
Modulation of Multiple Gene Clusters’ Expression by the PAS-LuxR Transcriptional Regulator PteF


 , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
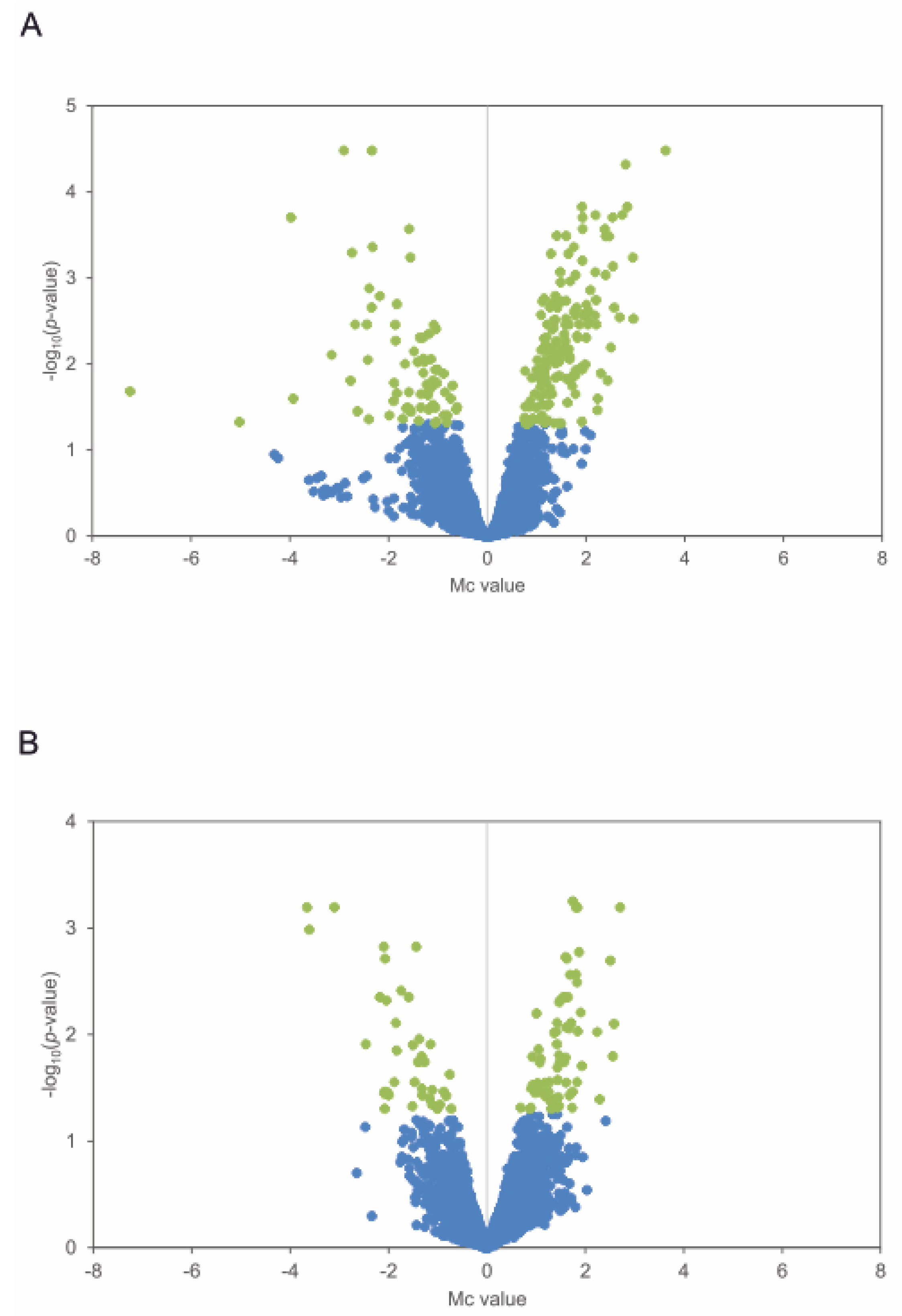
2.1. Identification of Genes with an Altered Expression Profile in S. avermitilis ΔpteF Mutant
2.1.1. Genes Involved in Genetic Information- and Protein-Processing and Amino Acid Metabolism
2.1.2. Genes Involved in Nucleotide and Vitamin Metabolism, and DNA Replication, Recombination, and Repair
2.1.3. Carbohydrate Metabolism Genes
2.1.4. Lipid Metabolism Genes
2.1.5. Energy Production Genes
2.1.6. Transport and External Signals Processing
2.1.7. Genes Involved in Cell Envelope Biosynthesis and Morphological Differentiation
2.1.8. Regulatory Genes
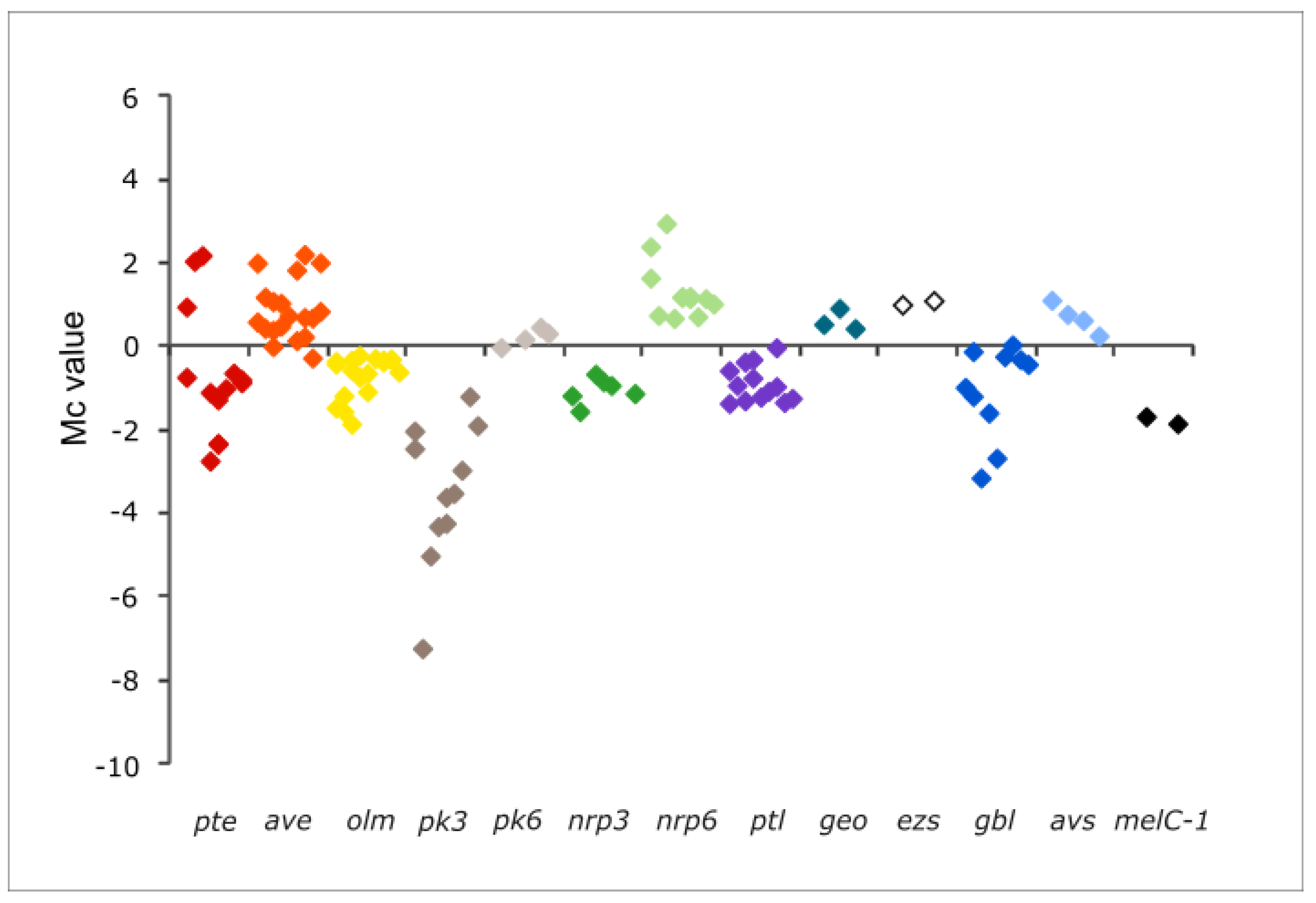
2.1.9. Secondary Metabolite Genes
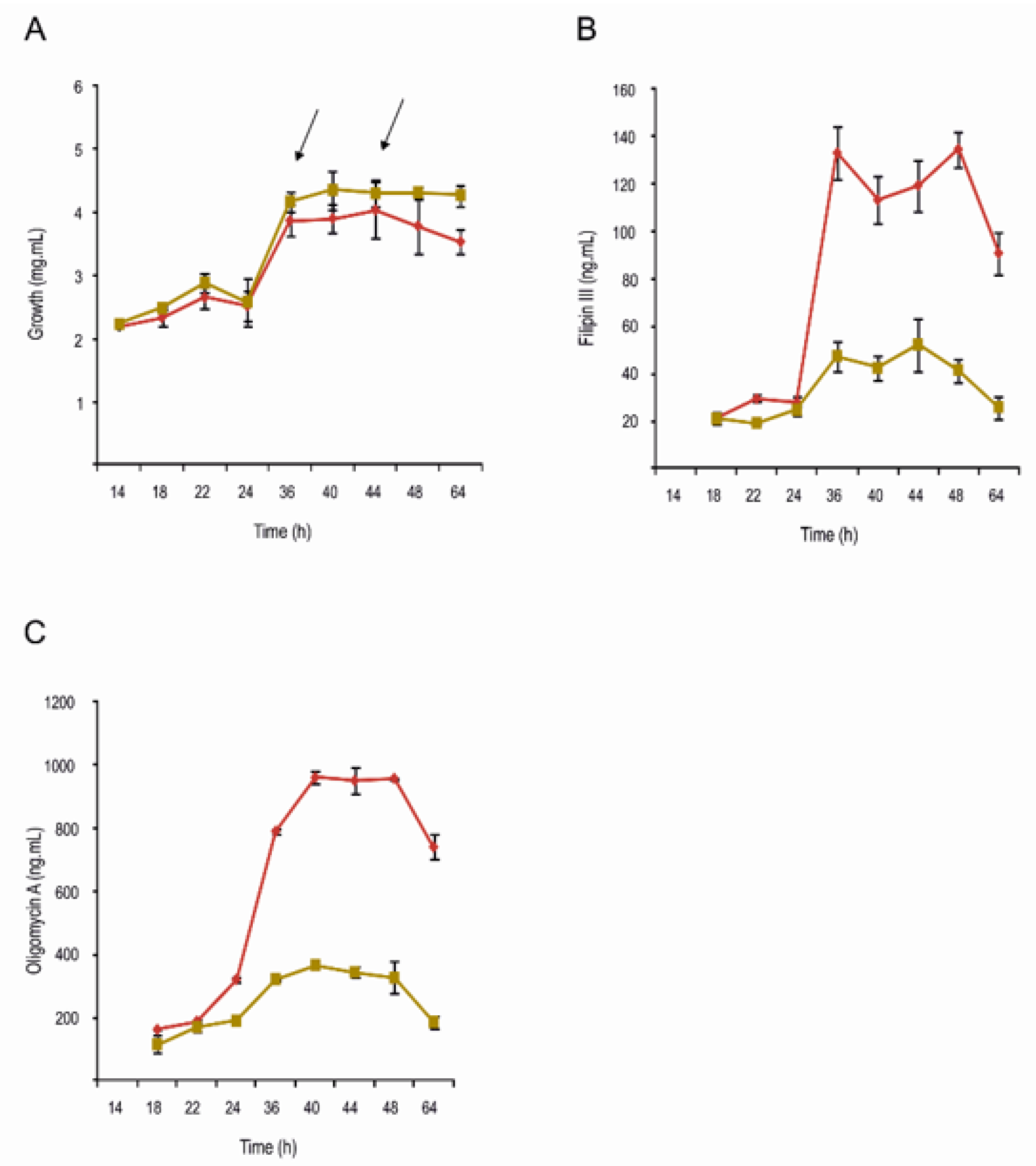
2.2. Filipin and Oligomycin Production Are Strongly Reduced in S. avermitilis ΔpteF
2.3. Validation of Microarray Results by Using Quantitative RT-PCR
2.4. Concluding Remarks
3. Materials and Methods
3.1. Strains and Cultivation
3.2. Nucleic Acid Extractions
3.3. Microarray Hybridizations
3.4. Identification of Differentially Transcribed Genes
3.5. Assessment of Filipin and Oligomycin Production
3.6. Reverse Transcription-Quantitative PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hefti, M.H.; Francoijs, K.-J.; de Vries, S.C.; Dixon, R.; Vervoort, J. The PAS fold: A redefinition of the PAS domain based upon structural prediction. Eur. J. Biochem. 2004, 271, 1198–1208. [Google Scholar] [CrossRef]
- Santos, C.L.; Correia-Neves, M.; Moradas-Ferreira, P.; Mendes, M.V. A walk into the LuxR regulators of Actinobacteria: Phylogenomic distribution and functional diversity. PLoS ONE 2012, 7, e46758. [Google Scholar] [CrossRef]
- Möglich, A.; Ayers, R.A.; Moffat, K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 2009, 17, 1282–1294. [Google Scholar] [CrossRef]
- Antón, N.; Santos-Aberturas, J.; Mendes, M.V.; Guerra, S.M.; Martín, J.F.; Aparicio, J.F. PimM, a PAS domain positive regulator of pimaricin biosynthesis in Streptomyces natalensis. Microbiology 2007, 53, 3174–3183. [Google Scholar] [CrossRef]
- Santos-Aberturas, J.; Vicente, C.M.; Guerra, S.M.; Payero, T.D.; Martín, J.F.; Aparicio, J.F. Molecular control of polyene macrolide biosynthesis: Direct binding of the regulator PimM to eight promoters of pimaricin genes and identification of binding boxes. J. Biol. Chem. 2011, 286, 9150–9161. [Google Scholar] [CrossRef]
- Santos-Aberturas, J.; Payero, T.D.; Vicente, C.M.; Guerra, S.M.; Cañibano, C.; Martín, J.F.; Aparicio, J.F. Functional conservation of PAS-LuxR transcriptional regulators in polyene macrolide biosynthesis. Metab. Engl. 2011, 13, 756–767. [Google Scholar] [CrossRef]
- Vicente, C.M.; Payero, T.D.; Santos-Aberturas, J.; Barreales, E.G.; de Pedro, A.; Aparicio, J.F. Pathway-specific regulation revisited: Cross-regulation of multiple disparate gene clusters by PAS-LuxR transcriptional regulators. Appl. Microbiol. Biotechnol. 2015, 99, 5123–5135. [Google Scholar] [CrossRef]
- Vicente, C.M.; Santos-Aberturas, J.; Payero, T.D.; Barreales, E.G.; De Pedro, A.; Aparicio, J.F. PAS-LuxR transcriptional control of filipin biosynthesis in S. avermitilis. Appl. Microbiol. Biotechnol. 2014, 98, 9311–9324. [Google Scholar] [CrossRef]
- Olano, C.; García, I.; González, A.; Rodríguez, M.; Rozas, D.; Rubio, J.; Sánchez-Hidalgo, M.; Braña, A.F.; Méndez, C.; Salas, J.A. Activation and identification of five clusters for secondary metabolites in Streptomyces albus J1074. Microb. Biotechnol. 2014, 7, 242–256. [Google Scholar] [CrossRef]
- Martínez-Burgo, Y.; Santos-Aberturas, J.; Rodríguez-García, A.; Barreales, E.G.; Tormo, J.R.; Truman, A.W.; Reyes, F.; Aparicio, J.F.; Liras, P. Activation of Secondary Metabolite Gene Clusters in Streptomyces clavuligerus by the PimM Regulator of Streptomyces natalensis. Front. Microbiol. 2019, 10, 580. [Google Scholar] [CrossRef]
- Fowler-Goldsworthy, K.; Gust, B.; Mouz, S.; Chandra, G.; Findlay, K.C.; Chater, K.F. The actinobacteria-specific gene wblA controls major developmental transitions in Streptomyces coelicolor A3(2). Microbiology 2011, 157, 1312–1328. [Google Scholar] [CrossRef]
- Bush, M. The actinobacterial WhiB-like (Wbl) family of transcription factors. Mol. Microbiol. 2018, 110, 663–676. [Google Scholar] [CrossRef]
- Menzel, R.; Roth, J. Purification of the putA gene product. A bifunctional membrane-bound protein from Salmonella typhimurium responsible for the two-step oxidation of proline to glutamate. J. Biol. Chem. 1981, 256, 9755–9761. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, W.; Zhang, Y.; Jiang, L.; Chen, Z.; Wen, Y.; Li, J. Deletion of ku homologs increases gene targeting frequency in Streptomyces avermitilis. J. Ind. Microbiol. Biotechnol. 2012, 39, 917–925. [Google Scholar] [CrossRef]
- Sola-Landa, A.; Rodríguez-García, A.; Apel, A.K.; Martín, J.F. Target genes and structure of the direct repeats in the DNA-binding sequences of the response regulator PhoP in Streptomyces coelicolor. Nucleic Acids Res. 2008, 36, 1358–1368. [Google Scholar] [CrossRef][Green Version]
- Yao, J.; Rock, C.O. Phosphatidic acid synthesis in bacteria. Biochim. Biophys. Acta 2013, 1831, 495–502. [Google Scholar] [CrossRef]
- Staudenmaier, H.; Van Hove, B.; Yaraghi, Z.; Braun, V. Nucleotide sequences of the fecBCDE genes and locations of the proteins suggest a periplasmic-binding-protein-dependent transport mechanism for iron (III) dicitrate in Escherichia coli. J. Bacteriol. 1989, 171, 2626–2633. [Google Scholar] [CrossRef]
- McCormick, J.R.; Flärdh, K. Signals and regulators that govern Streptomyces development. FEMS Microbiol. Rev. 2012, 36, 206–231. [Google Scholar] [CrossRef]
- Gordon, N.D.; Ottaviano, G.L.; Connell, S.E.; Tobkin, G.V.; Son, C.H.; Shterental, S.; Gehring, A.M. Secreted-protein response to sigmaU activity in Streptomyces coelicolor. J. Bacteriol. 2008, 190, 894–904. [Google Scholar] [CrossRef]
- Bellier, A.; Gominet, M.; Mazodier, P. Post-translational control of the Streptomyces lividans ClgR regulon by ClpP. Microbiology 2006, 152, 1021–1027. [Google Scholar] [CrossRef][Green Version]
- Campo, N.; Rudner, D.Z. SpoIVB and CtpB are both forespore signals in the activation of the sporulation transcription factor sigmaK in Bacillus subtilis. J. Bacteriol. 2007, 189, 6021–6027. [Google Scholar] [CrossRef]
- Kleinschnitz, E.-M.; Heichlinger, A.; Schirner, K.; Winkler, J.; Latus, A.; Maldener, I.; Wohlleben, W.; Muth, G. Proteins encoded by the mre gene cluster in Streptomyces coelicolor A3(2) cooperate in spore wall synthesis. Mol. Microbiol. 2011, 79, 1367–1379. [Google Scholar] [CrossRef]
- Wang, L.; Grau, R.; Perego, M.; Hoch, J.A. A novel histidine kinase inhibitor regulating development in Bacillus subtilis. Genes Dev. 1997, 11, 2569–2579. [Google Scholar] [CrossRef]
- Jacques, D.A.; Langley, D.B.; Hynson, R.M.G.; Whitten, A.E.; Kwan, A.; Guss, J.M.; Trewhella, J. A novel structure of an antikinase and its inhibitor. J. Mol. Biol. 2011, 405, 214–226. [Google Scholar] [CrossRef]
- Aparicio, J.F.; Barreales, E.G.; Payero, T.D.; Vicente, C.M.; de Pedro, A.; Santos-Aberturas, J. Biotechnological production and application of the antibiotic pimaricin: Biosynthesis and its regulation. Appl. Microbiol. Biotechnol. 2016, 100, 61–78. [Google Scholar] [CrossRef]
- Ikeda, H.; Kazuo, S.Y.; Omura, S. Genome mining of the Streptomyces avermitilis genome and development of genome-minimized hosts for heterologous expression of biosynthetic gene clusters. J. Ind. Microbiol. Biotechnol. 2014, 41, 233–250. [Google Scholar] [CrossRef]
- Kitani, S.; Miyamoto, K.T.; Takamatsu, S.; Herawati, E.; Iguchi, H.; Nishitomi, K.; Uchida, M.; Nagamitsu, T.; Omura, S.; Ikeda, H.; et al. Avenolide, a Streptomyces hormone controlling antibiotic production in Streptomyces avermitilis. Proc. Natl. Acad. Sci. USA 2011, 108, 16410–16415. [Google Scholar] [CrossRef]
- Wang, J.B.; Zhang, F.; Pu, J.Y.; Zhao, J.; Zhao, Q.F.; Tang, G.L. Characterization of AvaR1, an autoregulator receptor that negatively controls avermectins production in a high avermectin-producing strain. Biotechnol. Lett. 2014, 36, 813–819. [Google Scholar] [CrossRef]
- Zhu, J.; Sun, D.; Liu, W.; Chen, Z.; Li, J.; Wen, Y. AvaR2, a pseudo c-butyrolactone receptor homologue from Streptomyces avermitilis, is a pleiotropic repressor of avermectin and avenolide biosynthesis and cell growth. Mol. Microbiol. 2016, 102, 562–578. [Google Scholar] [CrossRef]
- Yu, Q.; Bai, L.; Zhou, X.; Deng, Z. Inactivation of the positive LuxR-type oligomycin biosynthesis regulators OlmRI and OlmRII increases avermectin production in Streptomyces avermitilis. Chinese Sci. Bull. 2012, 57, 869–876. [Google Scholar] [CrossRef]
- Payero, T.D.; Vicente, C.M.; Rumbero, Á.; Barreales, E.G.; Santos-Aberturas, J.; De Pedro, A.; Aparicio, J.F. Functional analysis of filipin tailoring genes from Streptomyces filipinensis reveals alternative routes in filipin III biosynthesis and yields bioactive derivatives. Microb. Cell Fact 2015, 14, 114. [Google Scholar] [CrossRef]
- Kitani, S.; Ikeda, H.; Sakamoto, T.; Noguchi, S.; Nihira, T. Characterization of a regulatory gene, aveR, for the biosynthesis of avermectin in Streptomyces avermitilis. Appl. Microbiol. Biotechnol. 2009, 82, 1089–1096. [Google Scholar] [CrossRef]
- Narva, K.E.; Feitelson, J.S. Nucleotide sequence and transcriptional analysis of the redD locus of Streptomyces coelicolor A3(2). J. Bacteriol. 1990, 172, 326–333. [Google Scholar] [CrossRef][Green Version]
- Hunt, A.C.; Servín-González, L.; Kelemen, G.H.; Buttner, M.J. The bldC developmental locus of Streptomyces coelicolor encodes a member of a family of small DNA-binding proteins related to the DNA-binding domains of the MerR family. J. Bacteriol. 2005, 187, 716–728. [Google Scholar] [CrossRef]
- Chang, H.M.; Chen, M.Y.; Shieh, Y.T.; Bibb, M.J.; Chen, C.W. The cutRS signal transduction system of Streptomyces lividans represses the biosynthesis of the polyketide antibiotic actinorhodin. Mol. Microbiol. 1996, 21, 1075–1085. [Google Scholar]
- StrepDB—The Streptomyces Annotation Server. Available online: http://strepdb.streptomyces.org.uk/ (accessed on 1 June 2022).
- McLean, T.C.; Hoskisson, P.A.; Seipke, R.F. Coordinate regulation of antimycin and candicidin biosynthesis. mSphere 2016, 1, e00305-16. [Google Scholar] [CrossRef]
- Zou, Z.; Du, D.; Zhang, Y.; Zhang, J.; Niu, G.; Tan, H. A γ-butyrolactone-sensing activator/repressor, JadR3, controls a regulatory mini-network for jadomycin biosynthesis. Mol. Microbiol. 2014, 94, 490–505. [Google Scholar] [CrossRef]
- Zhou, Z.X.; Xu, Q.Q.; Bu, Q.T.; Liu, S.P.; Yu, P.; Li, Y.Q. Transcriptome-guided identification of SprA as a pleiotropic regulator in Streptomyces chattanoogensis. Appl. Microbiol. Biotechnol. 2015, 99, 1287–1298. [Google Scholar] [CrossRef]
- Barreales, E.G.; Payero, T.D.; Jambrina, E.; Aparicio, J.F. The gamma-butyrolactone system from Streptomyces filipinensis reveals novel clues to understand secondary metabolism control. Appl. Environ. Microbiol. 2020, 86, e00443-20. [Google Scholar] [CrossRef]
- Kitani, S.; Doi, M.; Shimizu, T.; Maeda, A.; Nihira, T. Control of secondary metabolism by farX, which is involved in the gamma-butyrolactone biosynthesis of Streptomyces lavendulae FRI-5. Arch. Microbiol. 2010, 192, 211–220. [Google Scholar] [CrossRef]
- Guyet, A.; Gominet, M.; Benaroudj, N.; Mazodire, P. Regulation of the clpP1clpP2 operon by the pleiotropic regulator AdpA in Streptomyces lividans. Arch. Microbiol. 2013, 195, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Engels, S.; Ludwig, C.; Schweitzer, J.E.; Mack, C.; Bott, M.; Schaffer, S. The transcriptional activator ClgR controls transcription of genes involved in proteolysis and DNA repair in Corynebacterium glutamicum. Mol. Microbiol. 2005, 57, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Hengst, C.D.; Bush, M.J.; Le, T.B.K.; Tran, N.T.; Chandra, G.; Zeng, W.; Travis, B.; Brennan, R.G.; Buttner, M.J. The MerR-like protein BldC binds DNA direct repeats as cooperative multimers to regulate Streptomyces development. Nat. Commun. 2018, 9, 1139. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Pang, A.-P.; Zhang, Y.; Qiao, J.; Zhao, G.-R. Comparative transcriptomic analysis reveals the significant pleiotropic regulatory effects of LmbU on lincomycin biosynthesis. Microb. Cell Fact 2020, 19, 30. [Google Scholar] [CrossRef]
- Santamarta, I.; López-García, M.T.; Kurt, A.; Nárdiz, N.; Alvarez-Álvarez, R.; Pérez-Redondo, R.; Martín, J.F.; Liras, P. Characterization of DNA-binding sequences for CcaR in the cephamycin-clavulanic acid supercluster of Streptomyces clavuligerus. Mol. Microbiol. 2011, 81, 968–981. [Google Scholar] [CrossRef]
- Huang, J.; Shi, J.; Molle, V.; Sohlberg, B.; Weaver, D.; Bibb, M.J.; Karoonuthaisiri, N.; Lih, C.J.; Kao, C.M.; Buttner, M.J.; et al. Cross-regulation among disparate antibiotic biosynthetic pathways of Streptomyces coelicolor. Mol. Microbiol. 2005, 58, 1276–1287. [Google Scholar] [CrossRef]
- Santos-Beneit, F.; Rodríguez-García, A.; Sola-Landa, A.; Martín, J.F. Cross-talk between two global regulators in Streptomyces: PhoP and AfsR interact in the control of afsS, pstS and phoRP transcription. Mol. Microbiol. 2009, 72, 53–68. [Google Scholar] [CrossRef]
- Barreales, E.G.; Payero, T.D.; de Pedro, A.; Aparicio, J.F. Phosphate effect on filipin production and morphological differentiation in Streptomyces filipinensis and the role of the PhoP transcription factor. PLoS ONE 2018, 13, e0208278. [Google Scholar] [CrossRef]
- Kieser, T.; Bibb, M.J.; Buttner, M.J.; Chater, K.F.; Hopwood, D.A. Practical Streptomyces Genetics; John Innes Foundation: Norwich, UK, 2000. [Google Scholar]
- Gadgil, M.; Lian, W.; Gadgil, C.; Kapur, V.; Hu, W.S. An analysis of the use of genomic DNA as a universal reference in two channel DNA microarrays. BMC Genom. 2005, 6, 66. [Google Scholar] [CrossRef]
- Beites, T.; Rodríguez-García, A.; Santos-Beneit, F.; Moradas-Ferreira, P.; Aparicio, J.F.; Mendes, M.V. Genome-wide analysis of the regulation of pimaricin production in Streptomyces natalensis by reactive oxygen species. Appl. Microbiol. Biotechnol. 2014, 98, 2231–2241. [Google Scholar] [CrossRef]
- Guerra, S.M.; Rodríguez-García, A.; Santos-Aberturas, J.; Vicente, C.M.; Payero, T.D.; Martín, J.F.; Aparicio, J.F. LAL regulators SCO0877 and SCO7173 as pleiotropic modulators of phosphate starvation response and actinorhodin biosynthesis in Streptomyces coelicolor. PLoS ONE 2012, 7, e31475. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef]
- Smyth, G.K.; Michaud, J.; Scott, H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005, 21, 2067–2075. [Google Scholar] [CrossRef]
- Wu, W.; Xing, E.P.; Myers, C.; Mian, I.S.; Bissell, M.J. Evaluation of normalization methods for cDNA microarray data by k-NN classification. BMC Bioinform. 2005, 6, 191. [Google Scholar] [CrossRef]
- Smyth, G.K.; Speed, T.P. Normalization of cDNA microarray data. Methods 2003, 31, 265–273. [Google Scholar] [CrossRef]
- Mehra, S.; Lian, W.; Jayapal, K.P.; Charaniya, S.P.; Sherman, D.H.; Hu, W.S. A framework to analyze multiple time series data: A case study with Streptomyces coelicolor. J. Ind. Microbiol. Biotechnol. 2006, 33, 159–172. [Google Scholar] [CrossRef]
- Ritchie, M.; Diyagama, D.; Neilson, J.; Van Laar, R.; Dobrovic, A.; Holloway, A.; Smyth, G.K. Empirical array quality weights in the analysis of microarray data. BMC Bioinform. 2006, 7, 261. [Google Scholar] [CrossRef]
- Barreales, E.G.; Vicente, C.M.; de Pedro, A.; Santos-Aberturas, J.; Aparicio, J.F. Promoter engineering reveals the importance of heptameric direct repeats for DNA-binding by SARP-LAL regulators in Streptomyces natalensis. Appl. Environ. Microbiol. 2018, 84, e00246-18. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Rasmussen, R. Quantification on the LightCycler. In Rapid Cycle Real-Time PCR, Methods and Applications; Meuer, S., Wittwer, C., Nakagawara, K., Eds.; Springer Press: Heidelberg, Germany, 2000; pp. 21–34. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Genes Underexpressed a | Genes Overexpressed a | Total | |
|---|---|---|---|
| Exponential phase (t1) | 63 | 145 | 208 |
| Stationary phase (t2) | 35 | 64 | 99 |
| Identified Genes b | |||
| Function | t1 | t2 | |
| Genetic information- and protein-processing; amino acid metabolism | 20 (7 ↓; 13 ↑) | 11 (5 ↓; 6 ↑) | |
| Nucleotide and vitamin metabolism; DNA replication, recombination, and repair | 16 (4 ↓; 12 ↑) | 4 (1 ↓; 3 ↑) | |
| Carbohydrate metabolism | 13 (3 ↓; 10 ↑) | 1 (1 ↓) | |
| Lipid metabolism | 8 (1 ↓; 7 ↑) | 4 (4 ↑) | |
| Energy production | 2 (2 ↓) | 1 (1 ↓) | |
| Transport and external signals’ processing | 20 (12 ↓; 8 ↑) | 8 (4 ↓; 4 ↑) | |
| Cell envelope biosynthesis and morphological differentiation | 9 (4 ↓; 5 ↑) | 5 (4 ↓; 1 ↑) | |
| Regulation | 27 (12 ↓; 15 ↑) | 12 (3 ↓; 9 ↑) | |
| Secondary metabolism | 60 (34 ↓; 26 ↑) | 6 (2 ↓; 4 ↑) | |
| Miscellaneous | 38 (11 ↓; 27 ↑) | 19 (1 ↓; 18 ↑) | |
| Gene | Description | Fold-Change | Mc | Corrected p-Value | p-Value | |
|---|---|---|---|---|---|---|
| Filipin cluster (pte) | ||||||
| 407 | pteH | Thioesterase | 1.93 | 0.95 | 0.1395 | 0.0076 |
| 408 | pteG | cholesterol oxidase | 4.14 | 2.05 | 0.0025 | 0.0000 |
| 410 | pteR | SARP-family transcriptional regulator | 4.53 | 2.18 | 0.0009 | 0.0000 |
| 411 | pteE | Ferredoxin | 2.14 | −1.10 | 0.2533 | 0.0233 |
| 412 | pteD | cytochrome P450 monooxygenase | 6.68 | −2.74 | 0.0005 | 0.0000 |
| 413 | pteC | cytochrome P450 monooxygenase | 5.03 | −2.33 | 0.0004 | 0.0000 |
| 414 | pteB | Dehydrogenase | 5.06 | −2.34 | 0.0000 | 0.0000 |
| 415 | pteA5 | modular polyketide synthase | 2.01 | −1.01 | 0.1136 | 0.0054 |
| 416 | pteA4 | modular polyketide synthase | 2.43 | −1.28 | 0.0095 | 0.0002 |
| 417 | pteA3 | modular polyketide synthase | 1.56 | −0.64 | 0.3514 | 0.0457 |
| 418 | pteA2 | modular polyketide synthase | 1.83 | −0.87 | 0.3639 | 0.0506 |
| 419 | pteA1 | modular polyketide synthase | 1.74 | −0.80 | 0.4388 | 0.0752 |
| Non-ribosomal peptide-6 (nrp6) | ||||||
| 600 | fecC1 | ABC transporter iron(III)/siderophore transport system ATP-binding protein | 5.28 | 2.40 | 0.0003 | 0.0000 |
| 601 | fecD1 | ABC transporter iron(III)/siderophore permease | 1.68 | 0.75 | 0.5625 | 0.1290 |
| 602 | fecB | ABC transporter iron(III)/siderophore-binding protein | 7.73 | 2.95 | 0.0006 | 0.0000 |
| 603 | nrps6 | non-ribosomal peptide synthetase | 1.60 | 0.68 | 0.3119 | 0.0342 |
| 604 | hypothetical protein | 2.27 | 1.18 | 0.0224 | 0.0005 | |
| 605 | fadD2 | acyl-CoA synthetase | 2.30 | 1.20 | 0.0049 | 0.0001 |
| 606 | hypothetical protein | 1.64 | 0.71 | 0.3525 | 0.0464 | |
| 607 | taurine catabolism dioxygenase | 2.22 | 1.15 | 0.0017 | 0.0000 | |
| 608 | fabC2 | acyl carrier protein | 2.03 | 1.02 | 0.1136 | 0.0054 |
| 609 | fabH4 | 3-oxoacyl-ACP synthase III | 2.22 | 1.15 | 0.1182 | 0.0058 |
| 610 | MFS transporter protein | 2.28 | 1.19 | 0.0166 | 0.0004 | |
| 611 | beta-hydroxylase | 3.12 | 1.64 | 0.0005 | 0.0000 | |
| Avermectin cluster (ave) | ||||||
| 935 | aveR | LuxR-family transcriptional regulator | 4.00 | 2.00 | 0.0049 | 0.0001 |
| 936 | aveF | C-5 ketoreductase | 1.51 | 0.59 | 0.6016 | 0.1518 |
| 937 | aveD | C5-O-methyltransferase | 1.35 | 0.43 | 0.6148 | 0.1603 |
| 938 | aveA1 | type I polyketide synthase | 2.27 | 1.18 | 0.1820 | 0.0122 |
| 939 | aveA2 | type I polyketide synthase | 1.31 | 0.39 | 0.7264 | 0.2537 |
| 940 | aveC | post-polyketide modification protein | 1.00 | 0.00 | 0.9993 | 0.9950 |
| 941 | aveE | cytochrome P450 monooxygenase | 2.06 | 1.04 | 0.1376 | 0.0074 |
| 942 | aveA3 | type I polyketide synthase | 1.40 | 0.49 | 0.6328 | 0.1725 |
| 943 | aveA4 | type I polyketide synthase | 1.66 | 0.73 | 0.4435 | 0.0764 |
| 944 a | orf-1 | Reductase | 1.10 | 0.14 | 0.9330 | 0.6990 |
| 945 | aveBI | dTDP-L-oleandrose transferase (glycosyltransferase) | 1.62 | 0.70 | 0.6052 | 0.1543 |
| 946 | aveBII | dTDP-glucose 4.6-dehydratase | 1.17 | 0.23 | 0.8027 | 0.3477 |
| 947 | aveBIII | glucose-1-phosphate thymidyltransferase | 2.11 | 1.08 | 0.2027 | 0.0154 |
| 948 | aveBIV | dTDP-4-keto-6-deoxy-L-hexose 4-reductase | 1.21 | −0.28 | 0.8828 | 0.5032 |
| 949 | aveBV | dTDP-4-keto-6-deoxyhexose 3.5-epimerase | 1.60 | 0.68 | 0.5387 | 0.1160 |
| 950 | aveBVI | dTDP-4-keto-6-deoxy-L-hexose2.3-dehydratase | 1.79 | 0.84 | 0.4308 | 0.0711 |
| 951 | aveBVII | dTDP-6-deoxy-L-hexose 3-O-methyltransferase | 4.03 | 2.01 | 0.0020 | 0.0000 |
| 952 | aveBVIII | dTDP-4-keto-6-deoxy-L-hexose 2.3-reductase | 3.56 | 1.83 | 0.0049 | 0.0001 |
| 953 | aveG | Thioesterase | 4.59 | 2.20 | 0.0018 | 0.0000 |
| Melanin cluster (melC-1) b | ||||||
| 1136 | melC1 | tyrosinase co-factor protein | 3.20 | −1.68 | 0.0776 | 0.0015 |
| 1137 | melC2 | Tyrosinase | 3.61 | −1.85 | 0.0078 | 0.0000 |
| γ-butyrolactone cluster (gbl) | ||||||
| 2266 | avaC | Phosphatase | 1.97 | −0.98 | 0.0794 | 0.0030 |
| 2267 | avaB | Oxidoreductase | 2.30 | −1.20 | 0.2777 | 0.0279 |
| 2268 | avaL2 | TetR-family transcriptional regulator | 8.88 | −3.15 | 0.0078 | 0.0001 |
| 2269 | avaA | gamma-butyrolactone biosynthesis protein | 3.01 | −1.59 | 0.1456 | 0.0083 |
| 2270 | avaL1 | TetR-family transcriptional regulator | 6.41 | −2.68 | 0.0035 | 0.0000 |
| Polyketide-3 cluster (pk3) | ||||||
| 2272 | hypothetical protein | 5.46 | −2.45 | 0.2000 | 0.0150 | |
| 2273 | Isomerase | 151.17 | −7.24 | 0.0208 | 0.0005 | |
| 2274 | secreted protein | 32.45 | −5.02 | 0.0473 | 0.0014 | |
| 2275 | transmembrane efflux protein | 19.97 | −4.32 | 0.1124 | 0.0053 | |
| 2276 | 3-oxoacyl-ACP synthase III | 18.90 | −4.24 | 0.1242 | 0.0063 | |
| 2277 | Thioesterase | 12.30 | −3.62 | 0.2222 | 0.0183 | |
| 2278 | F420-dependent dehydrogenase | 11.47 | −3.52 | 0.3037 | 0.0327 | |
| 2279 | acyl-CoA synthetase | 7.84 | −2.97 | 0.3522 | 0.0462 | |
| 2280 | pks3-1 | modular polyketide synthase | 2.30 | −1.20 | 0.6288 | 0.1702 |
| 2281 | pks3-2 | modular polyketide synthase | 3.73 | −1.90 | 0.5893 | 0.1436 |
| 2282 | pks3-3 | acyl carrier protein | 4.08 | −2.03 | 0.3974 | 0.0593 |
| Oligomycin cluster (olm) | ||||||
| 2890 | ccrA1 | crotonyl-CoA reductase | 1.34 | −0.42 | 0.4368 | 0.0743 |
| 2891 | hypothetical protein | 2.27 | −1.18 | 0.2280 | 0.0193 | |
| 2892 | olmA4 | modular polyketide synthase | 1.26 | −0.33 | 0.4760 | 0.0913 |
| 2893 | olmA5 | modular polyketide synthase | 1.16 | −0.22 | 0.6421 | 0.1789 |
| 2894 | olmB | cytochrome P450 monooxygenase | 2.13 | −1.09 | 0.1512 | 0.0087 |
| 2895 | olmA7 | modular polyketide synthase | 1.56 | −0.64 | 0.0746 | 0.0028 |
| 2896 | olmA6 | modular polyketide synthase | 1.67 | −0.74 | 0.1443 | 0.0080 |
| 2897 | olmA3 | modular polyketide synthase | 1.25 | −0.32 | 0.3610 | 0.0493 |
| 2898 | olmA2 | modular polyketide synthase | 1.24 | −0.31 | 0.5785 | 0.1377 |
| 2899 | olmA1 | modular polyketide synthase | 1.53 | −0.61 | 0.1817 | 0.0121 |
| 2900 | P450-like protein | 1.48 | −0.57 | 0.2474 | 0.0224 | |
| 2901 | olmRII | LuxR-family transcriptional regulator | 2.77 | −1.47 | 0.0712 | 0.0026 |
| 2902 | olmRI | LuxR-family transcriptional regulator | 2.95 | −1.56 | 0.0006 | 0.0000 |
| 2903 | olmC | Thioesterase | 3.63 | −1.86 | 0.1235 | 0.0062 |
| Neopentalenolactone cluster (ptl) | ||||||
| 2989 | MarR-family transcriptional regulator | 2.08 | −1.06 | 0.0487 | 0.0015 | |
| 2990 | gap1 | glyceraldehyde-3-phosphate dehydrogenase | 2.57 | −1.36 | 0.2469 | 0.0222 |
| 2991 | ptlH | 1-deoxypentalenic acid 11-beta hydroxylase | 1.91 | −0.93 | 0.1529 | 0.0089 |
| 2992 | ptlG | transmembrane efflux protein | 1.29 | −0.37 | 0.9068 | 0.5988 |
| 2993 | ptlF | 1-deoxy-11beta-hydroxypentalenic acid dehydrogenase | 1.69 | −0.76 | 0.2639 | 0.0251 |
| 2994 | ptlE | Baeyer-Villiger monooxygenase | 2.46 | −1.30 | 0.0087 | 0.0001 |
| 2995 | ptlD | Dioxygenase | 2.31 | −1.21 | 0.0962 | 0.0041 |
| 2996 | ptlC | hypothetical protein | 2.10 | −1.07 | 0.5367 | 0.1153 |
| 2997 | ptlB | farnesyl diphosphate synthase | 1.95 | −0.96 | 0.5690 | 0.1322 |
| 2998 | ptlA | pentalenene synthase | 2.53 | −1.34 | 0.5592 | 0.1269 |
| 2999 | ptlI | pentalenene C13 hydroxylase; cytochrome P450 | 2.36 | −1.24 | 0.4943 | 0.0993 |
| 3000 | ptlR | AraC-family transcriptional regulator | 1.49 | −0.58 | 0.5379 | 0.1158 |
| 3001 | ptlJ | Lyase | 1.24 | −0.31 | 0.6109 | 0.1579 |
| 3002 | ptlL | hypothetical protein | 1.02 | −0.03 | 0.9754 | 0.8867 |
| Albaflavenol/albaflavenone cluster (ezs) | ||||||
| 3031 | cyp14 | epi-isozizaene hydroxylase (cytochrome P450 monooxygenase) | 2.00 | 1.00 | 0.2918 | 0.0301 |
| 3032 | ezs | epi-isozizaene synthase (sesquiterpene cyclase) | 2.14 | 1.10 | 0.1621 | 0.0097 |
| Non-ribosomal peptide-3 cluster (nrp3) | ||||||
| 3155 | MbtH-like protein | 2.27 | −1.18 | 0.2696 | 0.0261 | |
| 3156 | nrps3-1 | non-ribosomal peptide synthetase | 2.95 | −1.56 | 0.0867 | 0.0035 |
| 3157 | export protein | 1.58 | −0.66 | 0.6407 | 0.1773 | |
| 3158 | nrps3-2 | non-ribosomal peptide synthetase | 1.79 | −0.84 | 0.6149 | 0.1623 |
| 3159 | nrps3-3 | non-ribosomal peptide synthetase | 1.91 | −0.93 | 0.5290 | 0.1123 |
| 3160 | Aminotransferase | 2.19 | −1.13 | 0.4384 | 0.0749 | |
| 3161 | dapF2 | diaminopimelate epimerase | 2.04 | −1.03 | 0.5045 | 0.1029 |
| 3162 | hypothetical protein | 2.87 | −1.52 | 0.1668 | 0.0101 | |
| 3163 | hypothetical protein | 2.36 | −1.24 | 0.3227 | 0.0384 | |
| 3164 | hypothetical protein | 3.01 | −1.59 | 0.0003 | 0.0000 | |
| Polyketide-4 cluster (pk4) | ||||||
| 7184 | pks4 | modular polyketide synthase | 9.32 | −3.22 | 0.2980 | 0.0315 |
| 7185 | UDP-glucose:sterol glucosyltransferase | 8.28 | −3.05 | 0.2753 | 0.0271 | |
| 7186 | cyp26 | cytochrome P450 hydroxylase | 7.36 | −2.88 | 0.2419 | 0.0213 |
| Polyhydroxycarboxylate siderophore cluster (avs) | ||||||
| 7320 | avsA | siderophore synthetase component | 2.16 | 1.11 | 0.0295 | 0.0007 |
| 7321 | avsB | siderophore synthetase component | 1.71 | 0.77 | 0.2474 | 0.0223 |
| 7322 | avsC | siderophore synthetase component | 1.55 | 0.63 | 0.4654 | 0.0862 |
| 7323 | avsD | diaminopimelate decarboxylase | 1.19 | 0.25 | 0.8125 | 0.3606 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vicente, C.M.; Payero, T.D.; Rodríguez-García, A.; Barreales, E.G.; Pedro, A.d.; Santos-Beneit, F.; Aparicio, J.F. Modulation of Multiple Gene Clusters’ Expression by the PAS-LuxR Transcriptional Regulator PteF. Antibiotics 2022, 11, 994. https://doi.org/10.3390/antibiotics11080994
Vicente CM, Payero TD, Rodríguez-García A, Barreales EG, Pedro Ad, Santos-Beneit F, Aparicio JF. Modulation of Multiple Gene Clusters’ Expression by the PAS-LuxR Transcriptional Regulator PteF. Antibiotics. 2022; 11(8):994. https://doi.org/10.3390/antibiotics11080994
Chicago/Turabian StyleVicente, Cláudia M., Tamara D. Payero, Antonio Rodríguez-García, Eva G. Barreales, Antonio de Pedro, Fernando Santos-Beneit, and Jesús F. Aparicio. 2022. "Modulation of Multiple Gene Clusters’ Expression by the PAS-LuxR Transcriptional Regulator PteF" Antibiotics 11, no. 8: 994. https://doi.org/10.3390/antibiotics11080994
APA StyleVicente, C. M., Payero, T. D., Rodríguez-García, A., Barreales, E. G., Pedro, A. d., Santos-Beneit, F., & Aparicio, J. F. (2022). Modulation of Multiple Gene Clusters’ Expression by the PAS-LuxR Transcriptional Regulator PteF. Antibiotics, 11(8), 994. https://doi.org/10.3390/antibiotics11080994