Abstract
Land-use conversion changes soil properties and their microbial communities, which, combined with the overuse of antibiotics in human and animal health, promotes the expansion of the soil resistome. In this context, we aimed to profile the resistome and the microbiota of soils under different land practices. We collected eight soil samples from different locations in the countryside of São Paulo (Brazil), assessed the community profiles based on 16S rRNA sequencing, and analyzed the soil metagenomes based on shotgun sequencing. We found differences in the communities’ structures and their dynamics that were correlated with land practices, such as the dominance of Staphylococcus and Bacillus genera in agriculture fields. Additionally, we surveyed the abundance and diversity of antibiotic resistance genes (ARGs) and virulence factors (VFs) across studied soils, observing a higher presence and homogeneity of the vanRO gene in livestock soils. Moreover, three β-lactamases were identified in orchard and urban square soils. Together, our findings reinforce the importance and urgency of AMR surveillance in the environment, especially in soils undergoing deep land-use transformations, providing an initial exploration under the One Health approach of environmental levels of resistance and profiling soil communities.
1. Introduction
Antimicrobial resistance (AMR), one of the most serious health risks of the 21st century, is a common competition mechanism used by environmental bacteria to ensure their survival in their natural environment. Although evidence of antimicrobial resistance in bacteria dates back to the pre-antibiotic era, studies suggest that human activity has a significant impact on the extension and diversity of the bacterial resistome [1,2,3].
Even though the soil microbiota naturally presents a large and robust diversity of ARGs in its intrinsic resistome, land-use transformation due to anthropic activities, such as the excessive use of antibiotics in livestock production [4], antibiotic-enriched manure application [5,6], and excessive use of xenobiotics in crops [7,8], along with increasing levels of deforestation for farming or urban purposes, can alter bacterial communities and disseminate ARGs throughout the environment [9]. In these highly modified sites, soil bacteria more frequently are in close proximity to commensal and pathogenic bacteria, which could lead to an increased horizontal ARG transfer rate among them, followed by a dominance of organisms with acquired resistance in comparison to intrinsically resistant bacteria [10].
The “One Health” surveillance approach takes into consideration the interrelated link among people, non-human animals, and the environment [2]. The substantial role of the latter in AMR spread can be noted in the soil’s capacity to serve as a resistance gene reservoir, facilitating the spread of ARGs found in mobile genetic elements (MGEs), such as plasmids, integrons, and transposons, among different bacterial species, speeding the development of multidrug-resistant (MDR) pathogens [11]. According to Ghosh et al. (2021) [12], MDR pathogens are thought to be responsible for up to 10 million cases of fatalities annually worldwide, with a mortality rate of 392,000 in Latin America.
Brazil, the largest country in Latin America, presents a population of 192 million people. Although the National Health Regulatory Agency (ANVISA) in Brazil has compiled data regarding healthcare-associated infections and levels of antimicrobial resistance in clinical settings over the last decades [13], the country is not equipped with a central microbiology reference laboratory, increasing the difficulty in conducting national data analysis regarding bacterial resistance [14]. The São Paulo state, located in the southeastern region in Brazil, inhabited by over 45 million people [15], is a critical region in terms of high levels of resistance among important pathogens, such as non-fermenting Gram-negative bacilli and Gram-positive cocci, such as Staphylococcus aureus [15].
Recent estimates suggest that Brazil was responsible for almost 8% of all antibiotic consumption for veterinary purposes globally in 2017 and has an increased consumption projection of 11.8% in 2030 [16]. This is mainly due to a shift toward intensified livestock production systems that regularly use antimicrobial agents, which can directly affect the number of ARGs disseminated through the environment and, consequently, might contribute to increased levels of AMR in clinical settings [16].
Previous studies have shown the seriousness and urgent need to tackle AMR in the countryside of São Paulo, Brazil, especially after COVID-19 pandemic [17]. During this period, high rates of antibiotic use in hospitalized patients and prolonged time in invasive therapy have caused an alarming increase in polymyxin B-resistant Klebsiella pneumoniae isolates in 2021 [18] and a nosocomial outbreak of extensively drug-resistant (EDR) K. pneumoniae in 2022 [19].
Despite the extreme importance of monitoring healthcare infections associated with antibiotic-resistant bacteria to combat AMR, there is still a lack of data regarding environmental levels of resistance in northeastern soils of the São Paulo state, where there has been historical land use of soils for urban construction, agricultural, and livestock practices, leading to a population density of approximately 1.75 million inhabitants [20,21,22]. Considering the urgent need to tackle AMR, not only in clinical settings, but also taking into account the One Health approach, we used 16S rDNA and shotgun sequencing to profile the bacterial communities and resistome of eight sites in the countryside of São Paulo, to provide the first, to our knowledge, environmental AMR surveillance of soil samples in this region.
2. Results
2.1. Bacterial Community Dynamics
To understand the differences in bacterial communities across soils from different locations and land uses, we collected eight soils from different sites across São Paulo’s northeast region (Table 1 and Figure S1), and we assessed the community profiles based on 16S rRNA sequencing through an Oxford Nanopore MinION device, which allows full-length sequencing of the 16S gene (Figure 1A). In all sampling sites, Bacillus was observed as one of the most ubiquitous bacteria in the communities, with relative abundances of 1.7% in the campus lawn soils and in the PPA, 2% in the urban square, 2.4% in the orchard, 4.4% in the hen house, 11.7% in the cattle sites, 17.7% in the pasture area, and 31.1% in the agriculture soils. Two other frequent genera identified were Vicinamibacter, with relative abundances ranging from 1.9% (orchard) to 6.2% (pasture) and Rhodoplanes, ranging from 1.9% (hen house) to 4.4% (PPA soil) (Table S2).

Table 1.
Main characteristics and soil sample locations.
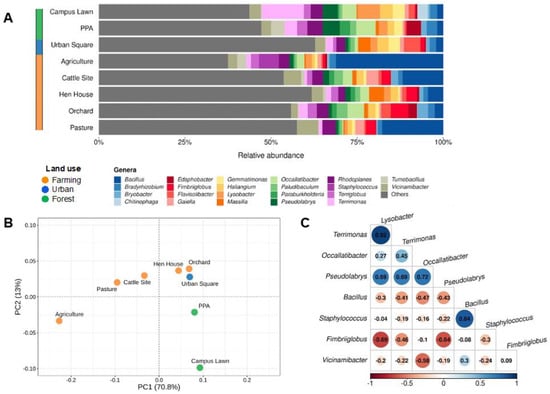
Figure 1.
Most abundant taxa and their distributions throughout the study sites at genera level. (A) Relative abundances of groups found in environmental samples according to 16S sequencing. All taxa found with relative abundances below 2.5% in each sample were labeled as “Others”. (B) Principal component analysis (PCA) of most abundant bacteria, clustered together per sampling site and colored according to land-use classification. (C) Correlogram indicating negative (in red) and positive (in blue) correlations among most abundant taxa (minimum relative abundance of 5%) in the studied sites.
Additionally, in order to comprehend bacterial diversity in each area, the Shannon index was calculated for each soil sample, indicating a smaller diversity in agricultural soils (3.781), followed by campus lawn (4.343) and PPA (4.504), and the highest diversity was found in the urban square (5.056) and hen house soils (5.087), in the analyzed conditions (Table S3). We next performed a principal component analysis (PCA) and a hierarchical clusterization (Figure 1B and Figure S2, respectively) with the resulting microbial profiles of each soil site sampled. In the former, the variance of sampling sites in two distinct groups was explained mostly by the first principal component (PC1 = 70.8%), which created a gradient of samples according to degree of land-use change. When this result was analyzed in combination with the PC2 (13%), three apparent clusters were formed, corroborating the land-use classification used. Additionally, statistical analyses indicated significant differences (F-value = 5.229, p-value < 0.001) in the relative abundance of genera across sampled sites.
To further understand the dynamics of bacterial communities, we performed a correlation analysis for taxa with a minimum relative abundance of 5% (Figure 1C). As shown in the figure, Terrimonas and Lysobacter displayed the strongest positive correlation (0.92), followed by Bacillus and Staphylococcus. On the other hand, Fimbriiglobus and Lysobacter displayed the strongest negative correlation (−0.69), followed by Fimbriiglobus and Pseudolabrys (−0.64) and Vicinamibacter with Occallatibacter (−0.58).
2.2. Abundance and Diversity of ARGs and VFs in Soils’ Metagenomes
Aiming to understand the abundance and diversity of antibiotic resistance genes (ARGs) and virulence factors (VFs) across studied samples, we proceeded to analyze the soil metagenomes based on shotgun sequencing through the Illumina platform (Figure 2). Using the CARD database, we detected ARGs in all sampled sites with a total of 254 ARGs identified, ranging from 17 (PPA) to 60 (cattle site) (Table S5). The identified ARGs conferred potential resistance to eight pharmacological classes of antibiotics, with glycopeptide (77.5%), rifamycin (12.2%), and macrolide/penam (5.9%) being the most frequent ARG types across all soils analyzed, followed by trimethoprim (1.1%), phenicol (0.8%), aminoglycoside (0.4%), and isoniazid/rifamycin (0.4%) (Figure 2A).
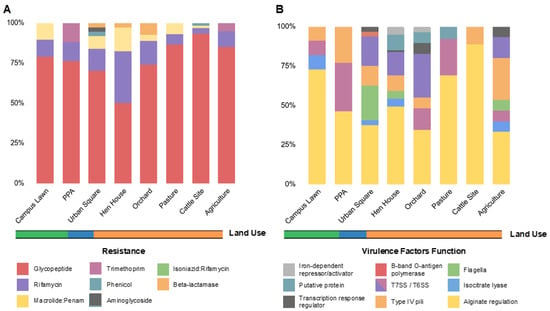
Figure 2.
Relative abundances of ARGs and VFs per soil metagenome. (A) Relative abundances of the identified ARGs per soil metagenome, colored by resistance category, as indicated in the legend. (B) Relative abundances of the identified VFs per soil metagenome, colored by function, as indicated in the legend. The ARGs were identified by CARD and VFs by VFDB, and the abundances were estimated by dividing the number of distinct resistance genes in the category (i.e., ARG potential resistance or VF function) by the total number of genes for all classes found in that site. Land use is indicated in orange (farming), blue (urban) and green (forest).
In total, 12 ARG types were identified in all soils, with vanRO and vanSO (glycopeptide), rbpA (rifamycin), and mtrA (macrolide/penam) being the most frequent genes, followed by dfrB3 and dfrB7 variants (trimethoprim), cpt (phenicol), aac2-lb (aminoglycoside), and efpA (isoniazid/rifamycin resistance). Additionally, three β-lactamase genes were identified, one being a serine-β-lactamase (SBL), identified as blaF, in urban square soils and two metallo-β-lactamase (MBL) encoding genes, identified as blaBJP-1 and blaLRA-9, in orchard soils (Figure 3A).
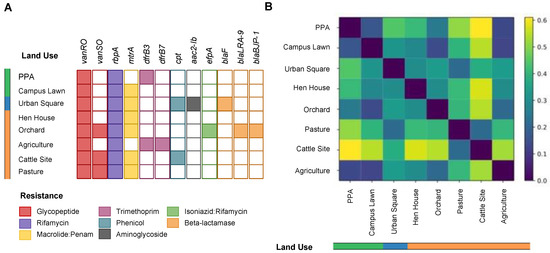
Figure 3.
Resistome profiles across studied soils. (A) Distribution of different ARGs per soil metagenome, colored by resistance category, as indicated in the legend. The absence of color indicates the absence of occurrence of the ARG in the soil. (B) Heatmap showing the (dis-)similarity between two soil resistome profiles using Bray–Curtis distance. The purple to yellow scale (0–0.6) indicates the degree of dissimilarity between the ARGs’ relative abundances in two soils, as indicated in the legend. Land use is indicated in orange (farming), blue (urban) and green (forest).
Our analyses indicated a higher dissimilarity between the resistome profiles of livestock soils (pasture and cattle sites) and forest soils (0.5), whereas other soils did not have such a pronounced dissimilarity in the relative abundances of ARGs (Figure 3B). Although dissimilarities were found, no statistical difference was observed in the resistome profiles across the studied sites (F-value = 1.06, p-value = 0.37) (Figure S3).
In order to visualize connections between the resistome (dis)-similarities of the selected soils, ARGs’ relative abundances across the studies sites were used to plot a chord diagram (Figure 4A). This indicates that even though vanRO, mtrA, and rbpA were widespread in all soils, a higher abundance of genes in cattle sites and pasture soils was observed. Notably, vanRO was the ARG with the most hits across soils, with 54 and 37 hits in cattle and pasture fields, respectively (Figure S4). Differently, a higher diversity of ARGs was identified in orchard and urban square soils, including in addition to the aforementioned β-lactamases genes, the two dfrB variants, cpt, aac2-lb, and efpA ARG types.
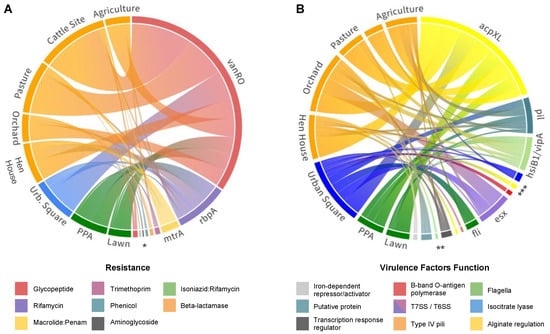
Figure 4.
Identified gene distributions across sequenced soil samples. (A) Chord diagram showing the distribution of different ARGs per soil metagenome. Resistance genes are colored by resistance category and soil sites are colored by land-use system as follows: orange—farming; blue—urban; green—forest. (B) Chord diagram showing the distribution of different VFs per soil metagenome. Virulence-related genes are colored by function, as indicated in the legend. (acpXL, alg, mucD) Acyl carrier proteins; (icl) isocitrate lyase; (hsiB1/vipA) type VI secretion system, T6SS; (flg, fli, cheW) Flagella; (pil) type IV pili; (esx) type VII secretion system, T7SS; (waaG) B-band O-antigen polymerase; (phoP) possible two-component system response transcriptional positive regulator; (mbtH) putative protein; (ideR) iron-dependent repressor and activator. * (vanSO, efpA, aac2-lb, cpt, bla, dfrB); ** (ideR, mbtH, phoP, mucD, waaG); *** (flgC, alg, icl). Resource: https://public.flourish.studio/visualisation/116771 & https://public.flourish.studio/visualisation/116970. Accessed on 22 November 2022.
From the VFDB database, 143 virulence genes were detected in all soil samples, divided into 25 different genes, assigned to 10 classes referring to their virulence functions (Figure 2B). Among them, the main virulence factor was an acyl carrier protein encoded by the acpXL gene (Figure 4B), which corresponded to 45.5% of all VFs identified throughout the soils. Types IV and VI secretion systems together composed 22.4% of the virulence genes found (Tables S6–S8), mostly involved in adaptation and manipulation of their environment and also in the aggravation of infectious conditions when present in pathogenic bacteria [23]. Nonetheless, genes associated with bacterial motility related to type IV pili, such as pilT, pilM, pilG, and pilH, and rotating flagella, such as flgC, fliE, fliQ, fliP, fliN, and fliA were also widespread in soils. When aligning our sequences against the PlasmidFinder database, no plasmid markers were identified in our metagenomic data.
3. Discussion
3.1. Bacterial Community Structure and Dynamics
The apparent clusterization of soils based on microbial composition in our analyses corroborated the land-use classification previously used. This could be observed, for instance, in the statistically significant difference between the agriculture field and PPA communities (adjusted p-value < 0.005) and the similarity between the PPA and campus lawn communities (adjusted p-value > 0.9). This result suggests that bacterial communities in soils are shaped and modified according to land use over the years, endorsing previous reports in the literature [24,25,26].
Vicinamibacter and Rhodoplanes genera, members of the Acidobacteriota and Pseudomonadota phyla, respectively, were ubiquitous and abundant in all soils, likely due to their essential roles in carbon, nitrogen, and sulfur cycling [27,28,29,30]. Forest soils presented smaller diversity indexes compared to urban and livestock soils (Table S3), which corroborates a previous hypothesis that higher taxonomic diversity is essential to stressed soils maintenance [31]. The main genera found in those soils were Lysobacter, Pseudolabris and Bradyrhizobium (phylum Pseudomonadota), Ocallatibacter (phylum Acidobacteriota), and Terrimonas (phylum Bacteroidota) [32,33,34], with a smaller abundance of Bacillus species compared to other sites (<2.5%), which goes in accordance to previous studies that profiled bacterial communities in preserved soils [9,10,35].
In the other hand, urban soils presented, along with hen house soils, the highest diversity indexes (Table S3), which could be explained by the accumulation of human activity wastes and by-products in the former, as well as the introduction of gastrointestinal microbiota members in the latter [36]. Both soils were abundant with Massilia (phylum Pseudomonadota), common environmental bacteria that have been shown to cause opportunistic infections in immunocompromised patients [37,38]. Farming soils presented a higher abundance of Bacillus, especially in livestock and agricultural soils (relative abundances of 12–31%), indicating a dominance of this group in farming land-use systems in São Paulo’s northeastern soils, corroborating previous reports [39,40]. This could be explained by a spore-forming characteristic of Bacillus, which facilitates their high resistance to most adverse environmental conditions on farming land-use systems, such as heat, desiccation, and high levels of UV radiation [33,41,42].
In agricultural soils, Staphylococcus and Bacillus represented the majority of sequenced members (Figure 1A), which could be connected to the 84% positive correlation observed (Figure 1C). In addition, the microbial diversity in agriculture fields was smaller compared to other soils, which could be attributed to soil microbial community homogenization due to the intensified land use of sugarcane crop soils [43,44], such as the one collected for this study. It is important to note that some environmental microorganisms, such as Staphylococcus and Bacillus identified in the studied soils, not only are commonly found in the environment due to their important interactions with other bacteria and functional maintenance in soils, but can also infect humans and other animals [45]. An imbalance caused by anthropic activities on soil microbial communities could favor Staphylococcus and Bacillus species due to their early proliferation characteristics [46], taking advantage of transient conditions to outgrow more fastidious microorganisms. Thus, the early proliferation of these bacteria, along with positive interactions between them, could explain their dominance in agricultural fields [47,48].
Members of the genus Bacillus are among the most abundant bacterial genera found in soils, with a widespread distribution through different ecological niches, which goes in accordance to our findings [49]. Although Bacillus species were sequenced in all studied soils, a higher prevalence of the genus was observed in farming soils, which corroborates previous reports [50,51]. In spite of the fact that the majority of Bacillus species are strictly environmental, common mechanisms used for environmental competition and cell survival can aid the infection process of vertebrate hosts in certain strains, allowing these bacteria to occupy an additional niche [52,53]. Bacillus anthracis, for instance, can persist for many decades in soils as endospores and, when inhaled by humans or grazer livestock animals, can result in the anthrax disease [54,55]. Nonetheless, human exposure to B. anthracis spores has been associated over the years with agricultural contact or along cattle trails, both in South and in North America [56,57].
On the other hand, Staphylococcus spp. Are common in the biosphere and possess the capacity to withstand extreme temperatures and pH variations, allowing them to occupy different niches, including soil, water, non-human animals, and humans [58,59]. Comparably, other studies that profiled soil microbiota have also shown a high abundance of this genus in the microbial community [60,61]. In this sense, Staphylococcus aureus is one of the most prevalent bacteria in the genus, being part of the microbiota as a commensal organism or the agent of several diseases, such as dermatitis and urinary, gastrointestinal, and respiratory tract infections [62,63]. Leung and collaborators have correlated a higher number of S. aureus in the environment with the severity and persistence of atopic dermatitis in the United States [64]. In that sense, the substantial abundance of Staphylococcus in agriculture fields (5.8%) could suggest a concerning scenario, due to the high circulation of people in the area and the broad range of niches the bacterium can occupy, reinforcing the importance of more studies with a One Health approach.
3.2. Resistance Genes and Virulence Factors Identification
Although no statistical difference was observed among the resistome profiles of soils with different land uses, dissimilarities were identified (Figure 3B). One of the most pronounced results was the notorious number of genes related to glycopeptide resistance (vanRO and vanSO) in livestock soils, totaling 91 ARG hits within these soils (Figure S4). Differently, forest soils presented a smaller number of hits for the same gene (28 hits), with a threefold difference compared to the total hits in livestock soils. It is also worth mentioning that vanRO was identified in all sampled soils and vanSO in 38% of them (Figure 3A). Both vanRO and vanSO, components of the same vanO gene cluster that can potentially confer glycopeptide resistance, were first identified in Rhodococcus equi soil isolates in Denmark [65]. Glycopeptide antibiotics, such as vancomycin, are a last-resort treatment option for methicillin-resistant S. aureus (MRSA) and vancomycin-resistant enterococci (VRE) infections [66,67]. In Brazil, several waves of resistance of S. aureus against antimicrobials have been reported, with increasing numbers of MRSA strains isolated in different hospitals in São Paulo, one of the most affected in recent decades [61,66,68]. Additionally, glycopeptides, such as avoparcin, have been historically used as growth promoters in the livestock industry, with worldwide reports of VRE in cattle, poultry, and swine samples [69,70]. As a result of this, glycopeptide-resistant genes are commonly reported as abundant in fecal samples [71,72], which could explain the higher number of vanRO hits in livestock and poultry soils in our study. Nonetheless, other environmental studies have reported the presence of these genes in permafrost samples from over 10,000 years ago and throughout the environment, suggesting an innate resistance reservoir in the soil microbiome [73,74]. Although vancomycin resistance genes are commonly found in soils worldwide [74], our findings highlight the importance of environmental surveillance, given that the Staphylococcus genus was one of the major bacteria present in agricultural soils, and this soil showed a high abundance of vanRO genes. This, added to the facilitated route of transmission of vanRO from livestock to humans, either through direct contact or by the food chain, reinforces the need for monitoring of these soils [70].
Macrolide antibiotics act by binding to the bacterial 50S ribosomal subunit, causing the cessation of bacterial protein synthesis, as a bacteriostatic agent [75]. The broad antibacterial activity of this antimicrobial has led to its widespread use in gastrointestinal and respiratory tracts, and in sexually transmitted infections [76]. In staphylococcal infections, there is an increasing cross-resistance to macrolides in MRSA strains, categorizing these bacteria as pathogens of great concern [77]. β-lactam antibiotics share the presence of a β-lactam ring in their structures, with a broad-spectrum activity due to the penicillin-binding protein (PBP) inactivation that hampers cell wall formation [77]. These are the most prescribed antibiotic classes in clinical settings worldwide, with annual expenses of approximately US $15 billion, representing 65% of the total antibiotic market [78,79]. In the last few years, the dissemination of Gram-negative bacteria resistant to β-lactams has been considered a public health threat, especially when considering the absence of new antibiotics with activity against these bacteria in the last 20 years [80]. The transcriptional activator of the mtrCDE multidrug efflux pump, mtrA (widespread through all sampled soils of this study), is responsible for expressing the operon that exports a wide variety of antimicrobial agents, including β-lactams and macrolides [81,82,83]. The aforementioned gene has been previously reported as abundant in soils, especially in those that have undergone land-use conversion [83,84,85].
Rifamycin resistance genes have been reported as abundant in both pristine and highly modified soils, which goes in accordance with our findings [86,87], although, to the best to our knowledge, no previous studies have reported such a robust presence of the rbpA gene in farming soils. Its genetic product is an RNA-binding protein that is responsible for conferring low resistance levels in the soil bacterium Streptomyces coelicolor [88]. In addition, Bortoluzzi and collaborators have pointed out that this gene could account for the transcriptional activity in Mycobacterium tuberculosis against rifamycin antibiotics [89]. In 2019, 73,000 new cases of tuberculosis (TB) and 4500 deaths due to this disease were reported, with several of them related to rifampicin-resistant strains [89,90]. Additionally, genomic characterization of the zoonotic and human-opportunistic pathogens R. equi and Mycolicibacterium peregrinum obtained from human, pig, and soil samples in Asia indicated the presence of the rbpA gene in all isolated genomes [91,92,93]. Nonetheless, the authors of the aforesaid study suggest that infections caused by these antibiotic-resistant bacteria might have an environmental source [93]. Moreover, in the farming soils of the present study (rbpA-enriched), the genera Rhodococcus, Mycolicibacterium, and Mycobacterium were identified as minor components of the soil microbiota, which represents a cause for concern. To the best of our knowledge, there have been no reports of the rbpA gene in Brazilian clinical settings, but the presence of the ARG in soils containing opportunistic pathogens or in close proximity to humans and livestock could pose a threat if the gene is transferred through the food chain or to pathogenic bacteria. Thus, studies under a One Health approach are of extreme importance when it comes to understanding possible environmental sources of ARGs and which opportunistic pathogens are present in the environment.
While fewer genes represent the majority of ARGs in livestock soils, the highest microbial and ARG diversity was identified in urban square and orchard soils, including β-lactamase-encoding genes. The products of these genes might confer resistance to most of the drugs included in the β-lactam class, which correspond to the vast majority of less toxic options used to treat bacterial infections [78,79]. These enzymes are capable of inactivating β-lactam antibiotics and can be classified either as SBLs, with an active site containing a catalytic serine residue [93], or as MBLs, which use zinc as a cofactor for catalyzation [94]. The two MBL-encoding genes (blaLRA-9 and blaBJP-1) identified in orchard soils are categorized in the B3 MBL subclass and were previously reported in environmental samples in China, Japan, and Alaska [95,96], conferring high levels of resistance when expressed in Escherichia coli clones [97,98].
Although no reports, to our knowledge, of the aforementioned MBLs have been performed in clinical settings, the occurrence of these genes in orchard soils could pose a threat to human health if they migrate to pathogens. For example, blaBJP-1 confers less sensibility to chelating agents compared to other MBLs and a high catalytic activity with meropenem—a watch group antibiotic [99]. Thus, the transfer of these ARGs could lead to a risk of selection of bacterial resistance that should be prioritized as targets of stewardship programs and monitoring [99]. Carbapenem antibiotics have a broad activity spectrum against the majority of pathogenic bacteria, thus their classification as a “last-resort” treatment option [100]. These antibiotics show strong performance against extended-spectrum β-lactamases, but may be more susceptible to MBLs [101,102]. Although intrinsic carbapenem resistance is presented by some bacterial species due to the production of endogenous MBLs, acquired resistance, caused by horizontal gene transfer, is more common in clinically important bacteria, which highlights the potential thread related to the presence of MBLs in the studied soils [103].
On the other hand, the SBL-coding gene identified in urban square soils, blaF, is a chromosomally encoded class A β-lactamase [104]. This ARG has been previously reported in China and Rio de Janeiro (Brazil), usually identified in nontuberculous mycobacteria, organisms commonly found in soils and water, also causing opportunistic infections in humans [105,106]. It has shown broad-spectrum activity against most β-lactam antibiotics, with the exception of third-generation cephalosporins [107,108]. Few studies have indicated the presence of this β-lactamase in soils, reinforcing the need for environmental surveillance in highly modified soils, aiming to provide further information regarding environmental β-lactamases and their associated risks to human health.
Considering the results related to virulence factor identification in the eight metagenomes, the acpXL gene was found in higher prevalence and it encoded an acyl carrier protein, required during the process of adding very long-chain fatty acid (VLCFA) to lipid A [109,110]. LPSs are known for their role in bacterial invasion, an essential function for the host infection process, and in bacterial adaptation in the environment, regardless of established mutualistic or pathogenic interactions [110]. The VLCFA attached to lipid A has been found in most members of the Rhizobiaceae family, as well as in the Bradyrhizobium genus, both found in the soils of our study. VLCFA presence could confer greater tolerance to stress and adaptation in distinct habitats [111,112] due to the stability conferred to the external membrane. In addition to its functions against stress, VLCFA can also be found in pathogenic or intracellular strains, such as the pathogen Brucella, for example, in which the linked lipid A ensures poor recognition by innate immunity [112].
Protein secretion systems, the second most common virulence class found in our study, are used for bacterial cells to interface with their environment through interaction and manipulation, where the secreted proteins can act as virulence factors, allowing these interactions [113,114]. The type VI secretion system (T6SS), with relative abundance of 11.2% in our samples (Figure 2B), is established by the VipA protein. This complex acts as a specialized bacterial nanomachinery that releases protein particles to other cells or to the environment, allowing bacteria to interact with their surrounding environment [115,116]. Thus, it consequently acts as an important determinant of the pathogenicity of eukaryotic cells, as well as in their competitive fitness in the community [116,117]. This indicates that secretion systems have a key role in shaping the microbiota of many ecological niches and explains the ubiquity of the T6SS-related genes across soils [117,118].
On the other hand, the second VF with the highest relative abundance found in our study was the secretion system type VII (T7SS). This system is a specialized protein secretion machinery that transports substrates through the cell envelope, widespread in Gram-positive members of the Actinomycetota and Bacillota phyla, abundant across all studied soils [119,120,121]. Finally, it is important to highlight that the overall abundance of the VF could be related to important housekeeping functions of T7SS, such as sporulation, conjugation, and cell wall stability, given that it is widespread among pathogenic and environmental microorganisms [122].
4. Materials and Methods
4.1. Study Area and Sample Collection
The samples used in this study were collected from different sites across São Paulo’s northeast region (Figure S1 and Table 1). Approximately 50 g of samples were aseptically taken from the upper 10 cm layer, after a 5 cm removal of litterfall, and placed in sterile Falcon tubes. For each selected site, 3 samples were collected and mixed for a better representation of the microbial community within. In total, eight samples were collected from: (i) a permanent preservation area (PPA) in the University of São Paulo campus—Ribeirão Preto, São Paulo; (ii) the University of São Paulo campus lawn—Ribeirão Preto, São Paulo; (iii) an agriculture field—Sertãozinho, São Paulo; (iv) a pasture field—São Carlos, São Paulo; (v) a livestock site—Taquaritinga, São Paulo; (vi) a hen house—São Carlos, São Paulo; (vii) an orchard field—São Carlos, São Paulo; and (viii) Urban square—Sertãozinho, São Paulo.
All soils were allocated in three categories, according to their land uses over the last decades, with those being (i) farming—sugarcane field, livestock site, hen house, pasture field, and orchard; (ii) urban—urban square; and (iii) forest—PPA and campus lawn.
4.2. DNA Extraction and Sequencing
The metagenomic DNA of each soil sample was extracted using DNAeasy Powersoil® Kit (QIAGEN), following manufacturer’s recommendations. The quantification and quality analysis of the extracted DNA was performed using Nanodrop™ One (Thermo Fisher Scientific) and by an agarose (1%) electrophoresis gel. Part of the extracted DNA was used for triplicate amplification of 16S rDNA, following Nanopore—16S Barcoding Kit 1-24 (SQK-16S024, Oxford Nanopore Technologies—ONT) recommendations, adding different barcodes for each replicate. All samples were individually purified, quantified by Nanodrop™ One, and mixed in proportioned amounts in order to make a representative pool of all soil samples. A single multiplex sequencing was performed using the aforementioned kit and Flongle flowcells in MinION model Mk1B. The remaining extracted DNA was submitted to metagenomic sequencing on an Illumina NovaSeq 6000 platform at Novogene (Sacramento, CA, USA), with a sequencing depth of 12 Gb/sample. Table S1 shows the data quality summary of raw data from shotgun sequencing.
4.3. Data Processing and Analysis
4.3.1. Amplicon Sequencing
Processing and analysis of the 16S rDNA reads were performed as recommended by de Siqueira and collaborators (2021) [123]. Briefly, reads were base-called using Guppy Base-calling Software (version 6.1.3) with the dna_r9.4.1_450bps_hac.cfg configuration file [124]. Base-called reads had their quality assessed by NanoStat (version 1.6) and NanoFilt (version 2.8) was used to select reads with quality scores above Q7 [125]. After the initial filtering step, demultiplexing of reads was performed by Porechop (version 2.4) using the barcodes from 16S Barcoding Kit 1-24 (SQK-16S024). Demultiplexed reads were mapped to a 16S rDNA NCBI reference database using minimap2 (version 2.17) [126].
4.3.2. Shotgun Sequencing
Shotgun sequencing raw data were processed with the fastp (version 23.1) tool (https://github.com/OpenGene/fastp, accessed on 17 November 2022) for adapter and low-quality reads removal [127]. High-quality reads assembly was carried out with the MegaHIT tool (https://github.com/voutcn/megahit, accessed on 17 November 2022) and the metagenome annotation was performed with Prokka (https://github.com/tseemann/prokka, accessed on 17 November 2022) [128], with the assembly statistics calculated with assembly_stats (https://github.com/sanger-pathogens/assembly-stats, accessed on 17 November 2022). The identification of ARGs, virulence factors (VFs), and plasmid markers was performed with the ABRICATE pipeline (https://github.com/tseemann/abricate, accessed on 17 November 2022), with an identity cut-off of 80%, by searching previously annotated genes in reference databases (ARG-ANNOT, CARD, PlasmidFinder, ResFinder and VFDB) [129,130,131,132,133].
4.4. Statistics and Graphical Representation
All statistical analyses were performed using R version 4.1.0. Differences between microbiota composition in the studied soils were measured using one-way ANOVA with Tukey post hoc test for determination of significance levels. A comparison was considered statistically significant at an adjusted p value < 0.05. Differences between resistome profiles in the studied soils were calculated using PERMANOVA. Vegan (version 2.5.7) [134] and Tidyverse (version 1.3.0) [135] packages were used for data manipulation and processing. Graphical representation was performed using ggplot2 [136] and Flourish Studio (https://flourish.studio, accessed on 22 November 2022).
5. Conclusions
The concept of One Health highlights that human health is interconnected to the health of other members on ecosystems, such as soils, animals, and plants. In that sense, microorganisms are crucial in One Health, given that they are the links among all these members, seen in the role of commensal bacteria in driving the organisms’ fitness, as well as maintaining key soil functions. Here, we showed that structure and composition of the microbial communities of soil samples correlate to its land use, along with concerning interactions among environmental and opportunistic pathogens in soils that have undergone land conversion. Our study has also shown several commonly found ARGs in soils that are also responsible for antibiotic resistance in potential pathogenic bacteria that are widespread in soils, with the highest abundances present in livestock soils, providing the first, to our knowledge, environmental AMR surveillance of soil samples in a crucial region of Brazil, in terms of population density and economic relevance. Although ARGs found in the soil samples in this study may confer resistance against competitors in these habitats, their gene products may also serve other functions in soils. Thus, we reinforce that ARG or VF hits within samples do not indicate actual antibiotic resistance or actual virulence determinants. Nevertheless, identifying β-lactamases in highly modified soils highlights the importance of environmental surveillance to pull the brakes and gather more information regarding resistance levels in regions at risk for higher selective pressure due to anthropic activities.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/antibiotics12020334/s1, Figure S1. Map of soil sample locations; Figure S2. Soil sample clusterization based on microbial composition; Figure S3. Permutation of the resistome profiles; Figure S4. Heatmap showing the presence of ARGs identified by CARD indicating the number of each resistance gene per soil; Table S1. Shotgun sequencing - Illumina NovaSeq 6000- data quality summary; Table S2. Relative abundances of most abundant genera (above 2.5%) identified in sequenced soils; Table S3. Alpha diversity indexes of sampled soils; Table S4. Data availability; Table S5. Summary table of the identified ARGs by CARD in sequenced metagenomes; Table S6. Summary table of the identified VFs by VFDB on forest land use system; Table S7. Summary table of the identified VFs by VFDB on urban land use system; Summary table of the identified VFs by VFDB on farming land use system.
Author Contributions
Contributed to the conception and design of the study—J.V.W.O., A.F.T.F. and M.-E.G. Sample collection and processing—J.V.W.O. and A.F.T.F. Performed the wet-lab procedures—J.V.W.O. and S.V. Bioinformatic data analysis (amplicon and shotgun, respectively)—G.T. and R.S.-R. Manuscript writing and paper design—J.V.W.O., G.M.d.S. and M.-E.G. Review and approval of the final version of the paper—all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the São Paulo State Foundation (FAPESP, award # 2021/01748-5). J.V.W.O. and A.F.T.F. were supported by FAPESP fellowships (award # 2020/02228-2 and 2019/18789-6). G.M.d.S. was beneficiary of a CAPES scholarship (grant #888887.666860/2022-00). M.E.G. was supported by CNPq Research Productivity Scholarship (award # 302750/2020-7).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Soil metagenomes were deposited in the SRA repository under the BioProject number PRJNA900430 (Table S4).
Acknowledgments
The authors thank their laboratory colleagues for their insightful comments and suggestions throughout this study. The authors also would like to thank the lab technician Thalita Riul Prado for her assistance in the course of this work and to Guilherme Marcelino Viana de Siqueira for his support on microbiota data analysis.
Conflicts of Interest
The authors declare that there is no conflict of interest that could be perceived as prejudicial to the impartiality of the reported research.
References
- Lawther, K.; Santos, F.G.; Oyama, L.B.; Rubino, F.; Morrison, S.; Creevey, C.J.; McGrath, J.W.; Huws, S.A. Resistome analysis of global livestock and soil microbiomes. Front. Microbiol. 2022, 13, 897905. [Google Scholar] [CrossRef]
- Aslam, B.; Khurshid, M.; Arshad, M.I.; Muzammil, S.; Rasool, M.; Yasmeen, N.; Shah, T.; Chaudhry, T.H.; Rasool, M.H.; Shahid, A.; et al. Antibiotic resistance: One health one world outlook. Frontiers in Cellular and Infection Microbiology. Front. Cell. Infect. Microbiol. 2021, 11, 1153. [Google Scholar] [CrossRef]
- Shafranskaya, D.; Chori, A.; Korobeynikov, A. Graph-based approaches significantly improve the recovery of antibiotic resistance genes from complex metagenomic datasets. Front. Microbiol. 2021, 12, 714836. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Huo, L.; Guo, Y.; Gao, M.; Wang, G.; Hu, D.; Li, C.; Wang, Z.; Liu, G.; Wang, X. Metagenomic assembly reveals hosts and mobility of common antibiotic resistome in animal manure and commercial compost. Environ. Microbiome 2022, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Giles, M.; Daniell, T.; Neilson, R.; Yang, X.R. Does reduced usage of antibiotics in livestock production mitigate the spread of antibiotic resistance in soil, earthworm guts, and the phyllosphere? Environ. Int. 2020, 136, 105359. [Google Scholar] [CrossRef] [PubMed]
- Hosain, M.Z.; Kabir, S.L.; Kamal, M.M. Antimicrobial uses for livestock production in developing countries. Vet. World 2021, 14, 210–221. [Google Scholar] [CrossRef]
- Sorinolu, A.J.; Tyagi, N.; Kumar, A.; Munir, M. Antibiotic resistance development and human health risks during wastewater reuse and biosolids application in agriculture. Chemosphere 2021, 265, 129032. [Google Scholar] [CrossRef]
- Wu, J.; Wang, J.; Li, Z.; Guo, S.; Li, K.; Xu, P.; Ok, Y.S.; Jones, D.L.; Zou, J. Antibiotics and antibiotic resistance genes in agricultural soils: A systematic analysis. Crit. Rev. Environ. Sci. Technol. 2022, 52, 1–18. [Google Scholar] [CrossRef]
- Lemos, L.N.; Pedrinho, A.; Vascondelos, A.T.R.; Tsai, S.M.; Mendes, L.W. Amazon Desforestation Enriches Antibiotic Resistance Genes. Soil Biol. Biochem. 2020, 153, 108110. [Google Scholar] [CrossRef]
- Santos, A.; Burgos, F.; Martinez-Urtaza, J.; Barrientos, L. Metagenomic Characterization of Resistance Genes in Deception Island and Their Association with Mobile Genetic Elements. Microorganisms 2022, 10, 1432. [Google Scholar] [CrossRef]
- Hobeika, W.; Gaschet, M.; Ploy, M.C.; Buelow, E.; Sarkis, D.K.; Dagot, C. Resistome Diversity and Dissemination of WHO Priority Antibiotic Resistant Pathogens in Lebanese Estuaries. Antibiotics 2022, 11, 306. [Google Scholar] [CrossRef]
- Ghosh, S.; Bornman, C.; Zafer, M.M. Antimicrobial Resistance Threats in the emerging COVID-19 pandemic: Where do we stand? J. Infect. Public Health 2021, 14, 555–560. [Google Scholar] [CrossRef]
- Pillonetto, M.; Jordão, R.T.S.; Andraus, G.S.; Bergamo, R.; Rocha, F.B.; Onishi, M.C.; Almeida, B.M.M.; Nogueira, K.S.; Dal Lin, A.; Dias, V.M.C.H.; et al. The Experience of Implementing a National Antimicrobial Resistance Surveillance System in Brazil. Front. Public Health 2021, 8, 575536. [Google Scholar] [CrossRef] [PubMed]
- Gledhill, J.; Schell, P.A. New Approaches to Resistance in Brazil and Mexico; Social Anthropology: Durham, NC, USA, 2012; p. 21. [Google Scholar] [CrossRef]
- Rossi, F. The Challenges of Antimicrobial Resistance in Brazil. Clin. Infect. Dis. 2011, 52, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Tiseo, K.; Huber, L.; Gilbert, M.; Robinson, T.P.; Van Boeckel, T.P. Global trends in antimicrobial use in food animals from 2017 to 2030. Antibiotics 2020, 9, 918. [Google Scholar] [CrossRef] [PubMed]
- Fuga, B.; Sellera, F.P.; Cerdeira, L.; Esposito, F.; Cardoso, B.; Fontana, H.; Moura, Q.; Cardenas-Arias, A.; Sano, E.; Ribas, R.M.; et al. WHO Critical Priority Escherichia coli as One Health Challenge for a PostPandemic Scenario: Genomic surveillance and analysis of current trends in brazil. Microbiol. Spectr. 2022, 10, e01256-21. [Google Scholar] [CrossRef]
- Gaspar, G.G.; Ferreira, L.R.; Feliciano, C.S.; Campos, C.P., Jr.; Molina, F.M.R.; Vendruscolo, A.C.S.; Bradan, G.M.A.; Lopes, N.A.P.; Martinez, R.; Bollela, V.R. Pre-and post-COVID-19 evaluation of antimicrobial susceptibility for healthcare-associated infections in the intensive care unit of a tertiary hospital. Revista Sociedade Brasileira Medicina Tropical 2021, 54, e00902021. [Google Scholar] [CrossRef]
- Gaspar, G.G.; Tamasco, G.; Abichabki, N.; Scaranello, A.F.T.; Auxiliadora-Martins, M.; Pocente, R.; Andrade, L.N.; Guazzaroni, M.E.; Silva-Rocha, R.; Bollela, V.R. Nosocomial Outbreak of Extensively Drug-Resistant (Polymyxin B and Carbapenem) Klebsiella pneumoniae in a Collapsed University Hospital Due to COVID-19 Pandemic. Antibiotics 2022, 11, 814. [Google Scholar] [CrossRef]
- Calaboni, A.; Tambosi, L.R.; Igari, A.T.; Farinaci, J.S.; Metzger, J.P.; Uriarte, M. The forest transition in São Paulo, Brazil. Ecol. Soc. 2018, 23, 4. [Google Scholar] [CrossRef]
- Caldarelli, C.E.; Gilio, L. Expansion of the sugarcane industry and its effects on land use in São Paulo: Analysis from 2000 through 2015. Land Use Policy 2018, 76, 264–274. [Google Scholar] [CrossRef]
- Carrasco, R.A.; Pinheiro, M.M.F.; Junior, J.M.; Cicerelli, R.E.; Silva, P.A.; Osco, L.P.; Ramos, A.P.M. Land use/land cover change dynamics and their effects on land surface temperature in the western region of the state of São Paulo, Brazil. Reg. Environ. Change 2020, 20, 1–12. [Google Scholar] [CrossRef]
- Aroney, S.T.N.; Poole, P.S.; Sánchez-Cañizares, C. Rhizobial Chemotaxis and Motility Systems at Work in the Soil. Front. Plant Sci. 2021, 12, 725338. [Google Scholar] [CrossRef]
- de Souza, L.C.; Procópio, L. The profile of the soil microbiota in the Cerrado is influenced by land use. Appl. Microbiol. Biotechnol. 2021, 105, 4791–4803. [Google Scholar] [CrossRef] [PubMed]
- Santorufo, L.; Memoli, V.; Panico, S.C.; Esposito, F.; Vitale, L.; Di Natale, G.; Trifuoggi, M.; Barile, R.; De Marco, A.; Maisto, G. Impact of Anthropic Activities on Soil Quality under Different Land Uses. Int. J. Environ. Res. Public Health 2021, 18, 8423. [Google Scholar] [CrossRef] [PubMed]
- Jangid, K.; Williams, M.A.; Franzluebbers, A.J.; Sanderlin, J.S.; Reeves, J.H.; Jenkins, M.B.; Endale, D.M.; Coleman, D.C.; Whitman, W.B. Relative impacts of land-use, management intensity and fertilization upon soil microbial community structure in agricultural systems. Soil Biol. Biochem. 2008, 40, 2843–2853. [Google Scholar] [CrossRef]
- Liu, X.; Fengfeng, D.; Shaozhou, C.; Naiwei, L.; Jian, C.; Yajun, C.; Linhe, S.; Jinfeng, L.; Dongrui, Y. Increased Diversity of Rhizosphere Bacterial Community Confers Adaptability to Coastal Environment for Sapium sebiferum Trees. Forests 2022, 13, 667. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, S.H.; Jo, H.Y.; Finneran, K.T.; Kwon, M.J. Diversity and composition of soil Acidobacteria and Proteobacteria communities as a bacterial indicator of past land-use change from forest to farmland. Sci. Total Environ. 2021, 797, 148944. [Google Scholar] [CrossRef] [PubMed]
- Flieder, M.; Buongiorno, J.; Herbold, C.W.; Bela Hausmann, B.; Rattei, T.; Lloyd, K.G.; Loy, A.; Wasmund, K. Novel taxa of Acidobacteriota implicated in seafloor sulfur cycling. ISME J. 2021, 15, 3159–3180. [Google Scholar] [CrossRef]
- Anil, K.; Srijana, M.; Rakshak, K. Microbial community dynamics from a fast-receding glacier of Western Himalayas highlight the importance of microbes in primary succession, nutrient recycling, and xenobiotics degradation. Ecol. Indic. 2022, 144, 109565. [Google Scholar] [CrossRef]
- Suleiman, A.K.A.; Manoeli, L.; Boldo, J.T.; Pereira, M.G.; Roesch, L.F.W. Shifts in soil bacterial community after eight years of land-use change. Syst. Appl. Microbiol. 2013, 36, 137–144. [Google Scholar] [CrossRef]
- Mendes, L.W.; Tsai, S.M.; Navarrete, A.A.; De Hollander, M.; van Veen, J.A.; Kuramae, E.E. Soil-borne microbiome: Linking diversity to function. Microb. Ecol. 2015, 70, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Banasiewicz, J.; Lisboa, B.B.; Costa, P.B.; Schlindwein, G.; Venter, S.N.; Steenkamp, E.T.; Vargas, L.K.; Passaglia, L.M.P.; Stepkowski, T. Culture-independent assessment of the diazotrophic Bradyrhizobium communities in the Pampa and Atlantic Forest Biomes localities in southern Brazil. Syst. Appl. Microbiol. 2021, 44, 126228. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.; Lee, H.; Kwon, S.; Yoo, Y.; Kim, D.; Han, S.; Lee, A.; Kim, C.; Kim, G.; Kim, J. Influence of Tree Vegetation on Soil Microbial Communities in Temperate Forests and Their Potential as a Proactive Indicator of Vegetation Shift Due to Climate Change. Sustainability 2020, 12, 10591. [Google Scholar] [CrossRef]
- Bindari, Y.R.; Moore, R.J.; Van, T.T.H.; Hilliar, M.; Wu, S.B.; Walkden-Brown, S.W.; Gerber, P.F. Microbial communities of poultry house dust, excreta and litter are partially representative of microbiota of chicken caecum and ileum. PLoS ONE 2021, 16, e0255633. [Google Scholar] [CrossRef] [PubMed]
- Ali, G.A.; Ibrahim, E.B.; Doiphode, S.H.; Goravey, W. Massilia timonae bacteremia: Na ususual pathogen of septic abortion. IDCases 2022, 29, e01592. [Google Scholar] [CrossRef]
- La Scola, B.; Birtles, R.J.; Mallet, M.N.; Raoult, D. Massilia timonae gen. nov., sp. nov., isolated from blood of an immunocompromised patient with cerebellar lesions. J. Clin. Microbiol. 1998, 36, 2847–2852. [Google Scholar] [CrossRef]
- Ali, N.; Lin, Y.; Jiang, L.; Ali, I.; Ahmed, I.; Akhtar, K.; He, B.; Wen, R. Biochar and Manure Applications Differentially Altered the Class 1 Integrons, Antimicrobial Resistance, and Gene Cassettes Diversity in Paddy Soils. Front. Microbiol. 2022, 13, 943880. [Google Scholar] [CrossRef]
- Rampelotto, P.H.; de Siqueira Ferreira, A.; Barboza, A.D.M.; Roesch, L.F.W. Changes in diversity, abundance, and structure of soil bacterial communities in Brazilian Savanna under different land use systems. Microb. Ecol. 2013, 66, 593–607. [Google Scholar] [CrossRef]
- Sabaté, D.C.; Petroselli, G.; Erra-Balsells, R.; Audisio, M.C.; Brandan, C.P. Beneficial effect of Bacillus sp. P12 on soil biological activities and pathogen control in common bean. Biol. Control. 2020, 141, 104131. [Google Scholar] [CrossRef]
- Li, H.; Cai, X.; Gong, J.; Xu, T.; Ding, G.C.; Li, J. Long-term organic farming manipulated rhizospheric microbiome and Bacillus antagonism against pepper blight (Phytophthora capsici). Front. Microbiol. 2019, 10, 342. [Google Scholar] [CrossRef]
- Gossner, M.M.; Lewinsohn, T.M.; Kahl, T.; Grassein, F.; Boch, S.; Prati, D.; Birkhofer, K.; Renner, S.C.; Sikorski, J.; Wubet, T.; et al. Land-use intensification causes multitrophic homogenization of grassland communities. Nature 2016, 540, 266–269. [Google Scholar] [CrossRef]
- Gámez-Virués, S.; Perović, D.J.; Gossner, M.M.; Börschig, C.; Blüthgen, N.; De Jong, H.; Simons, N.K.; Klein, A.M.; Krauss, J.; Maier, G.; et al. Landscape simplification filters species traits and drives biotic homogenization. Nat. Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef]
- Al Amin, M.; Hoque, M.N.; Siddiki, A.Z.; Saha, S.; Kamal, M.M. Antimicrobial resistance situation in animal health of Bangladesh. Vet. World 2020, 13, 2713–2727. [Google Scholar] [CrossRef]
- Schierstaedt, J.; Jechalke, S.; Nesme, J.; Neuhaus, K.; Sørensen, S.; Grosch, R.; Smalla, K.; Schikora, A. Salmonella persistence in soil depends on reciprocal interactions with indigenous microorganisms. Environ. Microbiol. 2020, 22, 2639–2652. [Google Scholar] [CrossRef]
- Qian, H.; Zhang, Q.; Lu TPeijnenburg, W.J.G.; Penuelas, J.; Zhu, Y.G. Lessons learned from COVID-19 on potentially pathogenic soil microorganisms. Soil Ecol. Lett. 2021, 3, 1–5. [Google Scholar] [CrossRef]
- Patil, S.M.; Suryavanshi, M.V.; VChandanshive, V.V.; Kurade, M.B.; Govindwara, S.P.; Byong-Hun, J. Regeneration of textile wastewater deteriorated microbial diversity of soil microcosm through bioaugmentation. Chem. Eng. J. 2020, 390, 122533. [Google Scholar] [CrossRef]
- Saxena, A.K.; Kumar, M.; Chakdar, H.; Anuroopa, N.; Bagyaraj, D.J. Bacillus species in soil as a natural resource for plant health and nutrition. J. Appl. Microbiol. 2020, 128, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Lüneberg, K.; Schneider, D.; Siebe, C.; Daniel, R. Drylands soil bacterial community is affected by land use change and different irrigation practices in the Mezquital Valley, Mexico. Sci. Rep. 2018, 8, 1413. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liao, H.L.; Boughton, E.H.; Martens-Habbena, W.; Qiu, J. Effects of land-use intensity, grazing and fire disturbances on soil bacterial and fungal communities in subtropical wetlands. Agric. Ecosyst. Environ. 2023, 345, 108314. [Google Scholar] [CrossRef]
- Messelhäußer, U.; Ehling-Schulz, M. Bacillus cereus—A Multifaceted Opportunistic Pathogen. Curr. Clin. Microbiol. Rep. 2018, 5, 120–125. [Google Scholar] [CrossRef]
- Stewart, G.C. Chapter 28—Bacillus. In Veterinary Microbiology, 4th ed.; Wiley: Hoboken, NJ, USA, 2022. [Google Scholar] [CrossRef]
- Van Ert, M.N.; Easterday, W.R.; Huynh, L.Y.; Okinaka, R.T.; Hugh-Jones, M.E.; Ravel, J.; Zanecki, S.R.; Pearson, T.; Simonson, T.S.; U’Ren, J.M.; et al. Global genetic population structure of Bacillus anthracis. PLoS ONE 2007, 2, e461. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.J.; Kracalik, I.T.; Ross, N.; Alexander, K.A.; Hugh-Jones, M.E.; Fegan, M.; Elkin, B.T.; Epp, T.; Shury, T.K.; Zhang, W.; et al. The global distribution of Bacillus anthracis and associated anthrax risk to humans, livestock and wildlife. Nat. Microbiol. 2019, 4, 1337–1343. [Google Scholar] [CrossRef]
- Blackburn, J.K.; McNyset, K.M.; Curtis, A.; Hugh-Jones, M.E. Modeling the geographic distribution of Bacillus anthracis, the causative agent of anthrax disease, for the contiguous United States using predictive ecologic niche modeling. Am. J. Trop. Med. Hyg. 2007, 77, 1103–1110. [Google Scholar] [CrossRef]
- Schild, A.L.; Sallis, E.S.V.; Soares, M.P.; Ladeira, S.R.; Schramm, R.; Priebe, A.P.; Almeida, M.B.; Riet-Correa, F. Anthrax in cattle in southern Brazil: 1978–2006. Pesquisa Veterinária Brasileira 2006, 26, 243–248. [Google Scholar] [CrossRef]
- Kosecka-Strojek, M.; Buda, A.; Miedzobrodzki, J. Chapter 2—Staphylococcal Ecology and Epidemiology. In Pet-To-Man Travelling Staphylococci; Academic Press: Cambridge, MA, USA, 2018; pp. 11–24. [Google Scholar] [CrossRef]
- Sonola, V.S.; Misinzo, G.; Matee, M.I. Occurrence of Multidrug-Resistant Staphylococcus aureus among Humans, Rodents, Chickens, and Household Soils in Karatu, Northern Tanzania. Int. J. Environ. Res. Public Health 2021, 18, 8496. [Google Scholar] [CrossRef]
- Al Johny, B.O. Characterization of Methicillin-resistant Staphylococcus aureus Isolated from Nearby Hospitals from two Different Countries. J. Pure Appl. Microbiol. 2019, 13, 1683–1689. [Google Scholar] [CrossRef]
- Carvalho, S.P.; Almeida, J.B.; Andrade, Y.M.F.S.; Silva, L.S.C.; Chamon, R.C.; Santos, K.R.N.; Marques, L.M. Molecular characteristics of methicillin-resistant Staphylococcus aureus isolates from hospital and community environments in northeastern Brazil. Braz. J. Infect. Dis. 2019, 23, 134–138. [Google Scholar] [CrossRef]
- Leung, A.D.; Schiltz, A.M.; Hall, C.F.; Liu, A.H. Severe atopic dermatitis is associated with a high burden of environmental Staphylococcus aureus. Clin. Exp. Allergy 2008, 38, 789–793. [Google Scholar] [CrossRef]
- Gudeta, D.D.; Moodley, A.; Bortolaia, V.; Guardabassi, L. vanO, a new glycopeptide resistance operon in environmental Rhodococcus equi isolates. Antimicrob. Agents Chemother. 2014, 58, 1768–1770. [Google Scholar] [CrossRef]
- James, R.C.; Pierce, J.G.; Okano, A.; Xie, J.; Boger, D.L. Redesign of glycopeptide antibiotics: Back to the future. ACS Chem. Biol. 2012, 7, 797. [Google Scholar] [CrossRef]
- Damasco, A.P.; Costa, T.M.D.; Morgado, P.G.M.; Guimarães, L.C.; Cavalcante, F.S.; Nouér, S.A.; Santos, K.R.N.D. Daptomycin and vancomycin non-susceptible methicillin-resistant Staphylococcus aureus clonal lineages from bloodstream infection in a Brazilian teaching hospital. Braz. J. Infect. Dis. 2019, 23, 139–142. [Google Scholar] [CrossRef]
- Sharma, R.; Hammerschlag, M.R. Treatment of methicillin-resistant Staphylococcus aureus (MRSA) infections in children: A reappraisal of vancomycin. Curr. Infect. Dis. Rep. 2019, 21, 1–8. [Google Scholar] [CrossRef]
- Andrade, M.M.; Luiz, W.B.; da Silva Oliveira Souza, R.; Amorim, J.H. The History of Methicillin-Resistant Staphylococcus aureus in Brazil. Can. J. Infect. Dis. Med. Microbiol. 2020, 7, 1721936. [Google Scholar] [CrossRef]
- van den Bogaard, A.E.; Jensen, L.B.; Stobberingh, E.E. Vancomycin-resistant enterococci in turkeys and farmers. N. Engl. J. Med. 1997, 337, 1558–1559. [Google Scholar] [CrossRef] [PubMed]
- Nesme, J.; Cécillon, S.; Delmont, T.O.; Monier, J.M.; Vogel, T.M.; Simonet, P. Large-Scale Metagenomic-Based Study of Antibiotic Resistance in the Environment. Curr. Biol. 2014, 24, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.M.; Keis, S.; Smith, J.M.B.; Cook, G.M. A clonal lineage of VanA-type Enterococcus faecalis predominates in vancomycin-resistant enterococci isolated in New Zealand. Antimicrob. Agents Chemother. 2003, 47, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Furlan, J.P.R.; Dos Santos, L.D.R.; Ramos, M.S.; Gallo, I.F.L.; Stehling, E.G. Fecal cultivable aerobic microbiota of dairy cows and calves acting as reservoir of clinically relevant antimicrobial resistance genes. Braz. J. Microbiol. 2020, 51, 1377–1382. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Kim, S.; Lee, Y.M.; Shin, S.C. Characterization of antimicrobial resistance genes and virulence factor genes in an Arctic permafrost region revealed by metagenomics. Environ. Pollut. 2022, 294, 118634. [Google Scholar] [CrossRef]
- Klare, I.; Heier, H.; Claus, H.; Böhme, G.; Marin, S.; Seltmann, G.; Hakenbeck, R.; Antanassova, V.; Witte, W. Enterococcus faecium strains with vanA-mediated high-level glycopeptide resistance isolated from animal foodstuffs and fecal samples of humans in the community. Microb. Drug Resist. 1995, 1, 265–272. [Google Scholar] [CrossRef]
- Schmithausen, R.M.; Schulze-Geisthoevel, S.V.; Stemmer, F.; El-Jade, M.; Reif, M.; Hack, S.; Meilaender, A.; Montabauer, G.; Fimmers, R.; Parcina, M.; et al. Analysis of transmission of MRSA and ESBL-E among pigs and farm personnel. PLoS ONE 2015, 10, e0138173. [Google Scholar] [CrossRef]
- Kricker, J.A.; Page, C.P.; Gardasson, F.R.; Baldursson, O.; Gudjonsson, T.; Parnaham, M.J. Nanontimicrobial Actions of Macrolides. Pharmacol. Rev. 2021, 73, 1404–1433. [Google Scholar] [CrossRef]
- Miklasińska-Majdanik, M. Mechanisms of Resistance to Macrolide Antibiotics among Staphylococcus aureus. Antibiotics 2021, 10, 1406. [Google Scholar] [CrossRef]
- Balsalobre, L.; Blanco, A.; Alarcón, T. Beta-lactams. Antibiot. Drug Resist. 2019, 3, 57–72. [Google Scholar] [CrossRef]
- Lima, L.M.; Silva, B.N.M.D.; Barbosa, G.; Barreiro, E.J. β-lactam antibiotics: An overview from a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 15, 112829. [Google Scholar] [CrossRef]
- Ewers, C.A.T.S.; Bethe, A.; Semmler, T.; Guenther, S.; Wieler, L.H. Extended-spectrum β-lactamase-producing and AmpC-producing Escherichia coli from livestock and companion animals, and their putative impact on public health: A global perspective. Clin. Microbiol. Infect. 2012, 18, 646–655. [Google Scholar] [CrossRef]
- MacNair, C.R.; Tsai, C.N.; Brown, E.D. Creative targeting of the Gram-negative outer membrane in antibiotic discovery. Ann. N. Y. Acad. Sci. 2020, 1459, 69–85. [Google Scholar] [CrossRef]
- Ohneck, E.A.; Zalucki, Y.M.; Johnson, P.J.; Dhulipala, V.; Golparian, D.; Unemo, M.; Jerse, A.E.; Shafer, W.M. A novel mechanism of high-level, broad-spectrum antibiotic resistance caused by a single base pair change in Neisseria gonorrhoeae. MBio 2011, 2, e00187-11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yan, B.; Mo, X.; Li, P.; Li, B.; Li, Q.; Li, N.; Mo, S.; Ou, Q.; Shen, P.; et al. Prevalence and proliferation of antibiotic resistance genes in the subtropical mangrove wetland ecosystem of South China Sea. MicrobiologyOpen 2019, 8, e871. [Google Scholar] [CrossRef] [PubMed]
- Salam, L.B.; Obayori, O.S.; Ilori, M.O.; Amund, O.O. Impact of spent engine oil contamination on the antibiotic resistome of a tropical agricultural soil. Ecotoxicology 2021, 30, 1251–1271. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, P.; Lu, T.; Wang, X.; Li, A.; Lu, Y.; Tao, M.; Pang, X. Impact of MtrA on phosphate metabolism genes and the response to altered phosphate conditions in Streptomyces. Environ. Microbiol. 2021, 23, 6907–6923. [Google Scholar] [CrossRef]
- Martín, J.F.; Liras, P. The balance metabolism safety net: Integration of stress signals by interacting transcriptional factors in Streptomyces and related Actinobacteria. Front. Microbiol. 2020, 10, 3120. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Lupoli, T.J. Modulation of a Mycobacterial ADP-Ribosyltransferase to Augment Rifamycin Antibiotic Resistance. ACS Infect. Dis. 2021, 7, 2604–2611. [Google Scholar] [CrossRef]
- Tupin, A.; Gualtieri, M.; Roquet-Banères, F.; Morichaud, Z.; Brodolin, K.; Leonetti, J.P. Resistance to rifampicin: At the crossroads between ecological, genomic and medical concerns. Int. J. Antimicrob. Agents 2010, 35, 519–523. [Google Scholar] [CrossRef]
- Newell, K.V.; Thomas, D.P.; Brekasis, D.; Paget, M.S. The RNA polymerase-binding protein RbpA confers basal levels of rifampicin resistance on Streptomyces coelicolor. Mol. Microbiol. 2006, 60, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, A.; Muskett, F.W.; Waters, L.C.; Addis, P.W.; Rieck, B.; Munder, T.; Schleier, S.; Forti, F.; Ghisotti, D.; Carr, M.D.; et al. Mycobacterium tuberculosis RNA polymerase-binding protein A (RbpA) and its interactions with sigma factors. J. Biol. Chem. 2013, 288, 14438–14450. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, M.L.; Silva, P.E.A.; Salvato, R.S.; Reis, A.J.; Schiefelbein, S.H.; Groll, A.V.; Barcellos, R.B.; Maschmann, R.; Esteves, L.S.; Spies, F.; et al. A highly rifampicin resistant Mycobacterium tuberculosis strain emerging in Southern Brazil. Tuberculosis 2020, 125, 102015. [Google Scholar] [CrossRef]
- Salvato, R.S.; Reis, A.J.; Schiefelbein, S.H.; Gómez, M.A.A.; Salvato, S.S.; Silva, L.V.; Costa, E.R.D.; Unis, G.; Dias, C.F.; Viveiros, M.; et al. Genomic-based surveillance reveals high ongoing transmission of multi-drug-resistant Mycobacterium tuberculosis in Southern Brazil. Int. J. Antimicrob. Agents 2021, 58, 106401. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Ohya, K.; Sawai, K.; Odoi, J.O.; Otsu, K.; Ota, A.; Ito, T.; Kawai, M.; Maruyama, F. Draft genome sequences of Mycolicibacterium peregrinum isolated from a pig with lymphadenitis and from soil on the same Japanese pig farm. BMC Res. Notes 2019, 12, 1–4. [Google Scholar] [CrossRef]
- Song, Y.; Xu, X.; Huang, Z.; Xiao, Y.; Yu, K.; Jiang, M.; Yin, S.; Zheng, M.; Meng, H.; Han, Y.; et al. Genomic Characteristics Revealed Plasmid-Mediated Pathogenicity and Ubiquitous Rifamycin Resistance of Rhodococcus equi. Front. Cell. Infect. Microbiol. 2022, 12, 807610. [Google Scholar] [CrossRef]
- van den Akker, F.; Bonomo, R.A. Exploring additional dimensions of complexity in inhibitor design for serine β-lactamases: Mechanistic and intra-and inter-molecular chemistry approaches. Front. Microbiol. 2018, 9, 622. [Google Scholar] [CrossRef]
- Salahuddin, P.; Kumar, A.; Khan, A.U. Structure, function of serine and metallo-β-lactamases and their inhibitors. Curr. Protein Pept. Sci. 2018, 19, 130–144. [Google Scholar] [CrossRef]
- Di Pisa, F.; Pozzi, C.; Benvenuti, M.; Docquier, J.D.; De Luca, F.; Mangani, S. Boric acid and acetate anion binding to subclass B3 metallo-β-lactamase BJP-1 provides clues for mechanism of action and inhibitor design. Inorg. Chim. Acta 2018, 470, 331–341. [Google Scholar] [CrossRef]
- Stoczko, M.; Frère, J.M.; Rossolini, G.M.; Docquier, J.D. Postgenomic scan of metallo-β-lactamase homologues in rhizobacteria: Identification and characterization of BJP-1, a subclass B3 ortholog from Bradyrhizobium japonicum. Antimicrob. Agents Chemother. 2006, 50, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.K.; Moe, L.A.; Rodbumrer, J.; Gaarder, A.; Handelsman, J. Functional metagenomics reveals diverse β-lactamases in a remote Alaskan soil. ISME J. 2009, 3, 243–251. [Google Scholar] [CrossRef]
- World Health Organization. 2019. Available online: https://apps.who.int/iris/handle/10665/327957 (accessed on 3 August 2022).
- Asaduzzamn, M.; Ullah, M.M.; Redwan, S.M.; Alam, M.J.; Juliana, F.M.; Hossain, N.; Das, B.; Asma, R.; Mandal, M.; Dutta, K.K. Emergence of Meropenem Resistance in Pathogens Recovered From Urine Cultures in Bangladesh. J. Pharm. Nd Biol. Sci. 2019, 13, 41–47. [Google Scholar] [CrossRef]
- Meletis, G. Carbapenem resistance: Overview of the problem and future perspectives. Ther. Adv. Infect. Dis. 2016, 3, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Aurilio, C.; Sansone, P.; Barbarisi, M.; Pota, V.; Giaccari, L.G.; Coppolino, F.; Barbarisi, A.; Passavanti, M.B.; Pace, M.C. Mechanisms of action of carbapenem resistance. Antibiotics 2022, 11, 421. [Google Scholar] [CrossRef] [PubMed]
- Bello-López, J.M.; Cabrero-Martínez, O.A.; Ibáñez-Cervantes, G.; Hernández-Cortez, C.; Pelcastre-Rodríguez, L.I.; Gonzalez-Avila, L.U.; Castro-Escarpulli, G. Horizontal gene transfer and its association with antibiotic resistance in the genus Aeromonas spp. Microorganisms 2019, 7, 363. [Google Scholar] [CrossRef] [PubMed]
- Timm, J.; Perilli, M.G.; Duez, C.; Trias, J.; Orefici, G.; Fattorini, L.; Amicosante, G.; Oratore, A.; Joris, B.; Frère, J.M.; et al. Transcription and expression analysis, using lacZ and phoA gene fusions, of Mycobacterium fortuitumβ-lactamase genes cloned from a natural isolate and a high-level β-lactamase producer. Mol. Microbiol. 1994, 12, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Liu, Z.; Xu, W.; Gao, Y.; Yang, S.; Grossart, H.P.; Li, M.; Luo, Z. Metagenomic exploration of antibiotic resistance genes and their hosts in aquaculture waters of the semi-closed Dongshan Bay (China). Sci. Total Environ. 2022, 838, 155784. [Google Scholar] [CrossRef]
- Morgado, S.; Ramos, N.D.V.; Freitas, F.; da Fonseca, É.L.; Vicente, A.C. Mycolicibacterium fortuitum genomic epidemiology, resistome and virulome. Memórias Instituto Oswaldo Cruz 2022, 116, e210247. [Google Scholar] [CrossRef]
- Pedro, H.D.S.P.; Nardi, S.M.T.; Belotti, N.C.U.; de Freitas, A.C.T.; de Souza, N.G.; Chimara, E. A laboratory-based analysis of rapidly growing mycobacteria in Northwest Paulista, Sao Paulo, Brazil. Int. J. Mycobacteriol. 2021, 10, 170. [Google Scholar] [CrossRef]
- Bansal, A.; Kar, D.; Pandey, S.D.; Matcha, A.; Kumar, N.G.; Nathan, S.; Ghosh, A.S. A Tyrosine Residue Along with a Glutamic Acid of the Omega-Like Loop Governs the Beta-Lactamase Activity of MSMEG_4455 in Mycobacterium smegmatis. Protein J. 2017, 36, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Bourassa, D.V.; Kannenberg, E.L.; Sherrier, D.J.; Buhr, R.F.; Carlon, R.W. The Lipopolysaccharide Lipid A Long-Chain Fatty Acid Is Important for Rhizobium leguminosarum Growth and Stress Adaptation in Free-Living and Nodule Environments. Mol. Plant-Microbe Interact. 2017, 30, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Caroff, M.; Novikov, A. LPS Structure, Function, and Heterogeneity. In Endotoxin Detection and Control in Pharma, Limulus, and Mammalian Systems; Williams, K., Ed.; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Tookmanian, E.M.; Belin, B.J.; Sáenz, J.P.; Newman, D.K. The role of hopanoids in fortifying rhizobia against a changing climate. Environ. Microbiol. 2021, 23, 2906–2918. [Google Scholar] [CrossRef] [PubMed]
- Roop, R.M.; Barton, I.S.; Hopersberger, D.; Martin, D.W. Uncovering the Hidden Credentials of Brucella Virulence. Microbiol. Mol. Biol. Rev. 2021, 85, e00021-19. [Google Scholar] [CrossRef]
- Smith, W.P.J.; Vettiger, A.; Winter, J.; Ryse, T.; Comstock, L.E.; Basier, M.; Foster, K.R. The evolution of the type VI secretion system as a disintegration weapon. PLoS Biol. 2020, 18, e3000720. [Google Scholar] [CrossRef]
- Allsopp, L.P.; Bernal, P.; Nolan, L.M.; Filloux, A. Causalities of war: The connection between type VI secretion system and microbiota. Cell. Microbiol. 2019, 22, 13153. [Google Scholar] [CrossRef]
- Cao, Z.; Casabona, M.G.; Kneuper, H.; Chalmers, J.D.; Palmer, T. The type VII secretion system of Staphylococcus aureus secretes a nuclease toxin that targets competitor bacteria. Nat. Microbiol. 2016, 2, 16183. [Google Scholar] [CrossRef]
- Gerc, A.J.; Diepold, A.; Trunk, K.; Porter, M.; Rickman, C.; Armitage, J.P.; Stanley-Wall, N.R.; Coulthurst, S.J. Visualization of the Serratia Type VI Secretion System Reveals Unprovoked Attacks and Dynamic Assembly. Cell Rep. 2015, 12, 2131–2142. [Google Scholar] [CrossRef]
- Gallegos-Monterrosa, R.; Coulthurst, S.J. The ecological impact of a bacterial weapon: Microbial interactions and the Type VI secretion system. FEMS Microbiol. Rev. 2021, 45, fuab033. [Google Scholar] [CrossRef]
- Sysoeva, T.A.; Zepeda-Rivera, M.A.; Huppert, L.A.; Burton, B.M. Dimer recognition and secretion by the ESX secretion system in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2014, 111, 7653–7658. [Google Scholar] [CrossRef]
- Lukaszczyk, M.; Pradhan, B.; Remaut, H. The Biosynthesis and Structures of Bacterial Pili. In Bacterial Cell Walls and Membranes; Kuhn, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 369–413. [Google Scholar] [CrossRef]
- Ellison, C.K.; Fei, C.; Dalia, T.N.; Wingreen, N.S.; Dalia, A.; Shaevitz, J.W.; Gitai, Z. Subcellular localization of type IV pili regulates bacterial multicellular development. Nat. Commun. 2022, 13, 6334. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.S.; Wong, G.C.L.; O’Toole, G.A. The Power of Touch: Type 4 Pili, the von Willebrand A Domain, and Surface Sensing by Pseudomonas aeruginosa. J. Bacteriol. 2022, 205, e00084-22. [Google Scholar] [CrossRef]
- Rumbaugh, K.P.; Sauer, K. Biofilm dispersion. Nat. Rev. Microbiol. 2020, 18, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Sauer, K.; Stoodley, P.; Goeres, D.M.; Hall-Stoodley, L.; Burmolle, M.; Stewart, P.S.; Bjarnsholt, T. The biofilm life cycle: Expanding the conceptual model of biofilm formation. Nat. Rev. Microbiol. 2022, 20, 608–620. [Google Scholar] [CrossRef]
- de Siqueira, G.M.; Marcelo, F.; Guazzaroni, M. Nanopore Sequencing Provides Rapid and Reliable Insight Into Microbial Profiles of Intensive Care Units. Front. Public Health 2020, 9, 710985. [Google Scholar] [CrossRef]
- Benton, M. Nanopore Guppy GPU Basecalling on Windows Using WSL2. 2021. Available online: https://hackmd.io/PrSp6UhqS2qxZ_rKOR18-g#Nanopore-Guppy-GPU-basecalling-on-Windows-using-WSL2 (accessed on 3 August 2022).
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Broeckhoven, C.V. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby, M.L.; Lund, O.; Villa, L.; Møller, F.A.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 7, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2013, 58, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2016, 45, D566–D573. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2015, 4, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Oksanei, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; Hara, R.B.O.; Simpson, G.L.; Solymos, P.; Steven, M.H.H.; Wagner, H.H. Vegan: Community Ecology Package. R Package Version 2. p.5–7. 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 3 August 2022).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).