
Natural and Man-Made Cyclic Peptide-Based Antibiotics


Abstract
1. Introduction
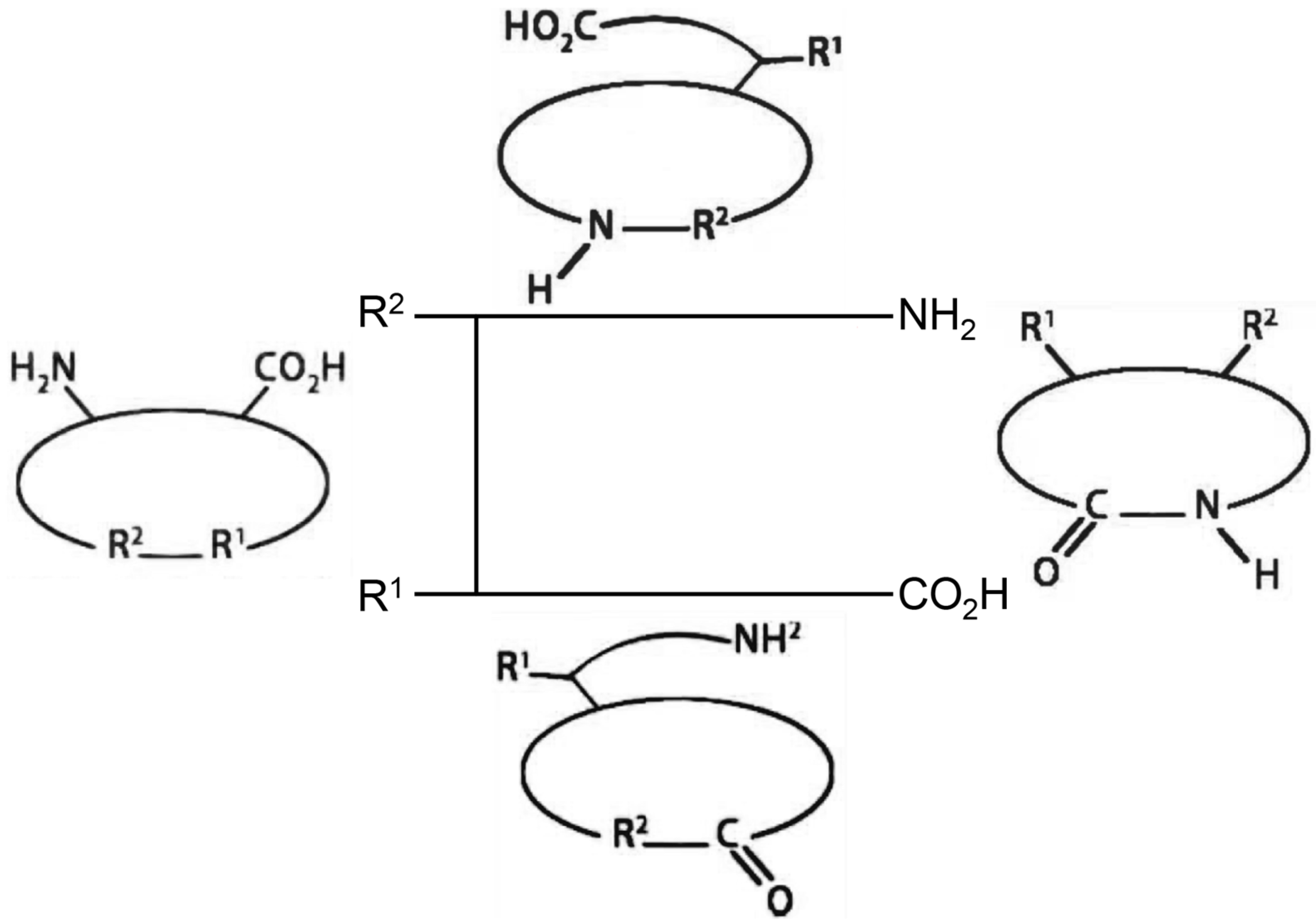
2. Advantages of Cyclic Peptides
3. Natural Cyclic Peptide-Based Antibiotics and Their Functions
4. Man-Made Cyclic Peptide-Based Antibiotics and Their Functions
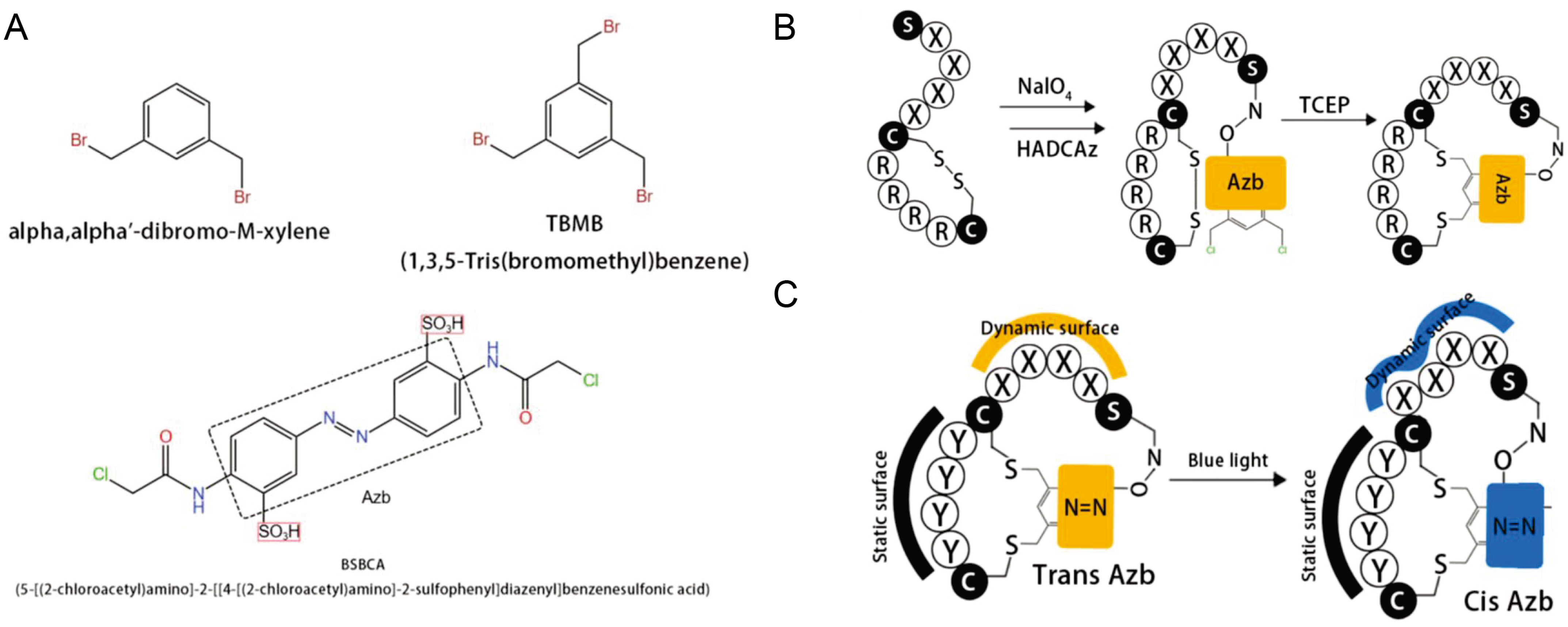
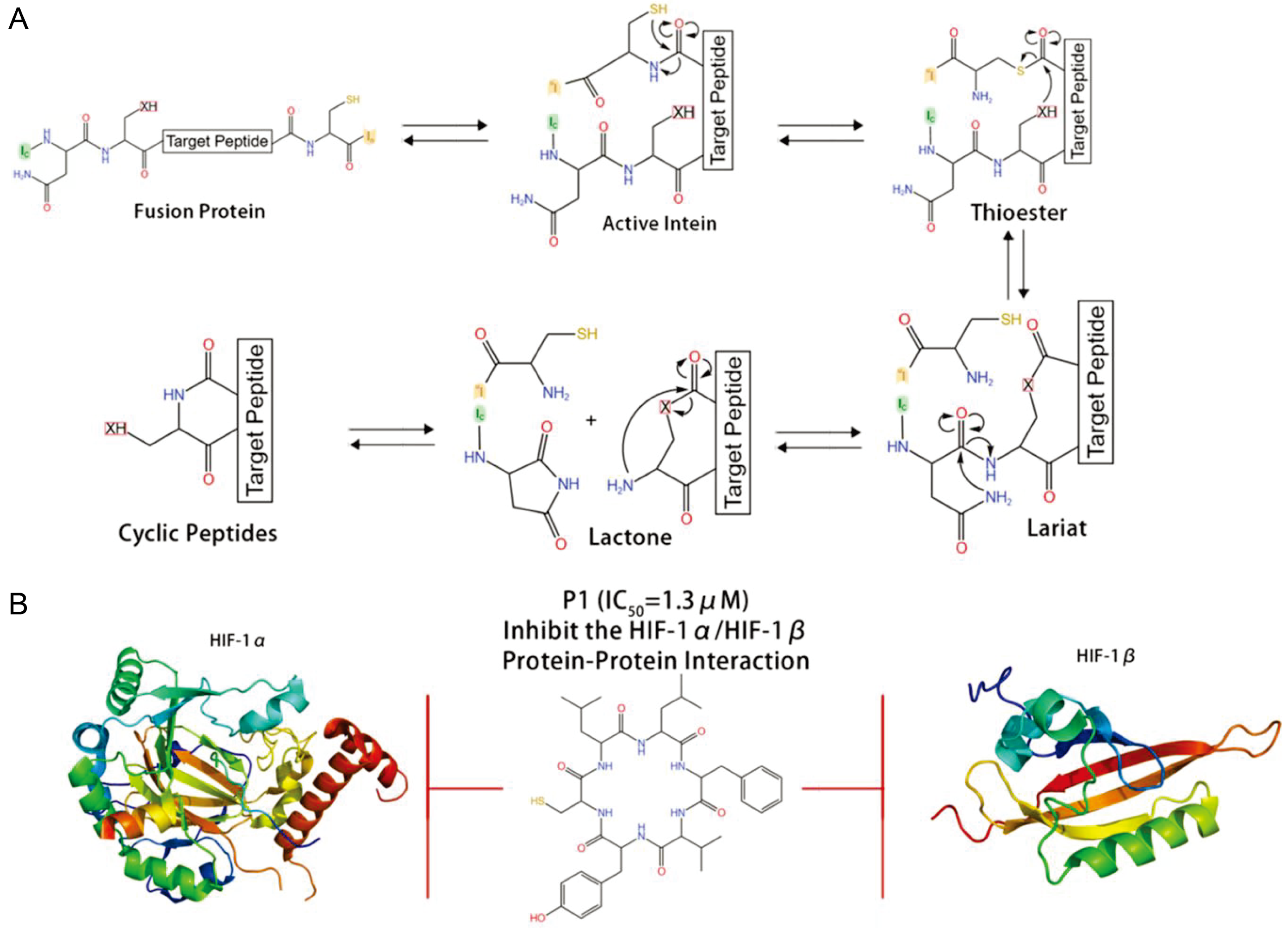
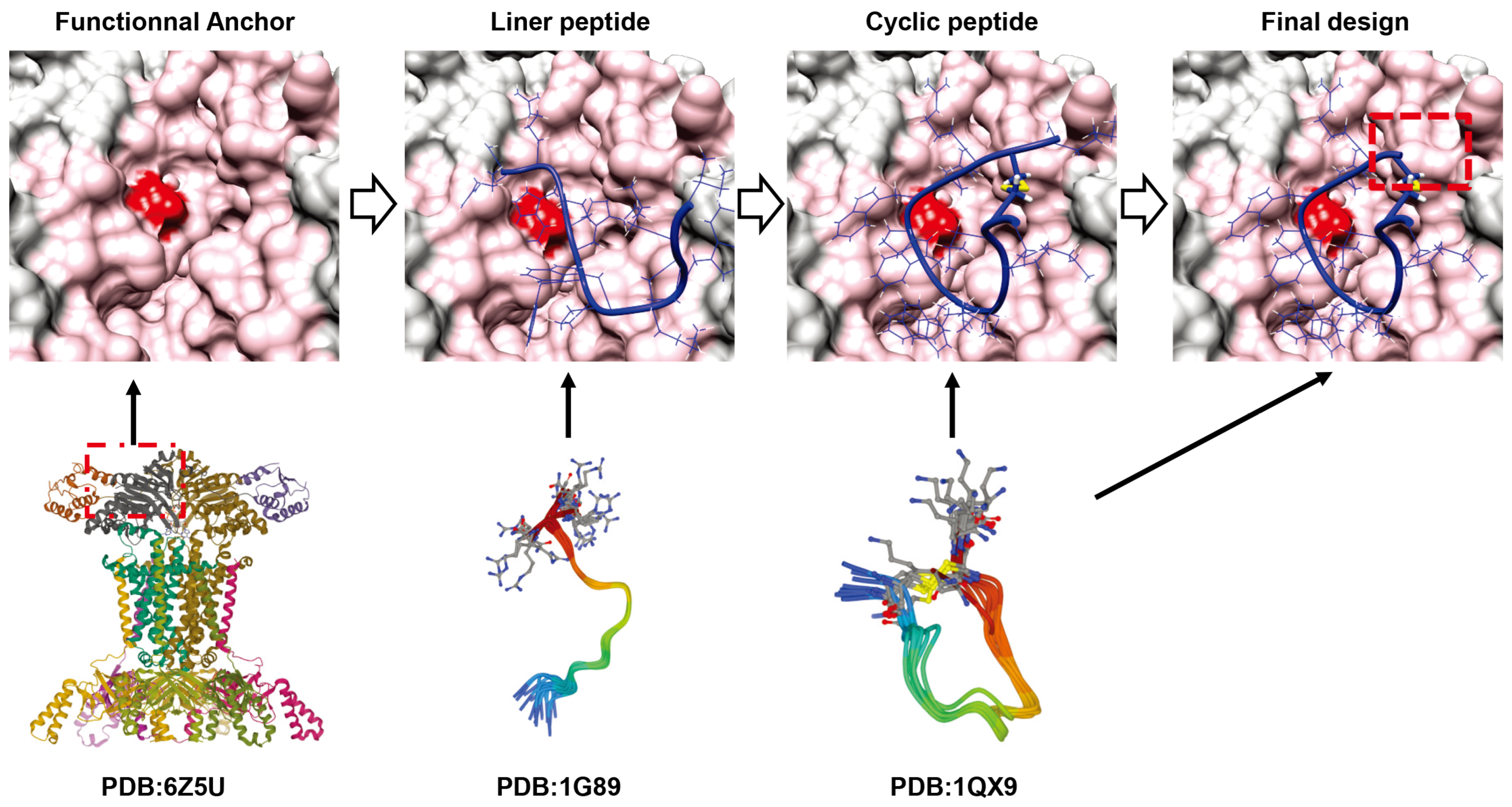
5. New Approaches for Cyclic Peptide Discovery
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mwangi, J.; Hao, X.; Lai, R.; Zhang, Z.Y. Antimicrobial peptides: New hope in the war against multidrug resistance. Zool. Res. 2019, 40, 488–505. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, M.; Lai, R.; Zhang, Z. Chemical modifications to increase the therapeutic potential of antimicrobial peptides. Peptides 2021, 146, 170666. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Improved methods for classification, prediction, and design of antimicrobial peptides. Methods Mol. Biol. 2015, 1268, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Wertz, P.W.; de Szalay, S. Innate Antimicrobial Defense of Skin and Oral Mucosa. Antibiotics 2020, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Amso, Z.; Hayouka, Z. Antimicrobial random peptide cocktails: A new approach to fight pathogenic bacteria. Chem. Commun. 2019, 55, 2007–2014. [Google Scholar] [CrossRef] [PubMed]
- Rathinakumar, R.; Walkenhorst, W.F.; Wimley, W.C. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening: The importance of interfacial activity. J. Am. Chem. Soc. 2009, 131, 7609–7617. [Google Scholar] [CrossRef]
- Abdalla, M.A.; McGaw, L.J. Natural Cyclic Peptides as an Attractive Modality for Therapeutics: A Mini Review. Molecules 2018, 23, 2080. [Google Scholar] [CrossRef]
- Ramadhani, D.; Maharani, R.; Gazzali, A.M.; Muchtaridi, M. Cyclic Peptides for the Treatment of Cancers: A Review. Molecules 2022, 27, 4428. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liao, X.; Zhong, S.; Zhao, B.; Xu, S. Synthesis of Marine Cyclopeptide Galaxamide Analogues as Potential Anticancer Agents. Mar. Drugs 2022, 20, 158. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.Y.; Chen, W.; Fan, J.T.; Song, R.; Wang, L.; Gu, Y.H.; Zeng, G.Z.; Shen, Y.; Wu, X.F.; Tan, N.H.; et al. Plant cyclopeptide RA-V kills human breast cancer cells by inducing mitochondria-mediated apoptosis through blocking PDK1-AKT interaction. Toxicol. Appl. Pharmacol. 2013, 267, 95–103. [Google Scholar] [CrossRef]
- Itokawa, H.; Takeya, K.; Mori, N.; Hamanaka, T.; Sonobe, T.; Mihara, K. Isolation and antitumor activity of cyclic hexapeptides isolated from Rubiae radix. Chem. Pharm. Bull. 1984, 32, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Bell, N.P.; Karp, C.L.; Alfonso, E.C.; Schiffman, J.; Miller, D. Effects of methylprednisolone and cyclosporine A on fungal growth in vitro. Cornea 1999, 18, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Cirigliano, A.M.; Rodriguez, M.A.; Gagliano, M.L.; Bertinetti, B.V.; Godeas, A.M.; Cabrera, G.M. Liquid chromatography coupled to different atmospheric pressure ionization sources-quadrupole-time-of-flight mass spectrometry and post-column addition of metal salt solutions as a powerful tool for the metabolic profiling of Fusarium oxysporum. J. Chromatogr. A 2016, 1439, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.A.; Al-Lihaibi, S.S.; Alarif, W.M.; Abdel-Lateff, A.; Nogata, Y.; Washio, K.; Morikawa, M.; Okino, T. Wewakazole B, a Cytotoxic Cyanobactin from the Cyanobacterium Moorea producens Collected in the Red Sea. J. Nat. Prod. 2016, 79, 1213–1218. [Google Scholar] [CrossRef]
- Pettit, G.R.; Hu, S.; Knight, J.C.; Chapuis, J.C. Antineoplastic agents. 571. Total synthesis of bacillistatin 2. J. Nat. Prod. 2009, 72, 372–379. [Google Scholar] [CrossRef]
- Steenbergen, J.N.; Alder, J.; Thorne, G.M.; Tally, F.P. Daptomycin: A lipopeptide antibiotic for the treatment of serious Gram-positive infections. J. Antimicrob. Chemother. 2005, 55, 283–288. [Google Scholar] [CrossRef]
- Tirilomis, T. Daptomycin and Its Immunomodulatory Effect: Consequences for Antibiotic Treatment of Methicillin-Resistant Staphylococcus aureus Wound Infections after Heart Surgery. Front. Immunol. 2014, 5, 97. [Google Scholar] [CrossRef]
- Molinelli, E.; Sapigni, C.; D’Agostino, G.M.; Brisigotti, V.; Rizzetto, G.; Bobyr, I.; Cirioni, O.; Giacometti, A.; Brescini, L.; Mazzanti, S.; et al. The Effect of Dalbavancin in Moderate to Severe Hidradenitis Suppurativa. Antibiotics 2022, 11, 1573. [Google Scholar] [CrossRef]
- Evans, B.J.; King, A.T.; Katsifis, A.; Matesic, L.; Jamie, J.F. Methods to Enhance the Metabolic Stability of Peptide-Based PET Radiopharmaceuticals. Molecules 2020, 25, 2314. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Yin, Y.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef]
- Gang, D.; Kim, D.W.; Park, H.S. Cyclic Peptides: Promising Scaffolds for Biopharmaceuticals. Genes 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed]
- Shinbara, K.; Liu, W.; van Neer, R.H.P.; Katoh, T.; Suga, H. Methodologies for Backbone Macrocyclic Peptide Synthesis Compatible with Screening Technologies. Front. Chem. 2020, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xie, J.; Wu, W.; Wang, M.; Chen, W.; Idres, S.B.; Rong, J.; Deng, L.W.; Khan, S.A.; Wu, J. Automated synthesis of prexasertib and derivatives enabled by continuous-flow solid-phase synthesis. Nat. Chem. 2021, 13, 451–457. [Google Scholar] [CrossRef]
- Jing, X.; Jin, K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev. 2020, 40, 753–810. [Google Scholar] [CrossRef]
- Knox, J.R.; Pratt, R.F. Different modes of vancomycin and D-alanyl-D-alanine peptidase binding to cell wall peptide and a possible role for the vancomycin resistance protein. Antimicrob. Agents Chemother. 1990, 34, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Nigro, E.; De Biasi, M.G.; Daniele, A.; Morelli, G.; Galdiero, S.; Scudiero, O. Cyclic Peptides as Novel Therapeutic Microbicides: Engineering of Human Defensin Mimetics. Molecules 2017, 22, 1217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef]
- Montali, A.; Berini, F.; Brivio, M.F.; Mastore, M.; Saviane, A.; Cappellozza, S.; Marinelli, F.; Tettamanti, G. A Silkworm Infection Model for In Vivo Study of Glycopeptide Antibiotics. Antibiotics 2020, 9, 300. [Google Scholar] [CrossRef]
- Shao, L.; Li, J.; Liu, A.; Chang, Q.; Lin, H.; Chen, D. Efficient bioconversion of echinocandin B to its nucleus by overexpression of deacylase genes in different host strains. Appl. Environ. Microbiol. 2013, 79, 1126–1133. [Google Scholar] [CrossRef]
- Davis, S.L.; Vazquez, J.A. Anidulafungin: An evidence-based review of its use in invasive fungal infections. Core Evid. 2008, 2, 241–249. [Google Scholar]
- Alarcon-Manoja, E.; Cardozo-Espinola, C.; Puerta-Alcalde, P.; Garcia-Vidal, C. Comments on practice guidelines for the diagnosis and management of aspergillosis made by the IDSA in 2016. Rev. Esp. Quimioter. 2017, 30 (Suppl. 1), 26–29. [Google Scholar] [PubMed]
- Kofla, G.; Ruhnke, M. Pharmacology and metabolism of anidulafungin, caspofungin and micafungin in the treatment of invasive candidosis: Review of the literature. Eur. J. Med. Res. 2011, 16, 159–166. [Google Scholar] [CrossRef]
- Hashimoto, S. Micafungin: A sulfated echinocandin. J. Antibiot. 2009, 62, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Wasmann, R.E.; Muilwijk, E.W.; Burger, D.M.; Verweij, P.E.; Knibbe, C.A.; Bruggemann, R.J. Clinical Pharmacokinetics and Pharmacodynamics of Micafungin. Clin. Pharmacokinet. 2018, 57, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Balkovec, J.M.; Hughes, D.L.; Masurekar, P.S.; Sable, C.A.; Schwartz, R.E.; Singh, S.B. Discovery and development of first in class antifungal caspofungin (CANCIDAS(R))—A case study. Nat. Prod. Rep. 2014, 31, 15–34. [Google Scholar] [CrossRef]
- Otsuka, Y. Potent Antibiotics Active against Multidrug-Resistant Gram-Negative Bacteria. Chem. Pharm. Bull. 2020, 68, 182–190. [Google Scholar] [CrossRef]
- Martin-Loeches, I.; Dale, G.E.; Torres, A. Murepavadin: A new antibiotic class in the pipeline. Expert Rev. Anti-Infect. Ther. 2018, 16, 259–268. [Google Scholar] [CrossRef]
- Amponnawarat, A.; Chompunud Na Ayudhya, C.; Ali, H. Murepavadin, a Small Molecule Host Defense Peptide Mimetic, Activates Mast Cells via MRGPRX2 and MrgprB2. Front. Immunol. 2021, 12, 689410. [Google Scholar] [CrossRef]
- Pernas, S.; Martin, M.; Kaufman, P.A.; Gil-Martin, M.; Gomez Pardo, P.; Lopez-Tarruella, S.; Manso, L.; Ciruelos, E.; Perez-Fidalgo, J.A.; Hernando, C.; et al. Balixafortide plus eribulin in HER2-negative metastatic breast cancer: A phase 1, single-arm, dose-escalation trial. Lancet Oncol. 2018, 19, 812–824. [Google Scholar] [CrossRef]
- Wong, R.S.M. Safety and efficacy of pegcetacoplan in paroxysmal nocturnal hemoglobinuria. Ther. Adv. Hematol. 2022, 13, 20406207221114673. [Google Scholar] [CrossRef]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-type TP53. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5236–5247. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2019. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2020, 25, 745. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Craven, T.W.; Levine, P.M. Cyclic Peptide Screening Methods for Preclinical Drug Discovery. J. Med. Chem. 2022, 65, 11913–11926. [Google Scholar] [CrossRef]
- Jafari, M.R.; Yu, H.; Wickware, J.M.; Lin, Y.S.; Derda, R. Light-responsive bicyclic peptides. Org. Biomol. Chem. 2018, 16, 7588–7594. [Google Scholar] [CrossRef] [PubMed]
- Delivoria, D.C.; Skretas, G. The Discovery of Peptide Macrocycle Rescuers of Pathogenic Protein Misfolding and Aggregation by Integrating SICLOPPS Technology and Ultrahigh-Throughput Screening in Bacteria. Methods Mol. Biol. 2022, 2371, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Miranda, E.; Nordgren, I.K.; Male, A.L.; Lawrence, C.E.; Hoakwie, F.; Cuda, F.; Court, W.; Fox, K.R.; Townsend, P.A.; Packham, G.K.; et al. A cyclic peptide inhibitor of HIF-1 heterodimerization that inhibits hypoxia signaling in cancer cells. J. Am. Chem. Soc. 2013, 135, 10418–10425. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.W.; Szostak, J.W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12297–12302. [Google Scholar] [CrossRef]
- Kamalinia, G.; Grindel, B.J.; Takahashi, T.T.; Millward, S.W.; Roberts, R.W. Directing evolution of novel ligands by mRNA display. Chem. Soc. Rev. 2021, 50, 9055–9103. [Google Scholar] [CrossRef]
- Yamagishi, Y.; Shoji, I.; Miyagawa, S.; Kawakami, T.; Katoh, T.; Goto, Y.; Suga, H. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 2011, 18, 1562–1570. [Google Scholar] [CrossRef]
- Johansen-Leete, J.; Ullrich, S.; Fry, S.E.; Frkic, R.; Bedding, M.J.; Aggarwal, A.; Ashhurst, A.S.; Ekanayake, K.B.; Mahawaththa, M.C.; Sasi, V.M.; et al. Antiviral cyclic peptides targeting the main protease of SARS-CoV-2. Chem. Sci. 2022, 13, 3826–3836. [Google Scholar] [CrossRef]
- Tsiamantas, C.; Otero-Ramirez, M.E.; Suga, H. Discovery of Functional Macrocyclic Peptides by Means of the RaPID System. Methods Mol. Biol. 2019, 2001, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, Y.; Seroski, D.T.; Wong, K.M.; Liu, R.; Paravastu, A.K.; Hudalla, G.A.; Hall, C.K. De novo design of peptides that coassemble into β sheet-based nanofibrils. Sci. Adv. 2021, 7, eabf7668. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, V.K. The emerging role of computational design in peptide macrocycle drug discovery. Expert Opin. Drug Discov. 2020, 15, 833–852. [Google Scholar] [CrossRef]
- Hosseinzadeh, P.; Watson, P.R.; Craven, T.W.; Li, X.; Rettie, S.; Pardo-Avila, F.; Bera, A.K.; Mulligan, V.K.; Lu, P.; Ford, A.S.; et al. Anchor extension: A structure-guided approach to design cyclic peptides targeting enzyme active sites. Nat. Commun. 2021, 12, 3384. [Google Scholar] [CrossRef]
- Mann, D.; Fan, J.; Somboon, K.; Farrell, D.P.; Muenks, A.; Tzokov, S.B.; DiMaio, F.; Khalid, S.; Miller, S.I.; Bergeron, J.R.C. Structure and lipid dynamics in the maintenance of lipid asymmetry inner membrane complex of A. baumannii. Commun. Biol. 2021, 4, 817. [Google Scholar] [CrossRef]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef] [PubMed]
- Rozek, A.; Powers, J.P.; Friedrich, C.L.; Hancock, R.E. Structure-based design of an indolicidin peptide analogue with increased protease stability. Biochemistry 2003, 42, 14130–14138. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Generic/Trade Name | Target | Indication |
|---|---|---|---|
| 2005 | Micafungin/Mycamine | 1,4-β-glucan synthase | antifungal |
| 2005 | Daptomycin/Cubicin | Membrane pore formation | antibacterial |
| 2006 | Anidualafungin/Eraxis | 1,5-β-glucan synthase | antifungal |
| 2009 | Telavancin/Vibative | Cell wall synthesis | antibacterial |
| 2014 | Dalbavancin/Dalvance | Cell wall synthesis | antibacterial |
| 2014 | Oritavancin/Orbactiv | Cell wall synthesis | antibacterial |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, S.; Zhang, Q.; Jin, L. Natural and Man-Made Cyclic Peptide-Based Antibiotics. Antibiotics 2023, 12, 42. https://doi.org/10.3390/antibiotics12010042
Lai S, Zhang Q, Jin L. Natural and Man-Made Cyclic Peptide-Based Antibiotics. Antibiotics. 2023; 12(1):42. https://doi.org/10.3390/antibiotics12010042
Chicago/Turabian StyleLai, Shian, Quan Zhang, and Lin Jin. 2023. "Natural and Man-Made Cyclic Peptide-Based Antibiotics" Antibiotics 12, no. 1: 42. https://doi.org/10.3390/antibiotics12010042
APA StyleLai, S., Zhang, Q., & Jin, L. (2023). Natural and Man-Made Cyclic Peptide-Based Antibiotics. Antibiotics, 12(1), 42. https://doi.org/10.3390/antibiotics12010042