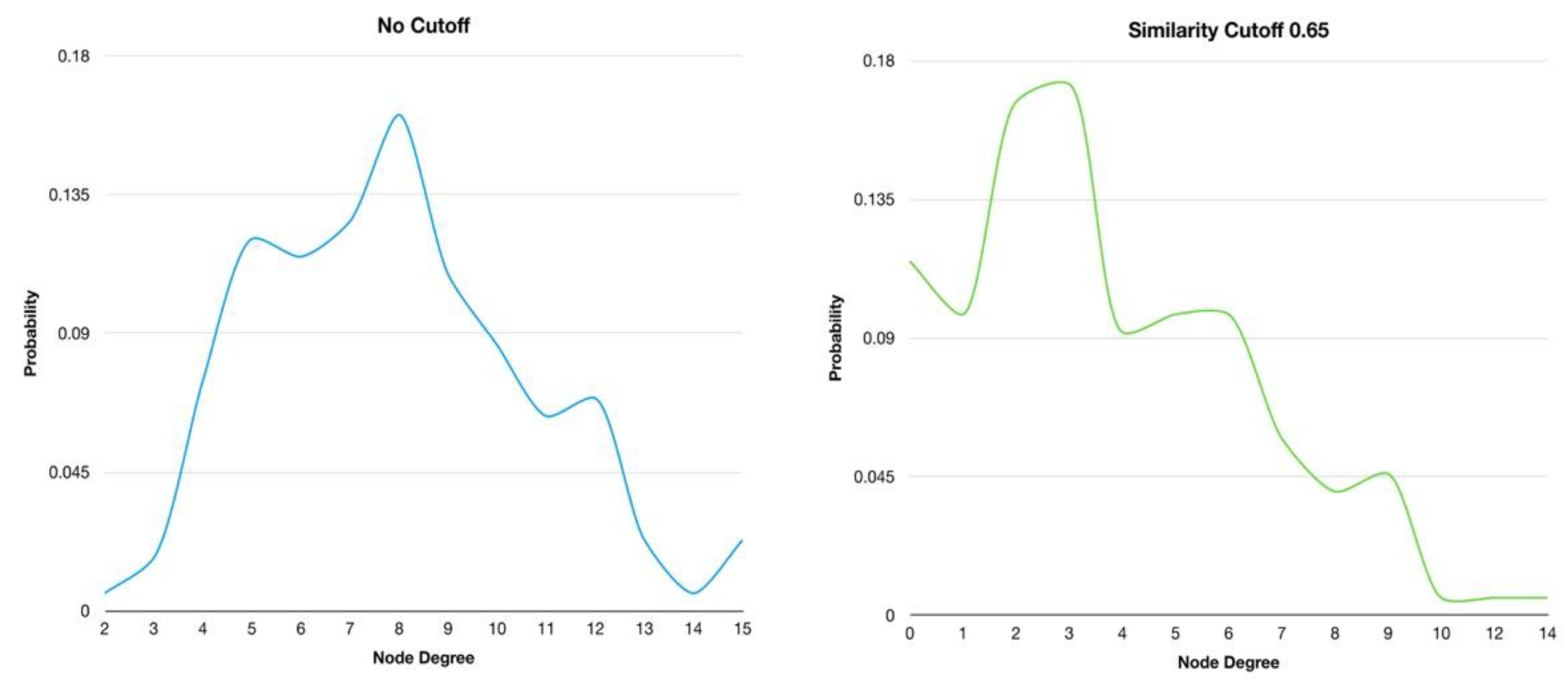
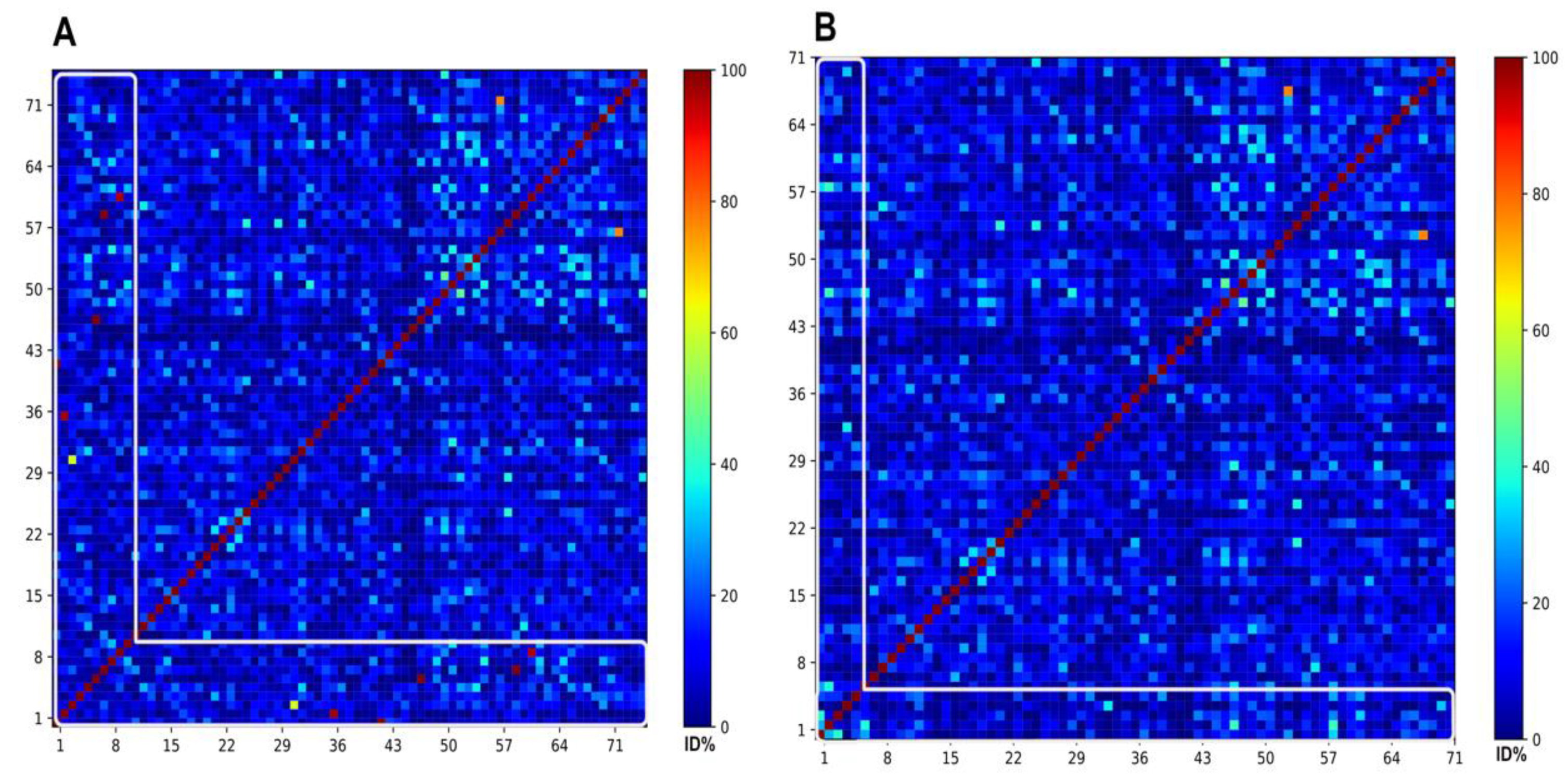
3.1. Half-Space Proximal Network Model
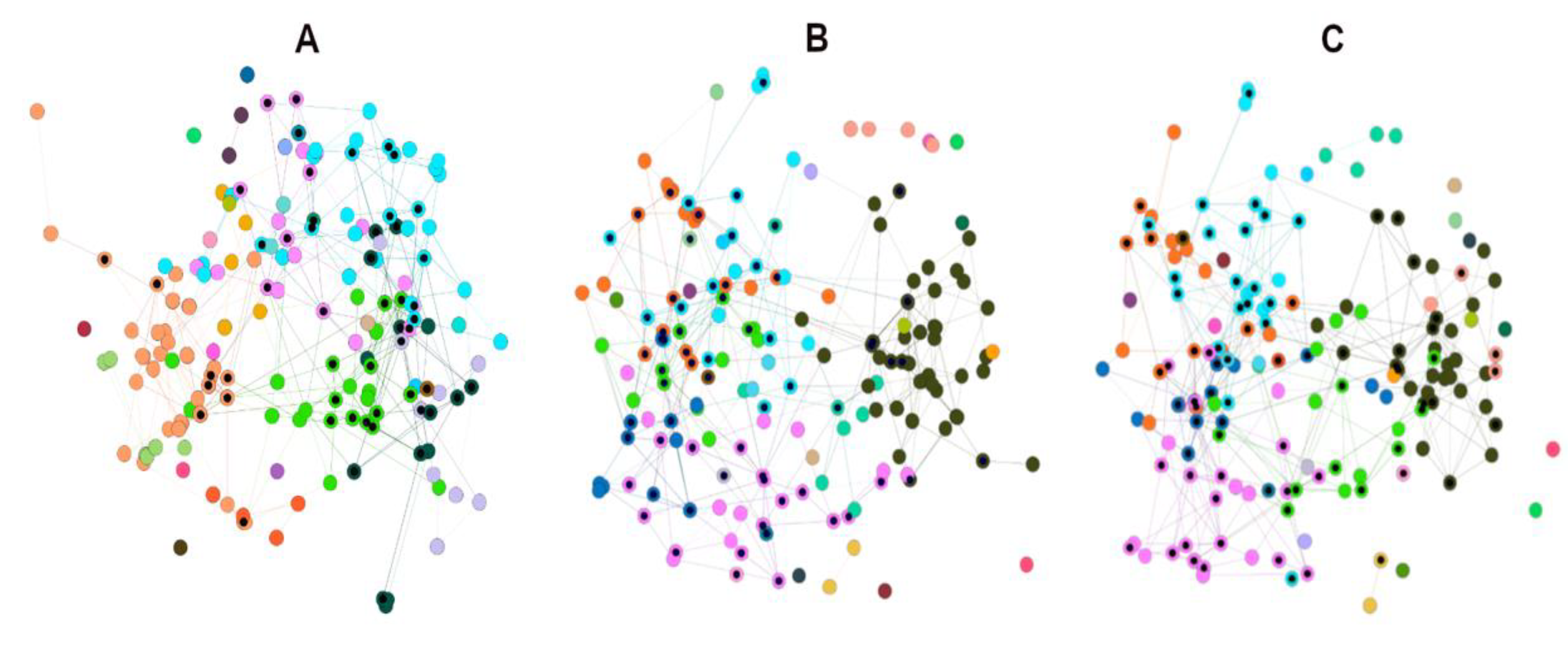
3.2. Network Visual Mining
3.2.1. Visual Mining of HSPNs; The Most Central and Atypical ABFPs
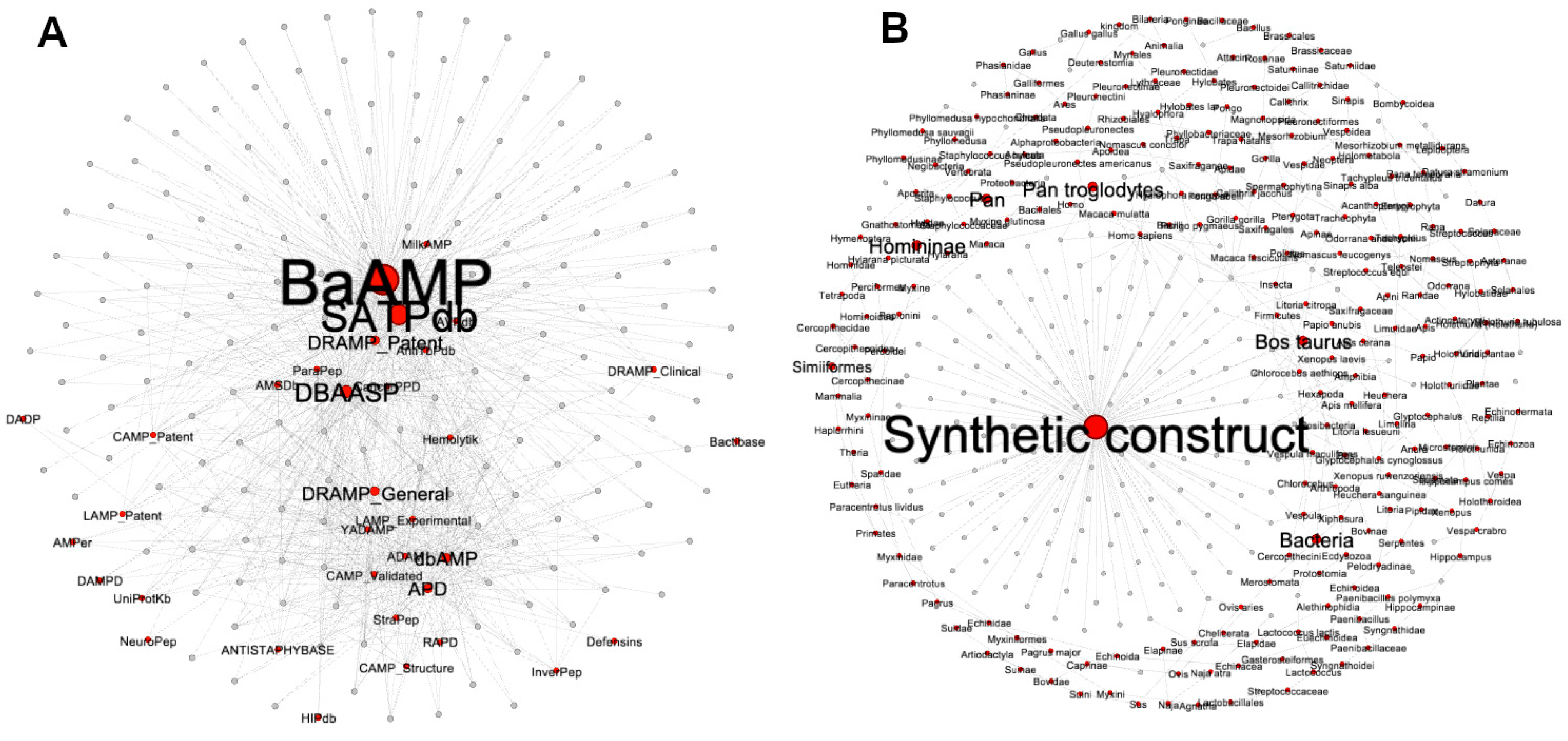
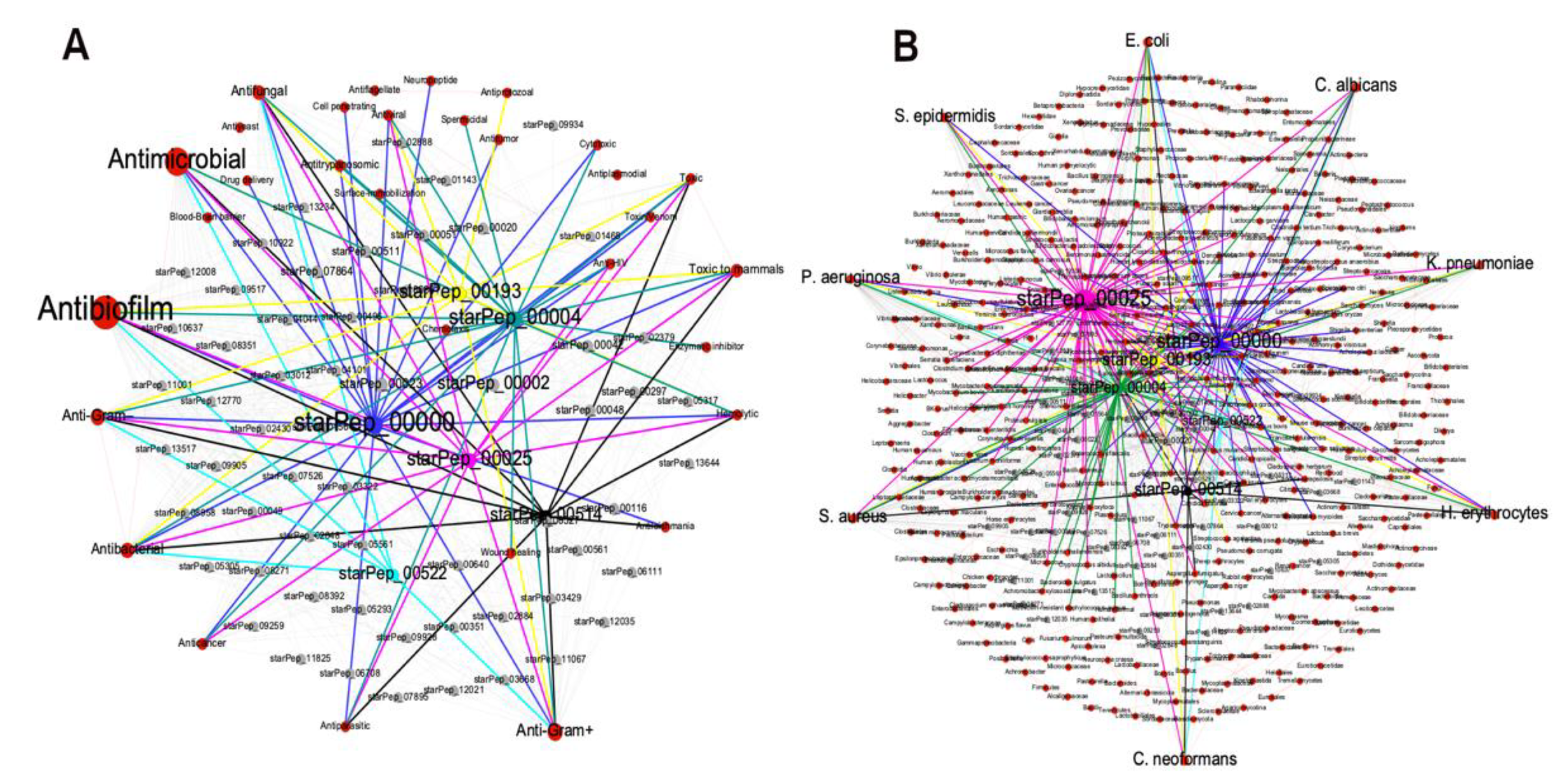
3.2.2. Metadata Analysis by Visual Mining
3.3. Representing the ABFPs with a Reduced Subset
3.3.1. The Selection of the Best Representative Subset
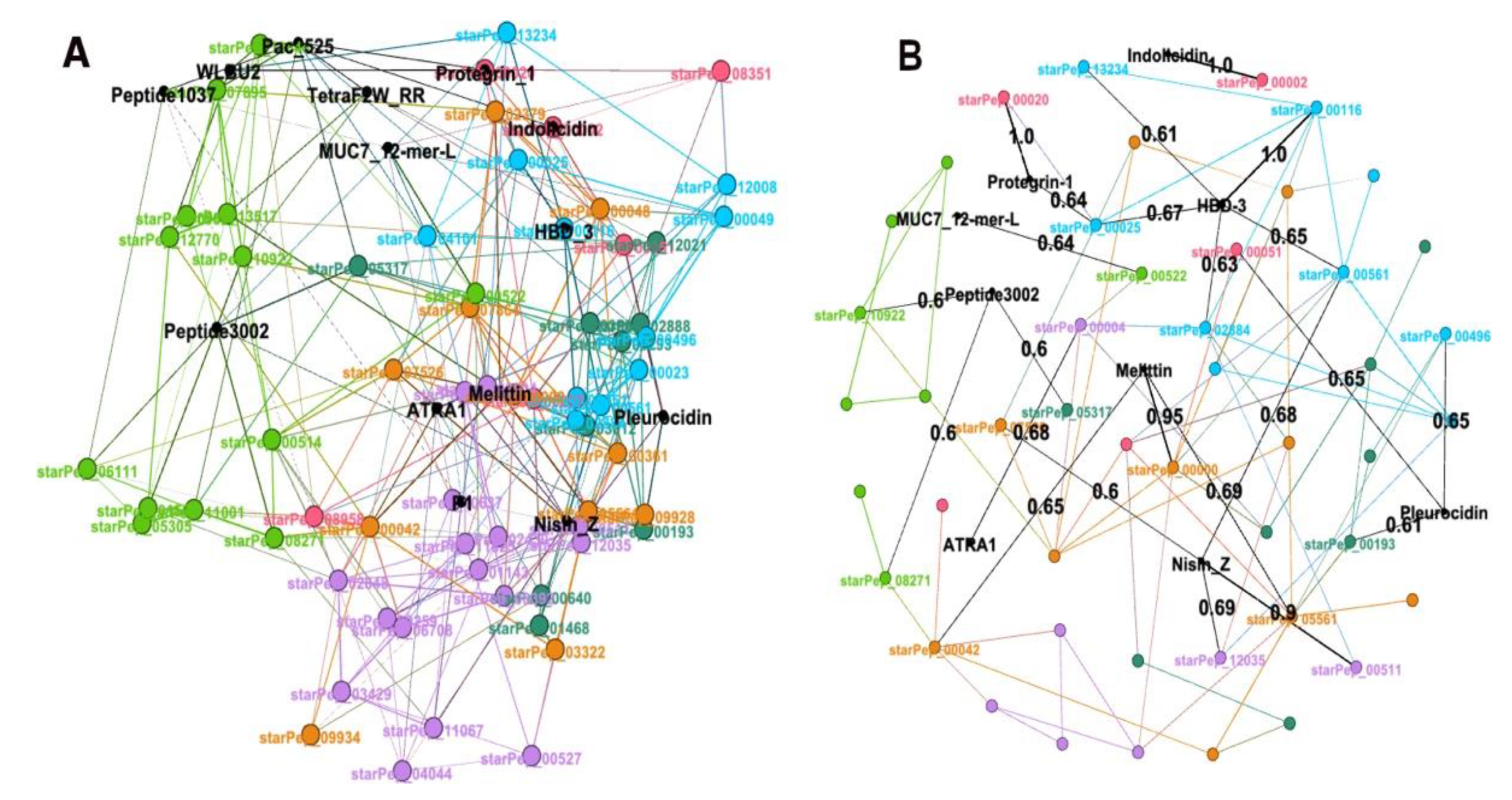
3.3.2. Visualizing/Analyzing the Best Representative Subset with HSPNs
3.3.3. Visualizing Mining of the METNs
3.4. External Representative ABFPs on the Representative Antibiofilm HSPN
3.5. Motif Discovery Assisted by Complex Networks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vestby, L.K.; Gronseth, T.; Simm, R.; Nesse, L.L. Bacterial Biofilm and its Role in the Pathogenesis of Disease. Antibiotics 2020, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Gloag, E.S.; Fabbri, S.; Wozniak, D.J.; Stoodley, P. Biofilm mechanics: Implications in infection and survival. Bioinformatics 2020, 2, 100017. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Nunez, C.; Reffuveille, F.; Fernandez, L.; Hancock, R.E. Bacterial biofilm development as a multicellular adaptation: Antibiotic resistance and new therapeutic strategies. Curr. Opin. Microbiol. 2013, 16, 580–589. [Google Scholar] [CrossRef]
- Sauer, K.; Stoodley, P.; Goeres, D.M.; Hall-Stoodley, L.; Burmolle, M.; Stewart, P.S.; Bjarnsholt, T. The biofilm life cycle: Expanding the conceptual model of biofilm formation. Nat. Rev. Microbiol. 2022, 20, 608–620. [Google Scholar] [CrossRef]
- An, A.Y.; Choi, K.G.; Baghela, A.S.; Hancock, R.E.W. An Overview of Biological and Computational Methods for Designing Mechanism-Informed Anti-biofilm Agents. Front. Microbiol. 2021, 12, 640787. [Google Scholar] [CrossRef]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial biofilm and associated infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef]
- Bryers, J.D. Medical biofilms. Biotechnol. Bioeng. 2008, 100, 1–18. [Google Scholar] [CrossRef]
- Veerachamy, S.; Yarlagadda, T.; Manivasagam, G.; Yarlagadda, P.K. Bacterial adherence and biofilm formation on medical implants: A review. Proc. Inst. Mech. Eng. H 2014, 228, 1083–1099. [Google Scholar] [CrossRef]
- Fleming, D.; Rumbaugh, K. The Consequences of Biofilm Dispersal on the Host. Sci. Rep. 2018, 8, 10738. [Google Scholar] [CrossRef]
- Rumbaugh, K.P.; Sauer, K. Biofilm dispersion. Nat. Rev. Microbiol. 2020, 18, 571–586. [Google Scholar] [CrossRef]
- Rather, M.A.; Gupta, K.; Bardhan, P.; Borah, M.; Sarkar, A.; Eldiehy, K.S.H.; Bhuyan, S.; Mandal, M. Microbial biofilm: A matter of grave concern for human health and food industry. J. Basic Microbiol. 2021, 61, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Breidenstein, E.B.; de la Fuente-Nunez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Romling, U.; Balsalobre, C. Biofilm infections, their resilience to therapy and innovative treatment strategies. J. Intern. Med. 2012, 272, 541–561. [Google Scholar] [CrossRef] [PubMed]
- Barraud, N.; Hassett, D.J.; Hwang, S.H.; Rice, S.A.; Kjelleberg, S.; Webb, J.S. Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 7344–7353. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.Q.; Estelles, A.; Li, L.; Abdelhady, W.; Gonzales, R.; Bayer, A.S.; Tenorio, E.; Leighton, A.; Ryser, S.; Kauvar, L.M. A Human Biofilm-Disrupting Monoclonal Antibody Potentiates Antibiotic Efficacy in Rodent Models of both Staphylococcus aureus and Acinetobacter baumannii Infections. Antimicrob. Agents Chemother. 2017, 61, e00904-17. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Nunez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef]
- Chavez de Paz, L.E.; Lemos, J.A.; Wickstrom, C.; Sedgley, C.M. Role of (p)ppGpp in biofilm formation by Enterococcus faecalis. Appl. Environ. Microbiol. 2012, 78, 1627–1630. [Google Scholar] [CrossRef]
- Hee, C.S.; Habazettl, J.; Schmutz, C.; Schirmer, T.; Jenal, U.; Grzesiek, S. Intercepting second-messenger signaling by rationally designed peptides sequestering c-di-GMP. Proc. Natl. Acad. Sci. USA 2020, 117, 17211–17220. [Google Scholar] [CrossRef]
- Verderosa, A.D.; Totsika, M.; Fairfull-Smith, K.E. Bacterial Biofilm Eradication Agents: A Current Review. Front. Chem. 2019, 7, 824. [Google Scholar] [CrossRef]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.; Rehm, B.H.; Hancock, R.E. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef]
- Di Somma, A.; Moretta, A.; Cane, C.; Cirillo, A.; Duilio, A. Antimicrobial and Antibiofilm Peptides. Biomolecules 2020, 10, 652. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ye, X.; Sun, P.; Xu, P.; Wang, L.; Liu, Z.; Huang, X.; Bai, Z.; Zhou, C. Antimicrobial and antibiofilm activity of the EeCentrocin 1 derived peptide EC1-17KV via membrane disruption. EBioMedicine 2020, 55, 102775. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct Target Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Shi, G.; Kang, X.; Dong, F.; Liu, Y.; Zhu, N.; Hu, Y.; Xu, H.; Lao, X.; Zheng, H. DRAMP 3.0: An enhanced comprehensive data repository of antimicrobial peptides. Nucleic Acids Res. 2022, 50, D488–D496. [Google Scholar] [CrossRef]
- Di Luca, M.; Maccari, G.; Maisetta, G.; Batoni, G. BaAMPs: The database of biofilm-active antimicrobial peptides. Biofouling 2015, 31, 193–199. [Google Scholar] [CrossRef]
- Sharma, A.; Gupta, P.; Kumar, R.; Bhardwaj, A. dPABBs: A Novel in silico Approach for Predicting and Designing Anti-biofilm Peptides. Sci. Rep. 2016, 6, 21839. [Google Scholar] [CrossRef]
- Gupta, S.; Sharma, A.K.; Jaiswal, S.K.; Sharma, V.K. Prediction of Biofilm Inhibiting Peptides: An In silico Approach. Front. Microbiol. 2016, 7, 949. [Google Scholar] [CrossRef]
- Fallah Atanaki, F.; Behrouzi, S.; Ariaeenejad, S.; Boroomand, A.; Kavousi, K. BIPEP: Sequence-based Prediction of Biofilm Inhibitory Peptides Using a Combination of NMR and Physicochemical Descriptors. ACS Omega 2020, 5, 7290–7297. [Google Scholar] [CrossRef]
- Bose, B.; Downey, T.; Ramasubramanian, A.K.; Anastasiu, D.C. Identification of Distinct Characteristics of Antibiofilm Peptides and Prospection of Diverse Sources for Efficacious Sequences. Front. Microbiol. 2021, 12, 783284. [Google Scholar] [CrossRef] [PubMed]
- Aguero-Chapin, G.; Galpert-Canizares, D.; Dominguez-Perez, D.; Marrero-Ponce, Y.; Perez-Machado, G.; Teijeira, M.; Antunes, A. Emerging Computational Approaches for Antimicrobial Peptide Discovery. Antibiotics 2022, 11, 936. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.; Marrero-Ponce, Y.; Rodriguez, H.; Aguero-Chapin, G.; Antunes, A.; Aguilera-Mendoza, L.; Martinez-Rios, F. A Novel Network Science and Similarity-Searching-Based Approach for Discovering Potential Tumor-Homing Peptides from Antimicrobials. Antibiotics 2022, 11, 401. [Google Scholar] [CrossRef]
- Ayala-Ruano, S.; Marrero-Ponce, Y.; Aguilera-Mendoza, L.; Perez, N.; Aguero-Chapin, G.; Antunes, A.; Aguilar, A.C. Network Science and Group Fusion Similarity-Based Searching to Explore the Chemical Space of Antiparasitic Peptides. ACS Omega 2022, 7, 46012–46036. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; Beltran, J.A.; Tellez Ibarra, R.; Guillen-Ramirez, H.A.; Brizuela, C.A. Graph-based data integration from bioactive peptide databases of pharmaceutical interest: Toward an organized collection enabling visual network analysis. Bioinformatics 2019, 35, 4739–4747. [Google Scholar] [CrossRef]
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; Garcia-Jacas, C.R.; Chavez, E.; Beltran, J.A.; Guillen-Ramirez, H.A.; Brizuela, C.A. Automatic construction of molecular similarity networks for visual graph mining in chemical space of bioactive peptides: An unsupervised learning approach. Sci. Rep. 2020, 10, 18074. [Google Scholar] [CrossRef]
- Chavez, E.; Dobrev, S.; Kranakis, E.; Opatrny, J.; Stacho, L.; Tejeda, H.; Urrutia, J. Half-space proximal: A new local test for extracting a bounded dilation spanner of a unit disk graph. In Proceedings of the Principles of Distributed Systems: 9th International Conference, OPODIS 2005, Revised Selected Papers 9, Pisa, Italy, 12–14 December 2005; pp. 235–245. [Google Scholar]
- Cherven, K. Network Graph Analysis and Visualization with Gephi; Packt Publishing: Birmingham, UK, 2013. [Google Scholar]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Fruchterman, T.M.; Reingold, E.M. Graph drawing by force-directed placement. Softw. Pract. Exp. 1991, 21, 1129–1164. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Bailey, T.L. STREME: Accurate and versatile sequence motif discovery. Bioinformatics 2021, 37, 2834–2840. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Grant, C.E. SEA: Simple enrichment analysis of motifs. BioRxiv 2021. [Google Scholar] [CrossRef]
- Newman, M. Networks; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Martinez, L.R.; Casadevall, A. Cryptococcus neoformans cells in biofilms are less susceptible than planktonic cells to antimicrobial molecules produced by the innate immune system. Infect. Immun. 2006, 74, 6118–6123. [Google Scholar] [CrossRef]
- Moazzezy, N.; Asadi Karam, M.R.; Rafati, S.; Bouzari, S.; Oloomi, M. Inhibition and eradication activity of truncated α-defensin analogs against multidrug resistant uropathogenic Escherichia coli biofilm. PLoS ONE 2020, 15, e0235892. [Google Scholar] [CrossRef]
- Eckert, R.; He, J.; Yarbrough, D.K.; Qi, F.; Anderson, M.H.; Shi, W. Targeted killing of Streptococcus mutans by a pheromone-guided “smart” antimicrobial peptide. Antimicrob. Agents Chemother. 2006, 50, 3651–3657. [Google Scholar] [CrossRef]
- de la Fuente-Nunez, C.; Reffuveille, F.; Mansour, S.C.; Reckseidler-Zenteno, S.L.; Hernandez, D.; Brackman, G.; Coenye, T.; Hancock, R.E. D-enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem. Biol. 2015, 22, 196–205. [Google Scholar] [CrossRef]
- Jamasbi, E.; Batinovic, S.; Sharples, R.A.; Sani, M.A.; Robins-Browne, R.M.; Wade, J.D.; Separovic, F.; Hossain, M.A. Melittin peptides exhibit different activity on different cells and model membranes. Amino Acids 2014, 46, 2759–2766. [Google Scholar] [CrossRef]
- Guha, S.; Ferrie, R.P.; Ghimire, J.; Ventura, C.R.; Wu, E.; Sun, L.; Kim, S.Y.; Wiedman, G.R.; Hristova, K.; Wimley, W.C. Applications and evolution of melittin, the quintessential membrane active peptide. Biochem. Pharmacol. 2021, 193, 114769. [Google Scholar] [CrossRef]
- Chen, X.; Hirt, H.; Li, Y.; Gorr, S.U.; Aparicio, C. Antimicrobial GL13K peptide coatings killed and ruptured the wall of Streptococcus gordonii and prevented formation and growth of biofilms. PLoS ONE 2014, 9, e111579. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, K.V.; Abdolhosseini, M.; Li, Y.; Chen, X.; Gorr, S.U.; Aparicio, C. Bio-inspired stable antimicrobial peptide coatings for dental applications. Acta. Biomater. 2013, 9, 8224–8231. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Chaudhary, K.; Dhanda, S.K.; Bhalla, S.; Usmani, S.S.; Gautam, A.; Tuknait, A.; Agrawal, P.; Mathur, D.; Raghava, G.P. SATPdb: A database of structurally annotated therapeutic peptides. Nucleic Acids Res. 2016, 44, D1119–D1126. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016, 44, 6503. [Google Scholar] [CrossRef]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2014, 42, D1147–D1153. [Google Scholar] [CrossRef]
- Mehta, D.; Anand, P.; Kumar, V.; Joshi, A.; Mathur, D.; Singh, S.; Tuknait, A.; Chaudhary, K.; Gautam, S.K.; Gautam, A.; et al. ParaPep: A web resource for experimentally validated antiparasitic peptide sequences and their structures. Database 2014, 2014, bau051. [Google Scholar] [CrossRef]
- Gautam, A.; Chaudhary, K.; Singh, S.; Joshi, A.; Anand, P.; Tuknait, A.; Mathur, D.; Varshney, G.C.; Raghava, G.P. Hemolytik: A database of experimentally determined hemolytic and non-hemolytic peptides. Nucleic Acids Res. 2014, 42, D444–D449. [Google Scholar] [CrossRef]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef]
- Seshadri Sundararajan, V.; Gabere, M.N.; Pretorius, A.; Adam, S.; Christoffels, A.; Lehvaslaiho, M.; Archer, J.A.; Bajic, V.B. DAMPD: A manually curated antimicrobial peptide database. Nucleic Acids Res. 2012, 40, D1108–D1112. [Google Scholar] [CrossRef] [PubMed]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, H.; Lu, H.; Li, G.; Huang, Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS ONE 2013, 8, e66557. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef] [PubMed]
- Reza, A.; Sutton, J.M.; Rahman, K.M. Effectiveness of Efflux Pump Inhibitors as Biofilm Disruptors and Resistance Breakers in Gram-Negative (ESKAPEE) Bacteria. Antibiotics 2019, 8, 229. [Google Scholar] [CrossRef]
- Li, J.; Chen, D.; Lin, H. Antibiofilm peptides as a promising strategy: Comparative research. Appl. Microbiol. Biotechnol. 2021, 105, 1647–1656. [Google Scholar] [CrossRef]
- Juretic, D. Designed Multifunctional Peptides for Intracellular Targets. Antibiotics 2022, 11, 1196. [Google Scholar] [CrossRef]
- Agüero-Chapin, G.; Galpert, D.; Molina-Ruiz, R.; Ancede-Gallardo, E.; Pérez-Machado, G.; De la Riva, G.A.; Antunes, A. Graph Theory-Based Sequence Descriptors as Remote Homology Predictors. Biomolecules 2020, 10, 26. [Google Scholar] [CrossRef]
- Vens, C.; Rosso, M.N.; Danchin, E.G. Identifying discriminative classification-based motifs in biological sequences. Bioinformatics 2011, 27, 1231–1238. [Google Scholar] [CrossRef]
- de Souza, C.M.; da Silva, Á.P.; Júnior, N.G.O.; Martínez, O.F.; Franco, O.L. Peptides as a therapeutic strategy against Klebsiella pneumoniae. Trends Pharmacol. Sci. 2022, 43, 335–348. [Google Scholar] [CrossRef]
- Edwards-Gayle, C.J.C.; Barrett, G.; Roy, S.; Castelletto, V.; Seitsonen, J.; Ruokolainen, J.; Hamley, I.W. Selective Antibacterial Activity and Lipid Membrane Interactions of Arginine-Rich Amphiphilic Peptides. ACS Appl. Bio. Mater. 2020, 3, 1165–1175. [Google Scholar] [CrossRef]
- Zarena, D.; Mishra, B.; Lushnikova, T.; Wang, F.; Wang, G. The pi Configuration of the WWW Motif of a Short Trp-Rich Peptide Is Critical for Targeting Bacterial Membranes, Disrupting Preformed Biofilms, and Killing Methicillin-Resistant Staphylococcus aureus. Biochemistry 2017, 56, 4039–4043. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Lushnikova, T.; Golla, R.M.; Wang, X.; Wang, G. Design and surface immobilization of short anti-biofilm peptides. Acta Biomater. 2017, 49, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Sidorczuk, K.; Gagat, P.; Pietluch, F.; Kala, J.; Rafacz, D.; Bakala, L.; Slowik, J.; Kolenda, R.; Rodiger, S.; Fingerhut, L.; et al. Benchmarks in antimicrobial peptide prediction are biased due to the selection of negative data. Brief Bioinform. 2022, 23, bbac343. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSPN—No Cutoff | |||
|---|---|---|---|
| Centrality Measure | Total | Peptide Name | Cluster |
| Node Degree Harmonic Betweenness Hub-Bridge | 1 | starPep_03668 | (1) |
| Node Degree Betweenness Hub-Bridge | 1 | starPep_00048 | (1) |
| Harmonic Hub-Bridge | 2 | starPep_00000 starPep_10922 | (1) (3) |
| Node Degree Betweenness | 7 | starPep_12469 starPep_07526 starPep_00145 starPep_06130 starPep_00042 starPep_08958 starPep_02281 | (3) (2)(2) (3) (0) (2) (2) |
| Node Degree | 1 | starPep_00517 | (0) |
| Harmonic | 7 | starPep_07864 starPep_07895 starPep_02907 starPep_12770 starPep_12531 starPep_13517 starPep_07893 | (0) (3)(3) (3)(3) (3) (2) |
| Betweenness | 1 | starPep_08001 | (3) |
| Hub-Bridge | 6 | starPep_00496 starPep_00561 starPep_00361 starPep_13515 starPep_00193 starPep_05561 | (1) (1) (1) (2) (1) (1) |
| HSPN—Cutoff 0.65 | |||
| Node Degree Harmonic Betweenness Hub-Bridge | 1 | starPep_00048 | (9) |
| Node Degree Betweenness Hub-Bridge | 1 | starPep_00042 | (11) |
| Harmonic Betweenness Hub-Bridge | 1 | starPep_10922 | (15) |
| Node Degree Harmonic Betweenness | 2 | starPep_00000 starPep_03668 | (15) (4) |
| Node Degree Harmonic | 1 | starPep_00361 | (9) |
| Node Degree Betweenness | 1 | starPep_07526 | (17) |
| Node Degree Hub-Bridge | 2 | starPep_00004 starPep_00193 | (11) (9) |
| Harmonic Betweenness | 1 | starPep_02379 | (17) |
| Harmonic Hub-Bridge | 3 | starPep_07895 starPep_02907 starPep_12531 | (15) (15) (15) |
| Node Degree | 2 | starPep_00561 starPep_05561 | (9) (4) |
| Harmonic | 1 | starPep_07893 | (14) |
| Betweenness | 3 | starPep_12530 starPep_04734 starPep_08958 | (15) (17) (15) |
| Hub-Bridge | 2 | starPep_12770 starPep_02908 | (15) (15) |
| HSPN—Cutoff 0.65 | |||
|---|---|---|---|
| Atypical Peptides | Total | Peptide Name | Cluster |
| Singletons | 20 | starPep_00002 starPep_00739 starPep_02281 starPep_02383 starPep_02400 starPep_02730 starPep_03693 starPep_04044 * starPep_05305 * starPep_05447 * starPep_05964 starPep_06255 starPep_06358 * starPep_08001 starPep_09934 * starPep_09989 * starPep_10637 * starPep_14812 * starPep_16445 * starPep_18706 * | (10) (18) (20) (25) (0) (5) (19) (13) (8) (27) (26) (21) (23) (24) (16) (29) (1) (12) (2) (3) |
| Isolated Community | 2 | starPep_04274–starPep_04424 starPep_13860 *–starPep_13861 * | (6) (28) |
| Harmonic (HC) | Hub-Bridge (HB) | ||||||
|---|---|---|---|---|---|---|---|
| Subsets | Cutoff | Edges | Nodes | Coverage 1 % | Edges | Nodes | Coverage 1 % |
| 1 | 0.90 | 227 | 154 | 89 | 276 | 154 | 89 |
| 2 | 0.80 | 230 | 138 | 79 | 235 | 137 | 79 |
| 3 | 0.70 | 199 | 122 | 70 | 201 | 125 | 72 |
| 4 | 0.60 | 167 | 103 | 59 | 162 | 104 | 60 |
| 5 | 0.50 | 128 | 80 | 46 | 112 | 80 | 46 |
| 6 | 0.45 | 115 | 74 | 43 | 88 | 68 | 39 |
| 7 | 0.40 | 74 | 54 | 31 | 63 | 51 | 29 |
| 8 | 0.35 | 62 | 45 | 26 | 44 | 40 | 23 |
| 9 | 0.30 | 40 | 32 | 22 | 35 | 34 | 20 |
| Class-Peptides | Action Mode | Sequence |
|---|---|---|
| Class1- HBD-3 | Influences icaAD and icaR genes’ transcription levels | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK |
| Class2- Nisin Z | Decreases adhesion, kills bacteria, reduces biofilm formation | ITSISLCTPGCKTGALM GCNMKTATCNCSIHVSK |
| Class3- MUC7 12-mer-L | Attracted to bacterial surfaces by the electrostatic bonding | RKSYKCLHKRCR |
| Class4- ATRA1 | Promotes biofilm dispersal | KRFKKFFKKLKNSVK KRFKKFFKKLKVIGVT FPF |
| Class5- Pleurocidin | Induces disturbance/permeabilization of the membranes and bindssss to bacterial DNA, causing interference with cellular functions | GWGSFFKKAAHVGK HVGKAALTHYL |
| Class6- Pac-525 | Able to enter membranes and to affect the lipopolysaccharides of Gram-negative bacteria | KWRRWVRWI |
| Class7- peptide 1037 | Decreases the attachment of bacterial cells, stimulates twitching motility, and influences two major quorum sensing systems | KRFRIRVRV |
| Class8- Indolicidin | Induces lipid removal and mixed indolicidin–lipid patches alongside membrane permeabilization | ILPWKWPWWPWRR |
| Class9- Protegrin-1 * | Forms amyloid fibers to associate with the bacterial membrane and produce transmembrane pores | RGGRLCYCRRRFCVCVGR |
| Class10- Peptide 3002 | Blocking (p)ppGpp | ILVRWIRWRIQW |
| Class11-TetraF2W-RR * | Disrupts membranes to kill bacteria rapidly | WWWLRRIW |
| Class12-P1 | Interferes with the proper secretion and/or intermolecular interaction of key extracellular polymers in the biofilm matrix | PARKARAATAATAATAATAATAAT |
| Class13-WLBU2 * | LPS-binding property interferes with bacterial attachment, destroying the bacterial membrane | RRWVRRVRRVWRRVVRVVRRWVRR |
| Class14-Melittin * | Inhibits the expression of biofilm-associated bap genes | GIGAVLKVLTTGLPALISWIKRKRQQ |
| No | Motif | EMBOSS Cons. | Cluster | Cluster Size | MSA Method | Enrichment Ratio * |
|---|---|---|---|---|---|---|
| 1 | RLFNR | xxxNR | 4 | 15 | MAFFT/ MUSCLE | - |
| 2 | GGG | GxG | MAFFT | - | ||
| 3 | GGGWK | xxGWK | MUSCLE | (−)/(2.25) | ||
| 4 | FKKA | xKKx | MAFFT | (−)/(4.0) | ||
| 5 | FWKWA | FWK | MAFFT | (3.0)/(2.83) | ||
| 6 | WGK | WxK | MAFFT | (−)/(1.41) | ||
| 7 | LLLLLKKK | LLLLLKKK | 6 | 2 | Pair-Aligned | - |
| 8 | LISWIK | lisxik | 7 | 8 | MAFFT | (−)/(2.71) |
| 9 | KNKRK | knkxk | MUSCLE | (3.0)/(2.22) | ||
| 10 | KRKQ | kxkQ | MAFFT | (3.0)/(−) | ||
| 11 | R[GP]RVS | rxRVS | MAFFT | (−)/(3.0) | ||
| 12 | RRPR | RRxR | MUSCLE | - | ||
| 13 | [GR]GG | xGG | MAFFT | (3.0)/(1.90) | ||
| 14 | GGRRRR | GGrrRR | MUSCLE | (−)/(2.0) | ||
| 15 | RRRRR | RRRRR | MAFFT/ MUSCLE | - | ||
| 16 | ISGI | Ixxx | 9 | 23 | MAFFT | - |
| 17 | FKKLL | xKKLL | 11 | 27 | MAFFT/ MUSCLE | (−)/(2.25) |
| 18 | KKLK | MAFFT | - | |||
| 19 | KKL | MUSCLE | - | |||
| 20 | LKK | LKK | MUSCLE | - | ||
| 21 | RIRVR | RIRVR | 14 | 23 | MAFFT | (−)/(1.58) |
| 22 | RVIR | xRVIR | MAFFT | (−)/(1.32) | ||
| 23 | VRVIR | MUSCLE | (−)/(2.83) | |||
| 24 | R[WL]R | RxR | MUSCLE | (1.57)/(−) | ||
| 25 | RIRRW | RIxRW | 15 | 26 | MAFFT/ MUSCLE | (−)/(4.0) |
| 26 | RI[VR]W | (−)/(1.67) | ||||
| 27 | WVV | WVV | MAFFT | (−)/(1.44) | ||
| 28 | I[IR]R | IIxR | MUSCLE | - | ||
| 29 | WLRK | Wxxx | 17 | 23 | MAFFT | (−)/(2.50) |
| 30 | RWK | Rxx | MUSCLE | - | ||
| 31 | KKL | Kxx | MAFFT | - | ||
| 32 | KR[AKL]RK | KRxRK | MUSCLE | (6.0)/(3.0) | ||
| 33 | WR[IV]R | xRWR[IV]R | 22 | 5 | MAFFT/ MUSCLE | |
| 32 | FRWRI | MAFFT | (3.0)/(−) | |||
| 33 | RWRVR | MUSCLE | (−)/(1.63) | |||
| 34 | YAPWYN | YAPWYN | 28 | 2 | Pair-Aligned | - |
| 35 | [FI][KW]RK | iKrK | Singletons | 20 | MAFFT/ MUSCLE | (−)/(1.46) |
| No | Motif | Cluster | Cluster Size | Matches in ABFPs | Matches in Control | Sites (%) | Score | Enrichment Ratio * |
|---|---|---|---|---|---|---|---|---|
| 1 | FKKA | 4 | 15 | 7 | 0 | 46.7 | 3.3e-003 | (−)/(3.33) |
| 2 | GGGR | 7 | 0 | 46.7 | 3.3e-003 | (−)/(2.11) | ||
| 3 | W[KR]WF | 7 | 0 | 46.7 | 3.3e-003 | (−)/(1.38) | ||
| 4 | FIH | 6 | 0 | 40.0 | 8.4e-002 | - | ||
| 5 | RLFNR | 5 | 0 | 33.3 | 2.1e-003 | - | ||
| 6 | KKK | 6 | 2 | 2 | 0 | 100 | 1.7e-001 | - |
| 7 | LLLLL | 2 | 0 | 100 | 1.7e-001 | - | ||
| 8 | RGG | 7 | 8 | 8 | 0 | 100 | 7.8e-005 | (3.0)/(1.56) |
| 9 | ISWIK | 4 | 0 | 50 | 3.8e-002 | (−)/(2.83) | ||
| 10 | NKRKQ | 4 | 0 | 50 | 3.8e-002 | - | ||
| 11 | RPRVS | 3 | 0 | 37.5 | 1.0e-001 | (−)/(3.71) | ||
| 12 | RRRRR | 3 | 0 | 37.5 | 1.0e-001 | - | ||
| 13 | SAC | 9 | 23 | 16 | 1 | 69.6 | 3.3e-006 | - |
| 14 | AKA | 5 | 0 | 21.7 | 2.85e-002 | - | ||
| 15 | CD[VI] | 5 | 0 | 21.7 | 2.85e-002 | - | ||
| 16 | IA[GVK] | 5 | 0 | 21.7 | 2.85e-002 | - | ||
| 17 | LFKKL | 11 | 27 | 9 | 0 | 33.3 | 8.8e-004 | (−)/(2.40) |
| 18 | KVLK | 8 | 0 | 29.6 | 2.1e-003 | (3.0)/(4.0) | ||
| 19 | KRFL | 6 | 0 | 22.2 | 1.1e-002 | (3.0)/(1.8) | ||
| 20 | VRLRI | 14 | 23 | 12 | 0 | 52.2 | 3.5e-005 | - |
| 21 | RVIR | 10 | 0 | 43.5 | 2.8e-004 | (−)/(1.32) | ||
| 22 | VWVI | 15 | 26 | 14 | 3 | 53.8 | 1.3e-003 | (3.0)/(3.0) |
| 23 | VIWRR | 8 | 0 | 30.8 | 2.1e-003 | (−)/(2.50) | ||
| 24 | LRK | 17 | 23 | 9 | 0 | 39.1 | 7.4e-004 | (3.0)/(1.27) |
| 25 | WRRK | 6 | 0 | 26.1 | 1.1e-002 | (−)/(1.67) | ||
| 26 | WRIR | 22 | 5 | 5 | 1 | 100 | 2.4e-002 | (−)/(3.25) |
| 27 | IRR | 2 | 3 | 40.0 | 9.0e-001 | (1.67)/(−) | ||
| 28 | APWTN | 28 | 2 | 2 | 0 | 100 | 1.7e-001 | (−)/(3.0) |
| 29 | KKRK | Singletons | 20 | 2 | 0 | 10.0 | 2.3e-001 | - |
| 30 | KKVVF | 2 | 0 | 10.0 | 2.4e-001 | - | ||
| 31 | LLKLL | 2 | 0 | 10.0 | 2.4e-001 | - | ||
| 32 | VKFK | 2 | 0 | 10.0 | 2.4e-001 | - | ||
| 33 | WRWR | 2 | 0 | 10.0 | 2.4e-001 | (−)/(1.64) |
| No | Motif | Cluster | Method | Enrichment Ratio |
|---|---|---|---|---|
| 1 | FWKWA | 4 | MAFFT | (3.0)/(2.83) |
| 2 | KNKRK | 7 | MUSCLE | (3.0)/(2.22) |
| 3 | [GR]GG | 7 | MAFFT/STREME | (3.0)/(1.90) |
| 4 | KVLK | 11 | STREME | (3.0)/(4.0) |
| 5 | KRFL | 11 | STREME | (3.0)/(1.8) |
| 6 | VWVI | 15 | STREME | (3.0)/(3.0) |
| 7 | KR[AKL]RK | 17 | MUSCLE | (6.0)/(3.0) |
| 8 | LRK | 17 | STREME | (3.0)/(1.27) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agüero-Chapin, G.; Antunes, A.; Mora, J.R.; Pérez, N.; Contreras-Torres, E.; Valdes-Martini, J.R.; Martinez-Rios, F.; Zambrano, C.H.; Marrero-Ponce, Y. Complex Networks Analyses of Antibiofilm Peptides: An Emerging Tool for Next-Generation Antimicrobials’ Discovery. Antibiotics 2023, 12, 747. https://doi.org/10.3390/antibiotics12040747
Agüero-Chapin G, Antunes A, Mora JR, Pérez N, Contreras-Torres E, Valdes-Martini JR, Martinez-Rios F, Zambrano CH, Marrero-Ponce Y. Complex Networks Analyses of Antibiofilm Peptides: An Emerging Tool for Next-Generation Antimicrobials’ Discovery. Antibiotics. 2023; 12(4):747. https://doi.org/10.3390/antibiotics12040747
Chicago/Turabian StyleAgüero-Chapin, Guillermin, Agostinho Antunes, José R. Mora, Noel Pérez, Ernesto Contreras-Torres, José R. Valdes-Martini, Felix Martinez-Rios, Cesar H. Zambrano, and Yovani Marrero-Ponce. 2023. "Complex Networks Analyses of Antibiofilm Peptides: An Emerging Tool for Next-Generation Antimicrobials’ Discovery" Antibiotics 12, no. 4: 747. https://doi.org/10.3390/antibiotics12040747
APA StyleAgüero-Chapin, G., Antunes, A., Mora, J. R., Pérez, N., Contreras-Torres, E., Valdes-Martini, J. R., Martinez-Rios, F., Zambrano, C. H., & Marrero-Ponce, Y. (2023). Complex Networks Analyses of Antibiofilm Peptides: An Emerging Tool for Next-Generation Antimicrobials’ Discovery. Antibiotics, 12(4), 747. https://doi.org/10.3390/antibiotics12040747