

Construction and Activity Testing of a Modular Fusion Peptide against Enterococcus faecalis
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
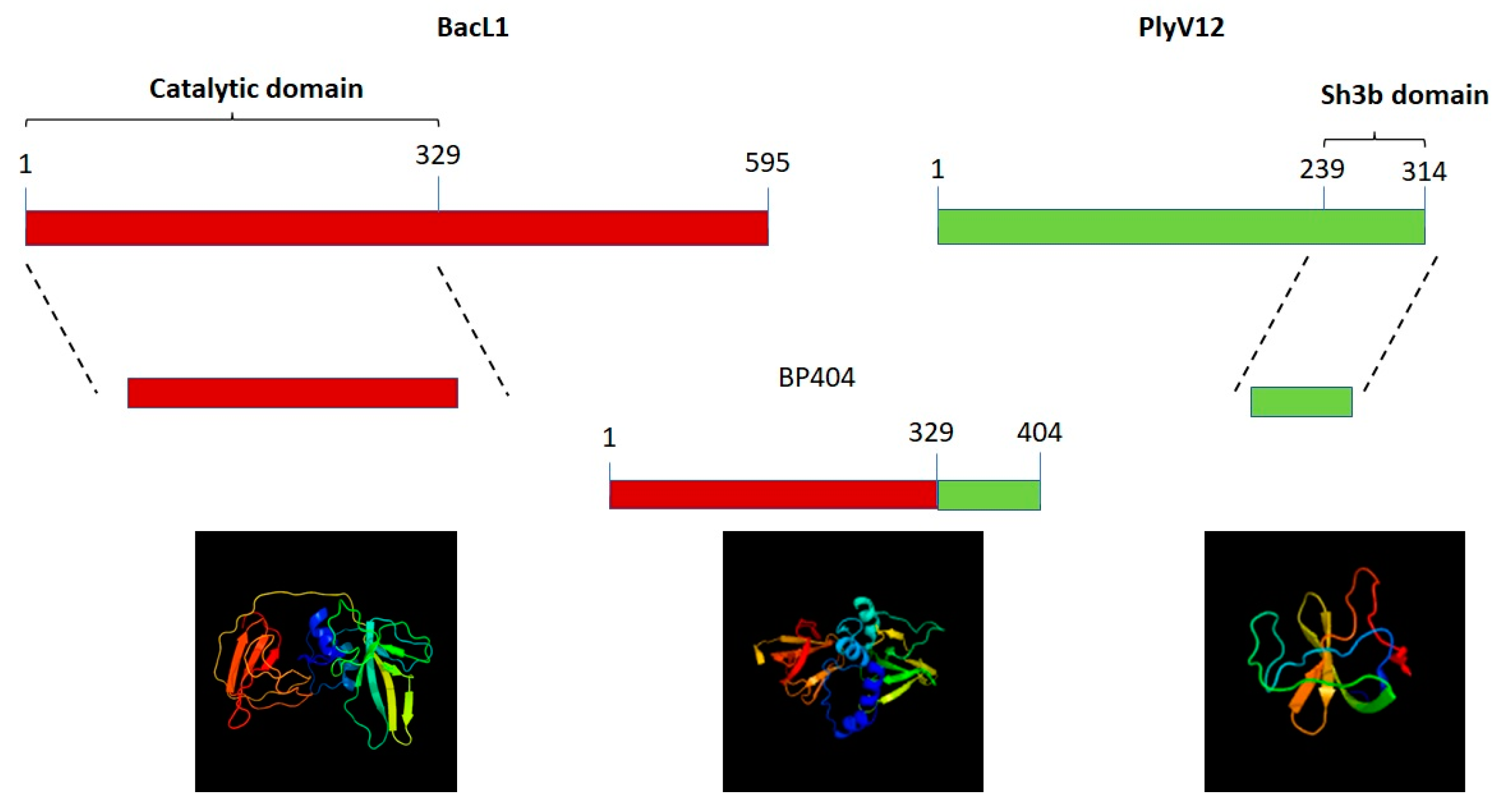
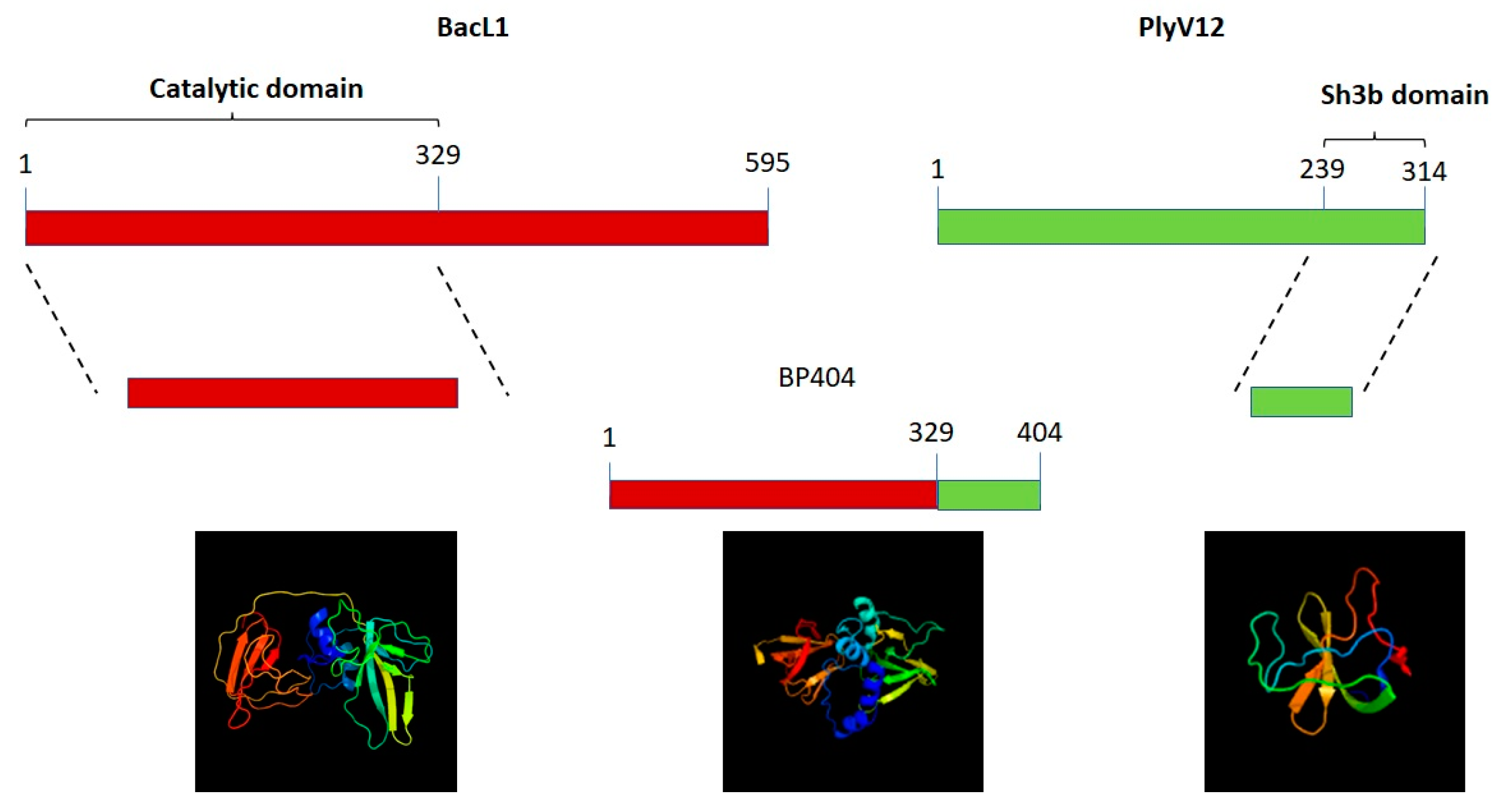
2.1. Construction of Protein BP404
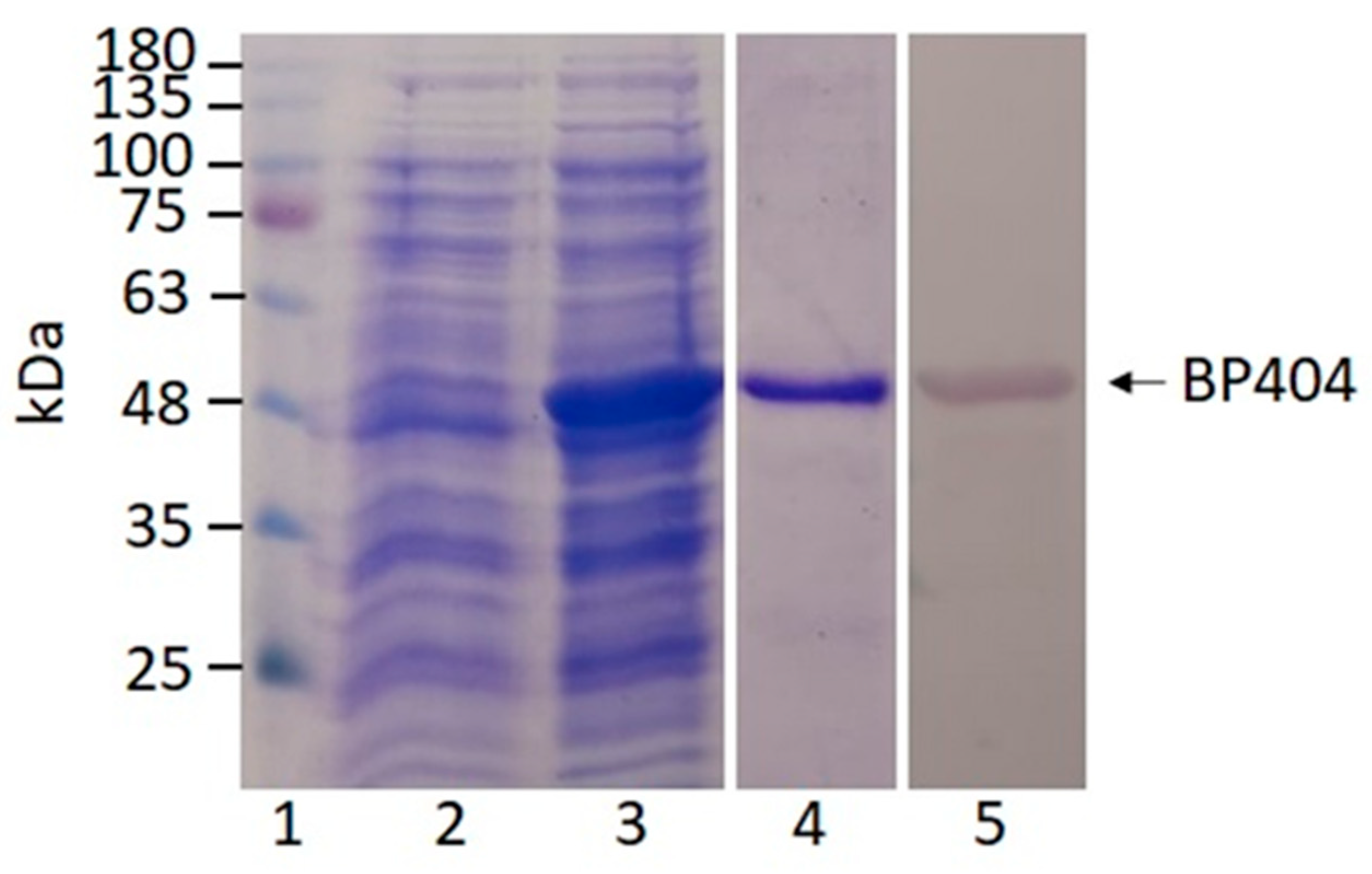
2.2. Synthesis and Purification of BP404
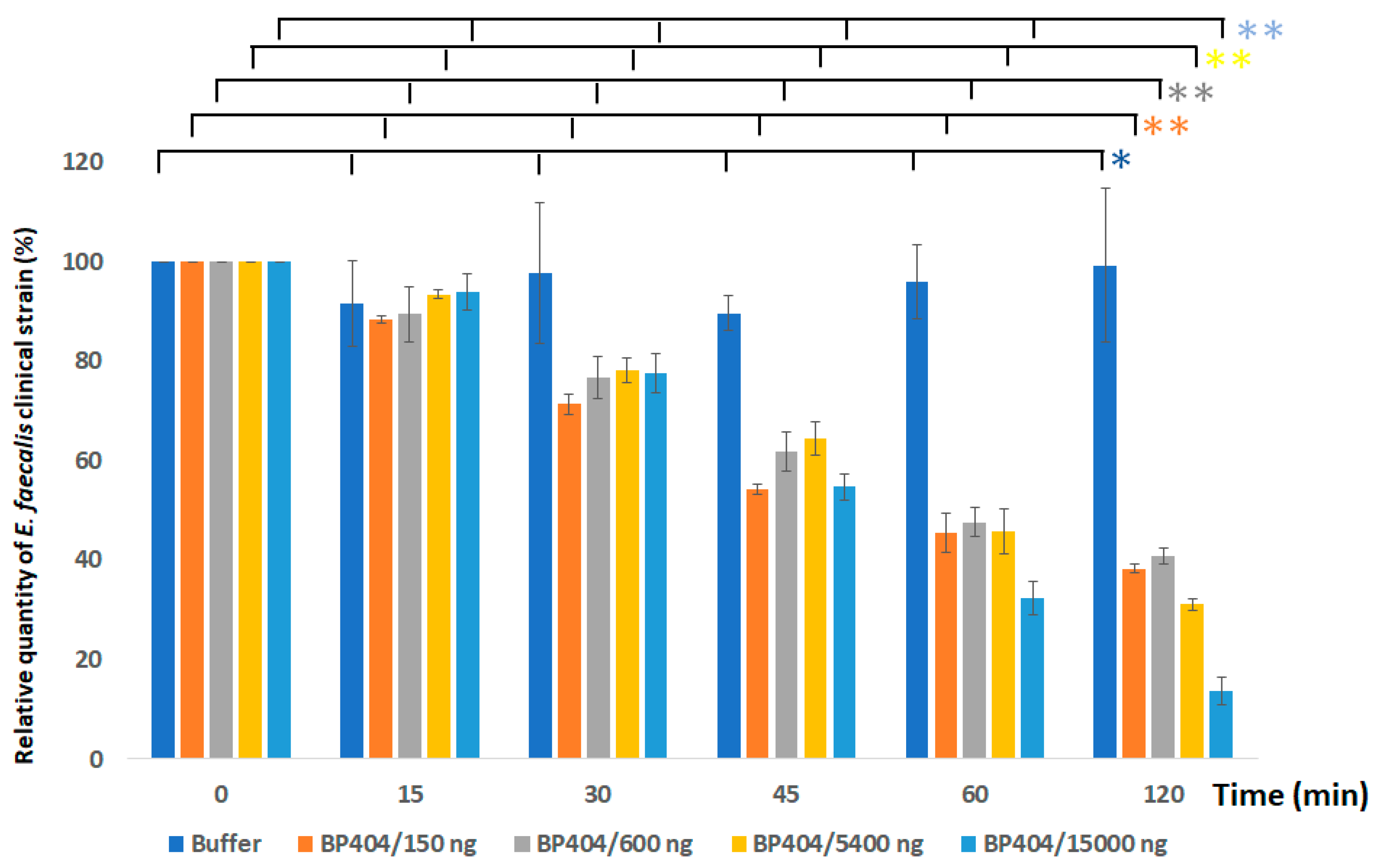
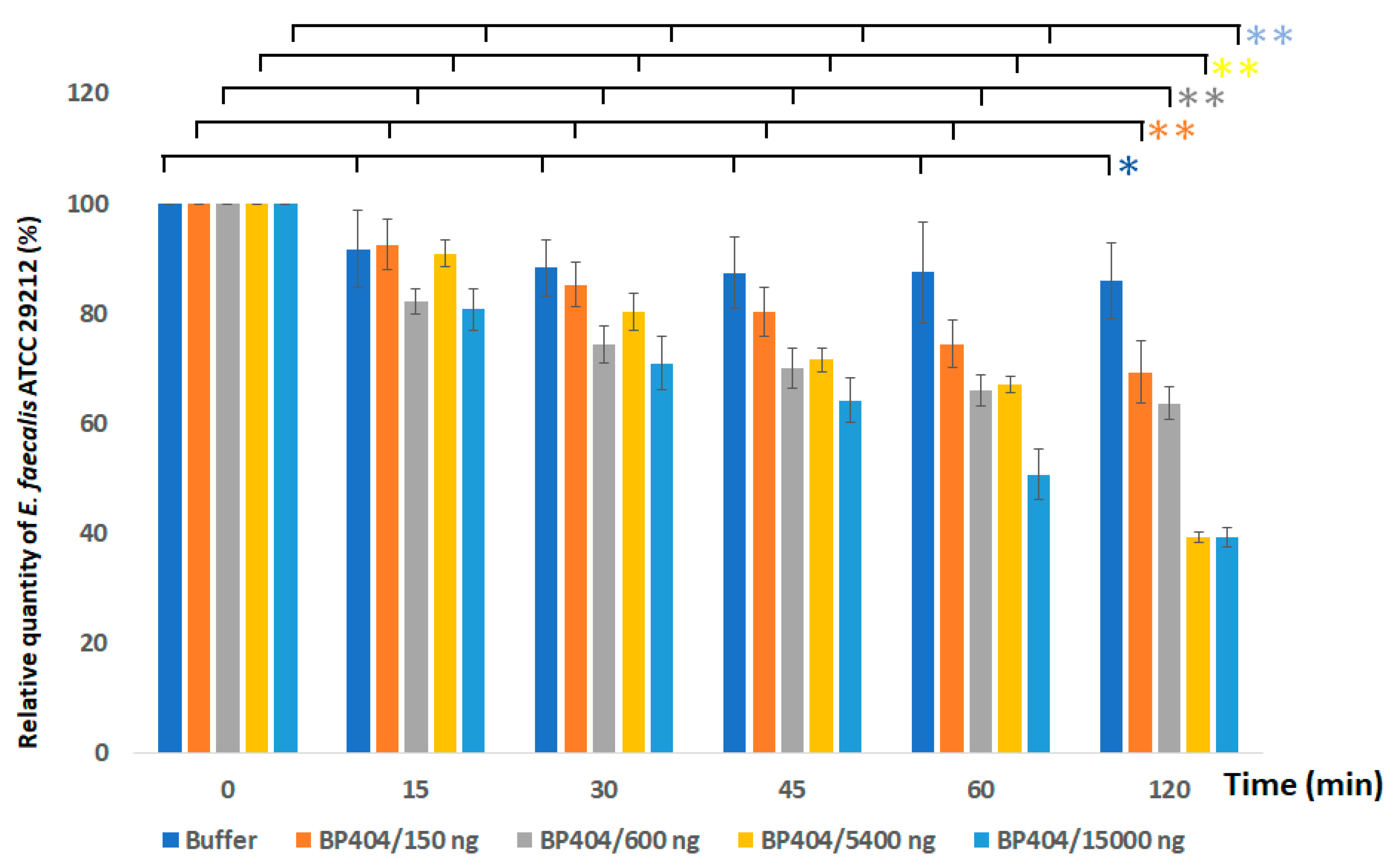
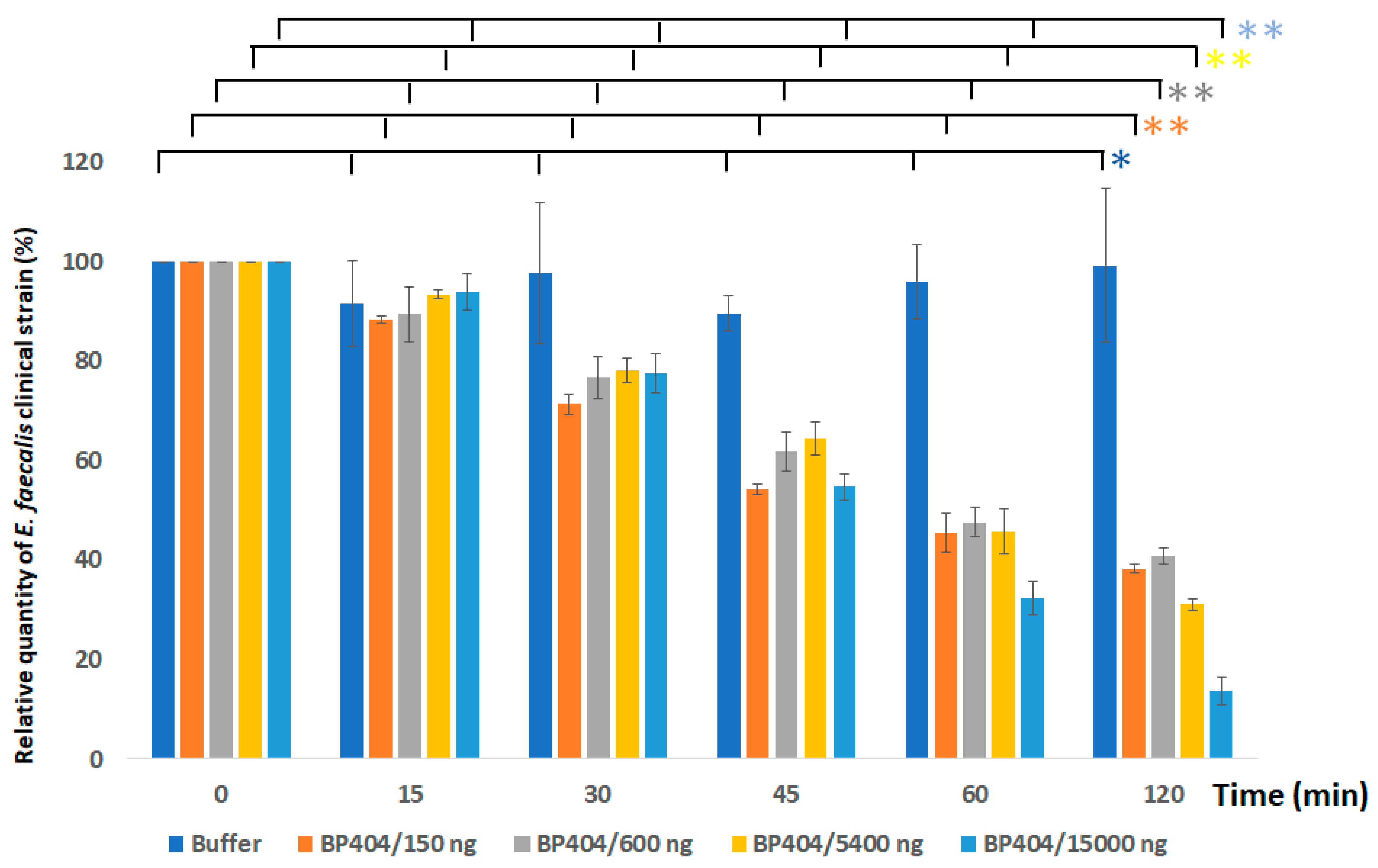
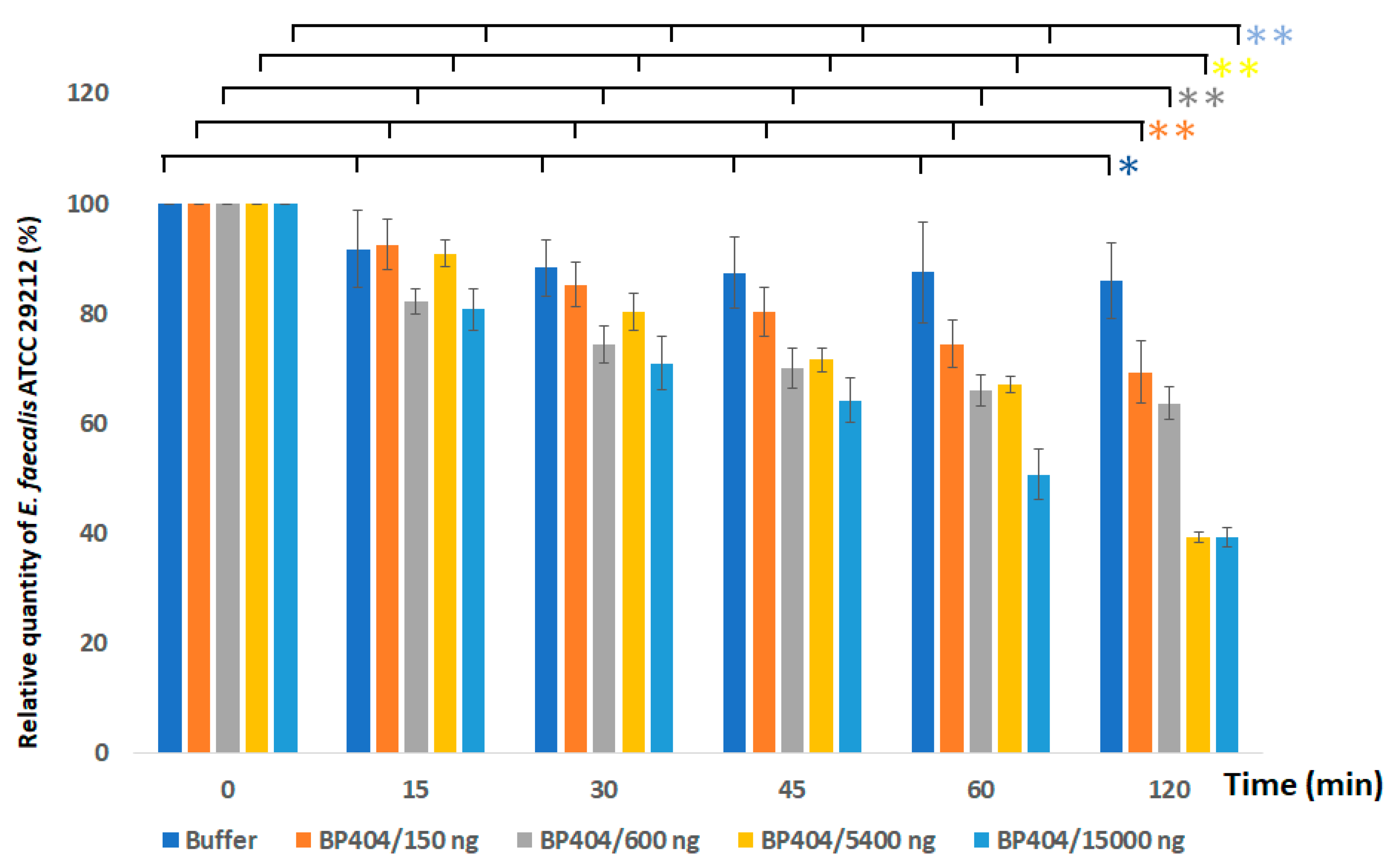
2.3. Protein BP404 Displays Activity against E. faecalis Strains
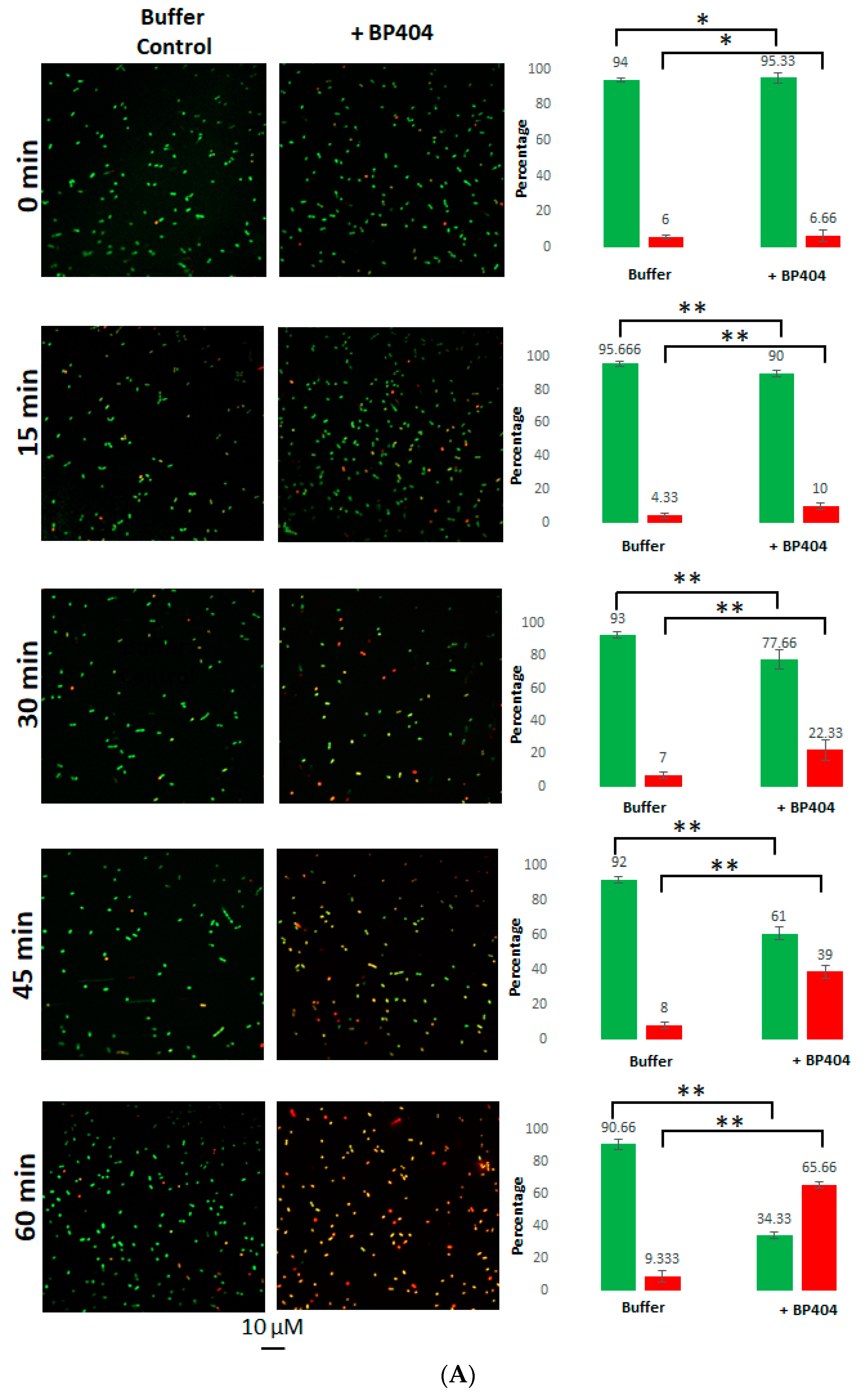
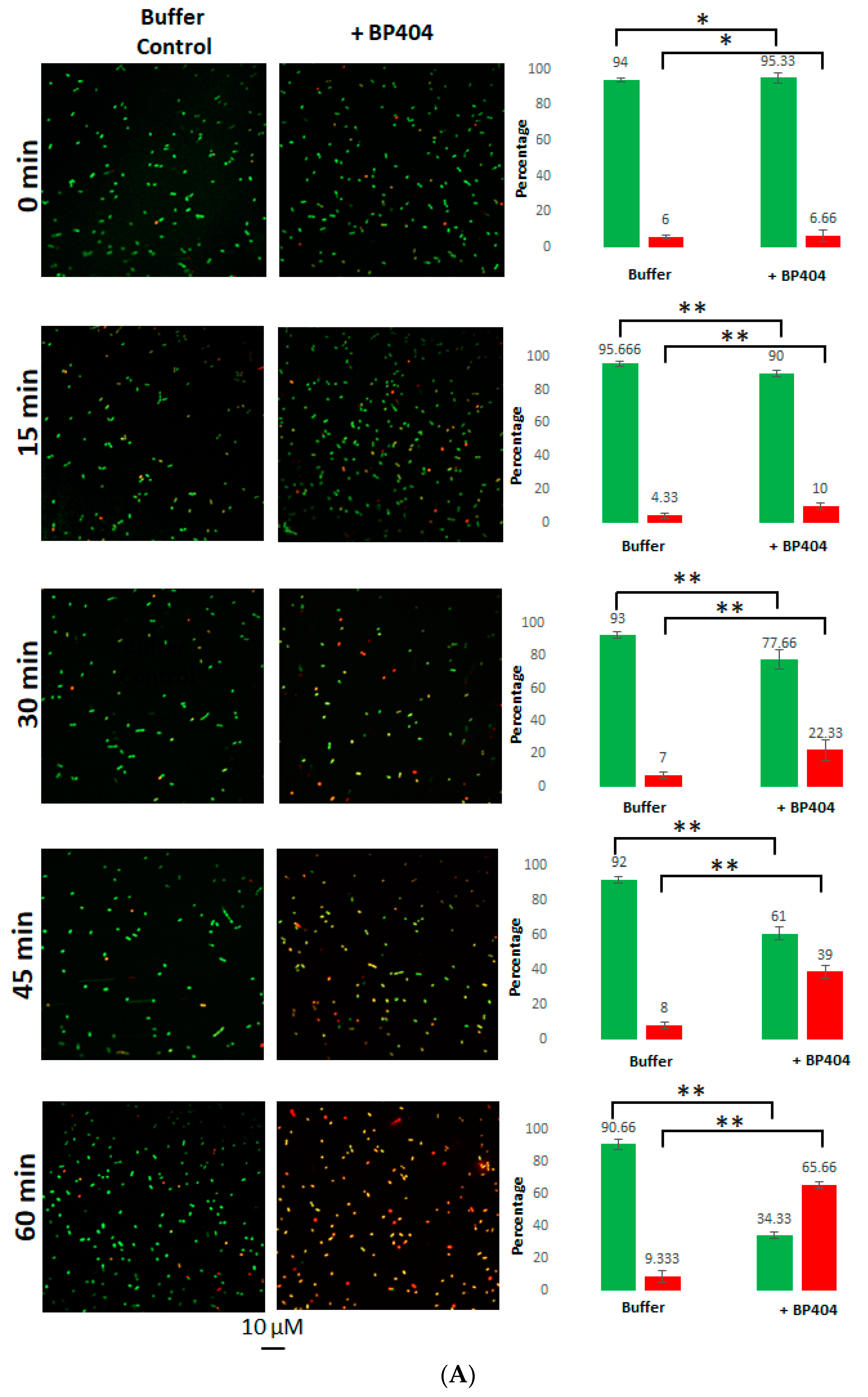
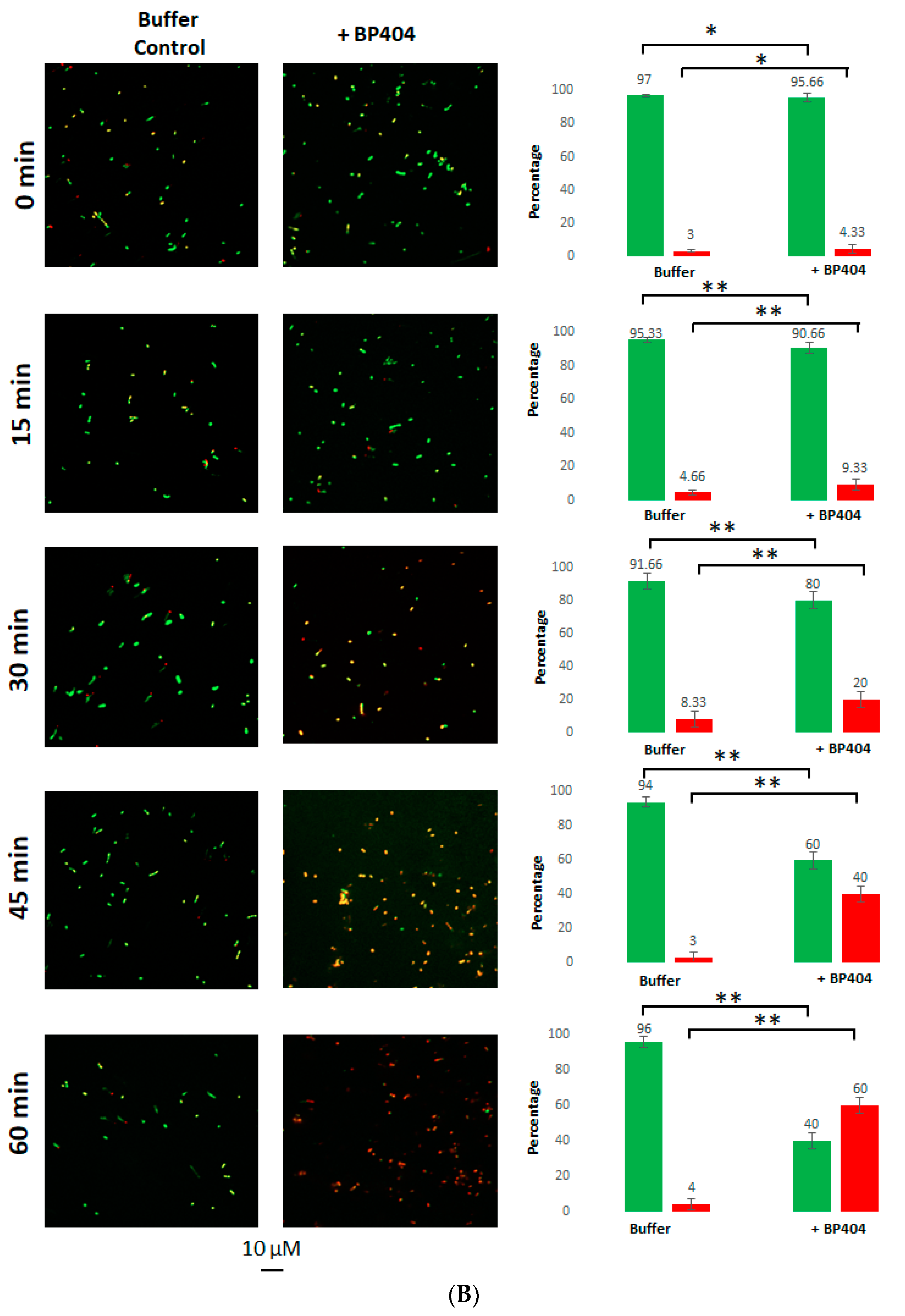
2.4. Live/Dead Staining of the E. faecalis Strains Treated with BP404
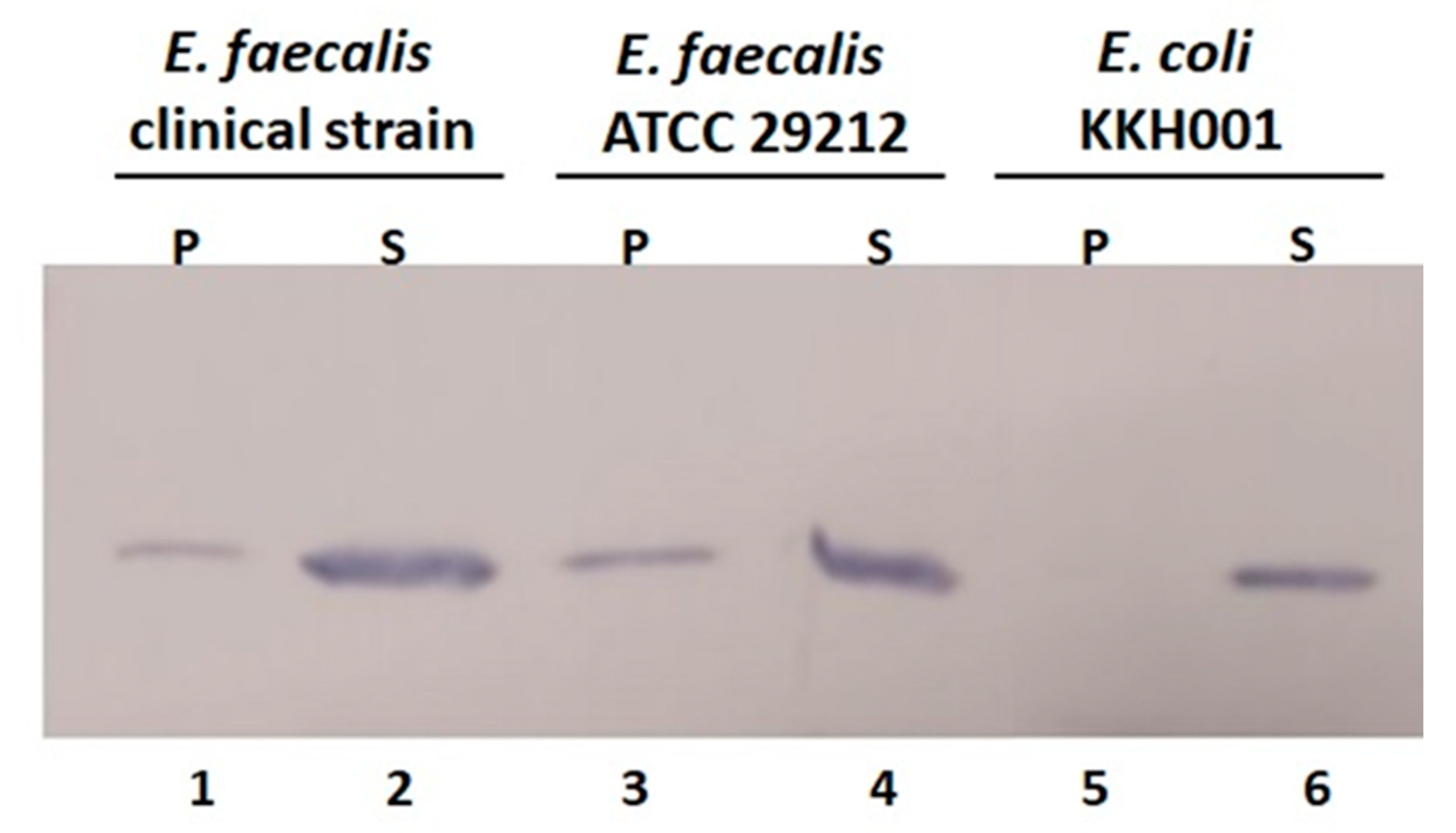
2.5. Cell Wall Binding Efficiency of BP404 towards the Tested E. faecalis Strains
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Software Used for Protein Structure Prediction
4.3. PCR Amplification of Genes
4.4. Gene Cloning and Expression
4.5. Purification of the Synthesized Protein
4.6. Activity Assessment of the Protein against Enterococcus faecalis Strains
4.7. Live/Dead Staining and Confocal Microscopy
4.8. Evaluation of Cell Binding Activity of the Protein
4.9. Software Used for Graph Preparation Statistical Analysis of the Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Tyne, D.; Gilmore, M.S. Friend Turned Foe: Evolution of Enterococcal Virulence and Antibiotic Resistance. Annu. Rev. Microbiol. 2014, 68, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, F.L.; Willems, R.J.; Leavis, H.L. Optimizing future treatment of enterococcal infections: Attacking the biofilm? Trends Microbiol. 2012, 20, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Rams, T.E.; Degener, J.E.; van Winkelhoff, A.J. Prevalence of beta-lactamase-producing bacteria in human periodontitis. J. Periodont. Res. 2013, 48, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Kouidhi, B.; Zmantar, T.; Mahdouani, K.; Hentati, H.; Bakhrouf, A. Antibiotic resistance and adhesion properties of oral Enterococci associated to dental caries. BMC Microbiol. 2011, 11, 155. [Google Scholar] [CrossRef]
- Arias, C.A.; Murray, B.E. Emergence and management of drug-resistant enterococcal infections. Expert Rev. Anti-Infect. Ther. 2008, 6, 637–655. [Google Scholar] [CrossRef]
- Hammerum, A.M.; Lester, C.H.; Neimann, J.; Porsbo, L.J.; Olsen, K.E.P.; Jensen, L.B.; Emborg, H.-D.; Wegener, H.C.; Frimodt-Moller, N. A vancomycin-resistant Enterococcus faecium isolate from a Danish healthy volunteer, detected 7 years after the ban of avoparcin, is possibly related to pig isolates. J. Antimicrob. Chemother. 2004, 53, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Arana, A.; Valle, J.; Solano, C.; Arrizubieta, M.J.; Cucarella, C.; Lamata, M.; Amorena, B.; Leiva, J.; Penadeés, J.R.; Lasa, I.; et al. The enterococcal surface protein, Esp, is involved in Enterococcus faecalis biofilm formation. Appl. Environ. Microbiol. 2001, 67, 4538–4545. [Google Scholar] [CrossRef]
- Kayaoglu, G.; Orstavik, D. Virulence factors of Enterococcus faecalis: Relationship to endodontic disease. Crit. Rev. Oral Biol. Med. 2004, 15, 308–320. [Google Scholar] [CrossRef]
- Sussmuth, S.D.; Muscholl-Silberhorn, A.; Wirth, R.; Susa, M.; Marre, R.; Rozdzinski, E. Aggregation substance promotes adherence, phagocytosis, and intracellular survival of Enterococcus faecalis within human macrophages and suppresses respiratory burst. Infect. Immun. 2000, 68, 4900–4906. [Google Scholar] [CrossRef] [PubMed]
- Chuang-Smith, O.N.; Wells, C.L.; Henry-Stanley, M.J.; Dunny, G.M. Acceleration of Enterococcus faecalis Biofilm Formation by Aggregation Substance Expression in an Ex Vivo Model of Cardiac Valve Colonization. PLoS ONE 2010, 5, e15798. [Google Scholar] [CrossRef]
- Ballering, K.S.; Kristich, C.J.; Grindle, S.M.; Oromendia, A.; Beattie, D.T.; Dunny, G.M. Functional Genomics of Enterococcus faecalis: Multiple Novel Genetic Determinants for Biofilm Formation in the Core Genome. J. Bacteriol. 2009, 191, 2806–2814. [Google Scholar] [CrossRef]
- Jack, R.W.; Tagg, J.R.; Ray, B. Bacteriocins of gram-positive bacteria. Microbiol. Rev. 1995, 59, 171–200. [Google Scholar] [CrossRef] [PubMed]
- Field, D.; Hill, C.; Cotter, P.D.; Ross, R.P. The dawning of a “Golden era” in lantibiotic bioengineering. Mol. Microbiol. 2010, 78, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Van Tyne, D.; Martin, M.J.; Gilmore, M.S. Structure, Function, and Biology of the Enterococcus faecalis Cytolysin. Toxins 2013, 5, 895–911. [Google Scholar] [CrossRef] [PubMed]
- Sawa, N.; Wilaipun, P.; Kinoshita, S.; Zendo, T.; Leelawatcharamas, V.; Nakayama, J.; Sonomoto, K. Isolation and Characterization of Enterocin W, a Novel Two-Peptide Lantibiotic Produced by Enterococcus faecalis NKR-4-1. Appl. Environ. Microbiol. 2012, 78, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Aymerich, T.; Holo, H.; Håvarstein, L.S.; Hugas, M.; Garriga, M.; Nes, I.F. Biochemical and genetic characterization of enterocin A from Enterococcus faecium, a new antilisterial bacteriocin in the pediocin family of bacteriocins. Appl. Environ. Microbiol. 1996, 62, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Bueno, M.; Maqueda, M.; Gálvez, A.; Samyn, B.; Van Beeumen, J.; Coyette, J.; Valdivia, E. Determination of the gene sequence and the molecular structure of the enterococcal peptide antibiotic AS-48. J. Bacteriol. 1994, 176, 6334–6339. [Google Scholar] [CrossRef] [PubMed]
- Kurushima, J.; Nakane, D.; Nishizaka, T.; Tomita, H. Bacteriocin Protein BacL1 of Enterococcus faecalis Targets Cell Division Loci and Specifically Recognizes l-Ala2-Cross-Bridged Peptidoglycan. J. Bacteriol. 2015, 197, 286–295. [Google Scholar] [CrossRef]
- Cotter, P.; Hill, C.; Ross, R. Bacteriocins: Developing innate immunity for food. Nat. Rev. Microbiol. 2005, 3, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.; Nes, I.F.; Holo, H. Enterolysin A, a cell wall-degrading bacteriocin from Enterococcus faecalis LMG 2333. Appl. Environ. Microbiol. 2003, 69, 2975–2984. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Kamei, E.; Ike, Y. Cloning and Genetic Analyses of the Bacteriocin 41 Determinant Encoded on the Enterococcus faecalis Pheromone-Responsive Conjugative Plasmid pYI14: A Novel Bacteriocin Complemented by Two Extracellular Components (Lysin and Activator). J. Bacteriol. 2008, 190, 2075–2085. [Google Scholar] [CrossRef]
- Kurushima, J.; Hayashi, I.; Sugai, M.; Tomita, H. Bacteriocin Protein BacL1 of Enterococcus faecalis Is a Peptidoglycan d-Isoglutamyl-l-lysine Endopeptidase. J. Biol. Chem. 2013, 288, 36915–36925. [Google Scholar] [CrossRef]
- Biziulevičius, G.A.; Biziulevičienė, G.; Kazlauskaitė, J. A list of enzyme preparations covered by the term enzybiotics should not be restricted to bacteriophage-encoded peptidoglycan hydrolases (lysins). J. Pharm. Pharmacol. 2008, 60, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Loessner, M.J. Bacteriophage Endolysins—Current state of research and applications. Curr. Opin. Microbiol. 2005, 8, 480–487. [Google Scholar] [CrossRef]
- Manoharadas, S.; Witte, A.; Bläsi, U. Antimicrobial activity of a chimeric enzybiotic towards Staphylococcus aureus. J. Biotechnol. 2009, 139, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [PubMed]
- Payne, C.M.; Resch, M.G.; Chen, L.; Crowley, M.F.; Himmel, M.E.; Taylor, L.E., II; Sandgren, M.; Ståhlberg, J.; Stals, I.; Tan, Z.; et al. Glycosylated linkers in multimodular lignocellulose-degrading enzymes dynamically bind to cellulose. Proc. Natl. Acad. Sci. USA 2013, 110, 14646–14651. [Google Scholar] [CrossRef]
- Manoharadas, S.; Altaf, M.; Alrefaei, A.F.; Ahmad, N.; Hussain, S.A.; Al-Rayes, B.F. An Engineered Multimodular Enzybiotic against Methicillin-Resistant Staphylococcus aureus. Life 2021, 11, 1384. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, A.W.; Collins, L.J.; Ackermann, H.W. A study of five bacteriophages of the Myoviridae family which replicate on different gram-positive bacteria. Arch. Virol. 1993, 133, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Yoong, P.; Schuch, R.; Nelson, D.; Fischetti, V.A. Identification of a Broadly Active Phage Lytic Enzyme with Lethal Activity against Antibiotic-Resistant Enterococcus faecalis and Enterococcus faecium. J. Bacteriol. 2004, 186, 4730–4739. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Montgomerie, S.; Cruz, J.A.; Shrivastava, S.; Arndt, D.; Berjanskii, M.; Wishart, D.S. PROTEUS2: A web server for comprehensive protein structure prediction and structure-based annotation. Nucleic Acids Res. 2008, 36, W202–W209. [Google Scholar] [CrossRef] [PubMed]
- Broendum, S.S.; Buckle, A.M.; McGowan, S. Catalytic diversity and cell wall binding repeats in the phage-encoded endolysins. Mol. Microbiol. 2018, 110, 879–896. [Google Scholar] [CrossRef] [PubMed]
- Kamitori, S.; Yoshida, H. Structure-Function Relationship of Bacterial SH3 Domains. In SH Domains: Structure, Mechanisms and Applications; Kurochkina, N., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 71–89. [Google Scholar]
- Yang, H.; Luo, D.; Etobayeva, I.; Li, X.; Gong, Y.; Wang, S.; Li, Q.; Xu, P.; Yin, W.; He, J.; et al. Linker Editing of Pneumococcal Lysin ClyJ Conveys Improved Bactericidal Activity. Antimicrob. Agents Chemother. 2020, 64, e01610-19. [Google Scholar] [CrossRef] [PubMed]
- Jai, H.S.B.M.; Dam, L.C.; Tay, L.S.; Koh, J.J.W.; Loo, H.L.; Kline, K.A.; Goh, B.C. Engineered Lysins with Customized Lytic Activities Against Enterococci and Staphylococci. Front. Microbiol. 2020, 11, 574739. [Google Scholar] [CrossRef]
- Catalao, M.J.; Gil, F.; Moniz-Pereira, J.; São-José, C.; Pimentel, M. Diversity in bacterial lysis systems: Bacteriophages show the way. FEMS Microbiol. Rev. 2013, 37, 554–571. [Google Scholar] [CrossRef]
- Rodríguez-Rubio, L.; Martinez, B.; Donovan, D.M.; García, P.; Rodriguez, A. Potential of the Virion-Associated Peptidoglycan Hydrolase HydH5 and Its Derivative Fusion Proteins in Milk Biopreservation. PLoS ONE 2013, 8, e54828. [Google Scholar] [CrossRef]
- Rodríguez-Rubio, L.; Gutiérrez, D.; Donovan, D.M.; Martínez, B.; Rodríguez, A.; García, P. Phage lytic proteins: Biotechnological applications beyond clinical antimicrobials. Crit. Rev. Biotechnol. 2016, 36, 542–552. [Google Scholar] [CrossRef]
- Shen, Y.; Mitchell, M.S.; Donovan, D.M.; Nelson, D.C. 15 Phage-based enzybiotics. Bacteriophages Health Dis. 2012, 24, 217. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed. Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Zhang, Y.; Li, X.; Liang, J.; Hu, L.; Gong, P.; Zhang, L.; Cai, R.; Zhang, H.; Ge, J.; et al. Endolysin LysEF-P10 shows potential as an alternative treatment strategy for multidrug-resistant Enterococcus faecalis infections. Sci Rep. 2017, 7, 10164. [Google Scholar] [CrossRef] [PubMed]
- Swift, S.M.; Rowley, D.T.; Young, C.; Franks, A.; Hyman, P.; Donovan, D.M. The endolysin from the Enterococcus faecalis bacteriophage VD13 and conditions stimulating its lytic activity. FEMS Microbiol Lett. 2016, 363, fnw216. [Google Scholar] [CrossRef] [PubMed]
- Navarre, W.W.; Schneewind, O. Surface Proteins of Gram-Positive Bacteria and Mechanisms of Their Targeting to the Cell Wall Envelope. Microbiol. Mol. Biol. Rev. 1999, 63, 174–229. [Google Scholar] [CrossRef] [PubMed]
- Schleifer, K.H.; Kandler, O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 1972, 36, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Hancock, L.E.; Murray, B.E.; Sillanpää, J. Enterococcal Cell Wall Components and Structures. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection [Internet]; Gilmore, M.S., Clewell, D.B., Ike, Y., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014. Available online: https://www.ncbi.nlm.nih.gov/books/NBK190431/ (accessed on 4 February 2023).
- Manoharadas, S.; Altaf, M.; Alrefaei, A.W.F.; Devasia, R.M.; Hadj, A.Y.M.B.; Abuhasil, M.S. Concerted dispersion of Staphylococcus aureus biofilm by bacteriophage and ‘green synthesized’ silver nanoparticles. RSC Adv. 2021, 11, 1420–1429. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Amplified | Primer Name | Primer Sequence | PCR Product Size (bp) |
|---|---|---|---|
| BacL1-Catalytic domain | BACFP | 5′AATTCGGATCCATGAATTACAG TCAAAAAGCAATCG3′ | 987 |
| BacL1-Catalytic domain | BACRP | 5′ATTCGGTACCATTCACTGAATCTCC TTTTGAACCAGA3′ | 987 |
| PlyV12- Cell wall binding domain | PLYFP | 5′GACCGGTACCGGATTTACGAAGGAA GAAGCTA3′ | 225 |
| PlyV12-Cell wall binding domain | PLYRP | 5′TTACCTGCAGTTACTTAAATGTAC CCCATGCTTCC3′ | 225 |
| Plasmid Name | Notes on the plasmid | Synthetized protein | |
| pQE30 | Expression vector. Induction of promoter with IPTG. N-terminal 6X His-tag for purification. | NA | |
| pQE-BAC | The catalytic domain encoding gene from BacL1 cloned as BamHI/KpnI into pQE30 vector. | NA | |
| pQE-BACPLY | The 225 bp cell wall binding gene from PlyV12 of phage ϕ1 was cloned as KpnI/PstI into pQE-BAC | 46 kDa | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manoharadas, S.; Altaf, M.; Ahmad, N.; Alrefaei, A.F.; Al-Rayes, B.F. Construction and Activity Testing of a Modular Fusion Peptide against Enterococcus faecalis. Antibiotics 2023, 12, 388. https://doi.org/10.3390/antibiotics12020388
Manoharadas S, Altaf M, Ahmad N, Alrefaei AF, Al-Rayes BF. Construction and Activity Testing of a Modular Fusion Peptide against Enterococcus faecalis. Antibiotics. 2023; 12(2):388. https://doi.org/10.3390/antibiotics12020388
Chicago/Turabian StyleManoharadas, Salim, Mohammad Altaf, Naushad Ahmad, Abdulwahed Fahad Alrefaei, and Basel F. Al-Rayes. 2023. "Construction and Activity Testing of a Modular Fusion Peptide against Enterococcus faecalis" Antibiotics 12, no. 2: 388. https://doi.org/10.3390/antibiotics12020388
APA StyleManoharadas, S., Altaf, M., Ahmad, N., Alrefaei, A. F., & Al-Rayes, B. F. (2023). Construction and Activity Testing of a Modular Fusion Peptide against Enterococcus faecalis. Antibiotics, 12(2), 388. https://doi.org/10.3390/antibiotics12020388