
Mining Amphibian and Insect Transcriptomes for Antimicrobial Peptide Sequences with rAMPage

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Results
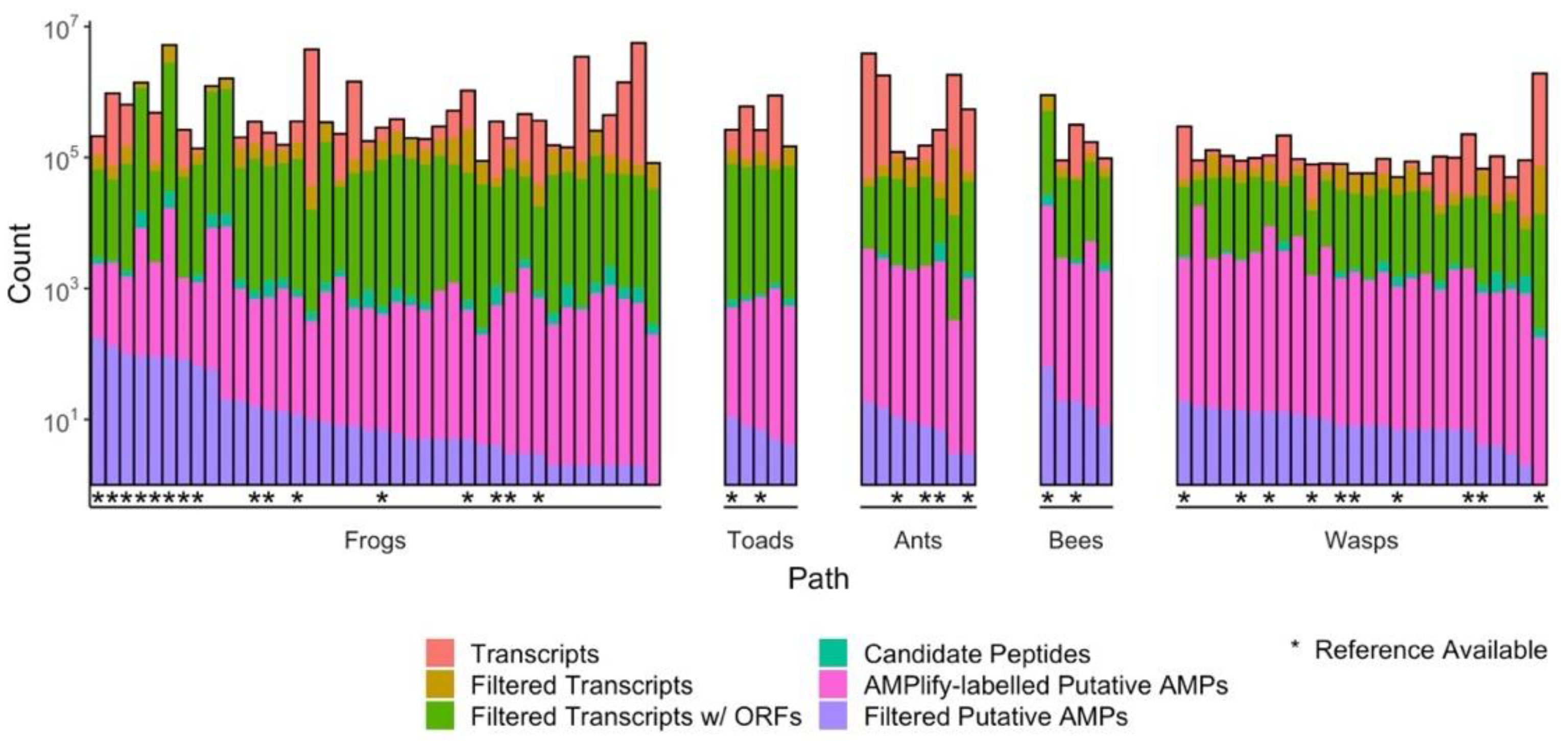
2.1. Identification of Putative AMPs
2.2. Antimicrobial Susceptibility Testing (AST) Results
2.3. Novelty of Discovered AMPs
3. Discussion
4. Materials and Methods
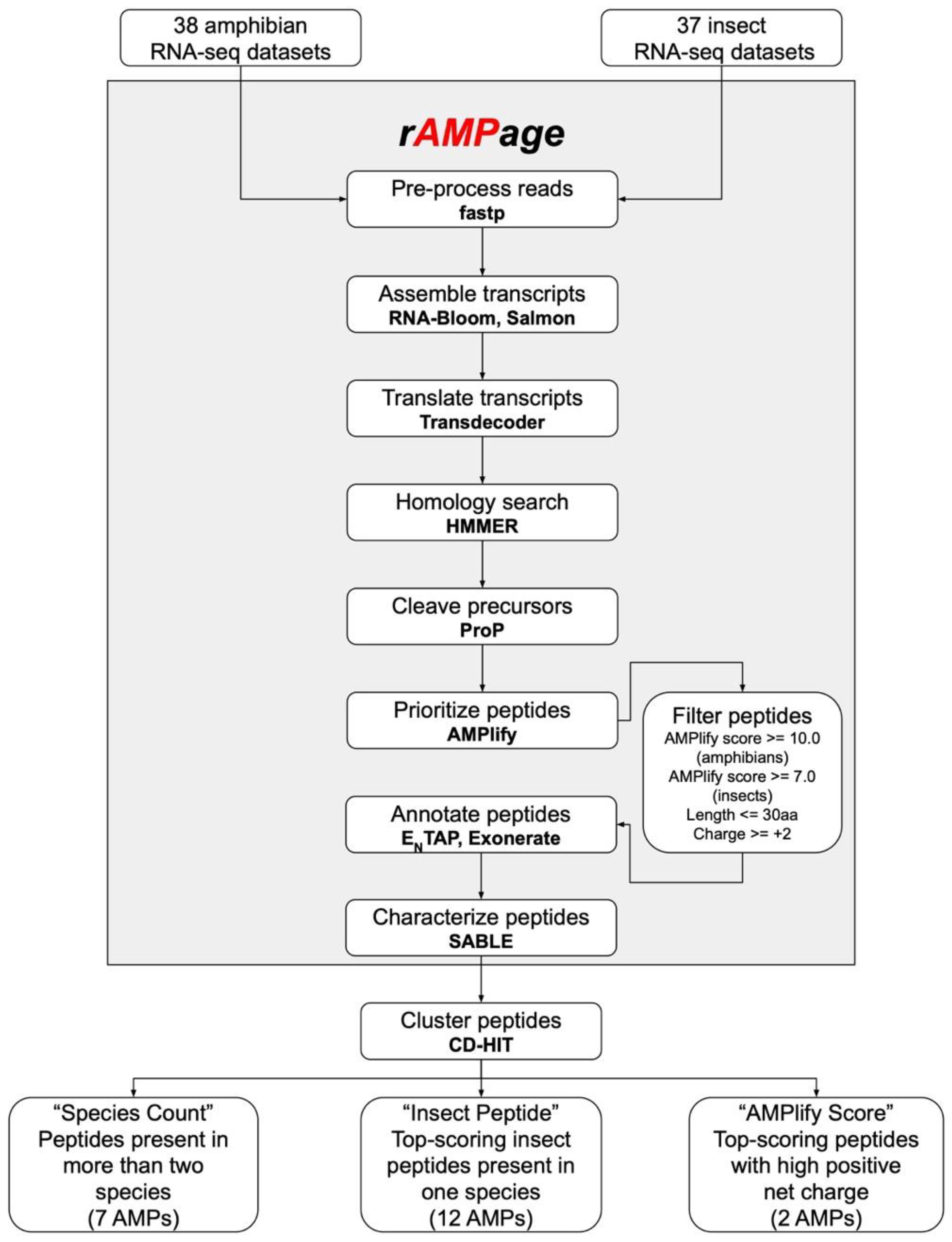
4.1. Collating Input RNA-Seq Datasets
4.2. Collating Reference AMP Datasets
4.3. rAMPage Pipeline
4.4. Selecting Filtered Putative AMPs for Validation
4.5. Antimicrobial Susceptibility Testing (AST)
4.6. Hemolysis Experiments
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hede, K. Antibiotic Resistance: An Infectious Arms Race. Nature 2014, 509, S2–S3. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.B.; Seo, J. Antimicrobial Peptides under Clinical Investigation. Pept. Sci. 2019, 111, e24122. [Google Scholar] [CrossRef]
- Zhang, L.; Gallo, R.L. Antimicrobial Peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and Consequences of Bacterial Resistance to Antimicrobial Peptides. Drug Resist. Updat. 2016, 26, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K.; Heinbockel, L.; Correa, W.; Lohner, K. Peptides with Dual Mode of Action: Killing Bacteria and Preventing Endotoxin-Induced Sepsis. Biochim. Biophys. Acta BBA-Biomembr. 2016, 1858, 971–979. [Google Scholar] [CrossRef]
- Klotman, M.E.; Chang, T.L. Defensins in Innate Antiviral Immunity. Nat. Rev. Immunol. 2006, 6, 447–456. [Google Scholar] [CrossRef] [PubMed]
- De Lucca, A.J.; Walsh, T.J. Antifungal Peptides: Novel Therapeutic Compounds against Emerging Pathogens. Antimicrob. Agents Chemother. 1999, 43, 1–11. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and Host-Defense Peptides as New Anti-Infective Therapeutic Strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Moravej, H.; Moravej, Z.; Yazdanparast, M.; Heiat, M.; Mirhosseini, A.; Moosazadeh Moghaddam, M.; Mirnejad, R. Antimicrobial Peptides: Features, Action, and Their Resistance Mechanisms in Bacteria. Microb. Drug Resist. 2018, 24, 747–767. [Google Scholar] [CrossRef]
- Vanhoye, D.; Bruston, F.; Nicolas, P.; Amiche, M. Antimicrobial Peptides from Hylid and Ranin Frogs Originated from a 150-Million-Year-Old Ancestral Precursor with a Conserved Signal Peptide but a Hypermutable Antimicrobial Domain. Eur. J. Biochem. 2003, 270, 2068–2081. [Google Scholar] [CrossRef]
- Helbing, C.C.; Hammond, S.A.; Jackman, S.H.; Houston, S.; Warren, R.L.; Cameron, C.E.; Birol, I. Antimicrobial Peptides from Rana [Lithobates] Catesbeiana: Gene Structure and Bioinformatic Identification of Novel Forms from Tadpoles. Sci. Rep. 2019, 9, 1529. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Mechkarska, M. Host-Defense Peptides with Therapeutic Potential from Skin Secretions of Frogs from the Family Pipidae. Pharmaceuticals 2014, 7, 58–77. [Google Scholar] [CrossRef]
- Wu, Q.; Patočka, J.; Kuča, K. Insect Antimicrobial Peptides, a Mini Review. Toxins 2018, 10, 461. [Google Scholar] [CrossRef]
- Sheehan, G.; Farrell, G.; Kavanagh, K. Immune Priming: The Secret Weapon of the Insect World. Virulence 2020, 11, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Novković, M.; Simunić, J.; Bojović, V.; Tossi, A.; Juretić, D. DADP: The Database of Anuran Defense Peptides. Bioinformatics 2012, 28, 1406–1407. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.-W.; Liu, W.-T.; Geng, L.-L.; Chen, X.-H.; Bi, K.-S. Quantitative Analysis of a Novel Antimicrobial Peptide in Rat Plasma by Ultra Performance Liquid Chromatography–Tandem Mass Spectrometry. J. Pharm. Anal. 2011, 1, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Chen, Y.; Yao, H.; Du, C.; Luan, N.; Yan, X. A Novel Defensin-like Antimicrobial Peptide from the Skin Secretions of the Tree Frog, Theloderma Kwangsiensis. Gene 2016, 576, 136–140. [Google Scholar] [CrossRef]
- Pei, J.; Feng, Z.; Ren, T.; Sun, H.; Han, H.; Jin, W.; Dang, J.; Tao, Y. Purification, Characterization and Application of a Novel Antimicrobial Peptide from Andrias Davidianus Blood. Lett. Appl. Microbiol. 2018, 66, 38–43. [Google Scholar] [CrossRef]
- Chen, W.; Hwang, Y.Y.; Gleaton, J.W.; Titus, J.K.; Hamlin, N.J. Optimization of a Peptide Extraction and LC–MS Protocol for Quantitative Analysis of Antimicrobial Peptides. Future Sci. OA 2019, 5, FSO348. [Google Scholar] [CrossRef]
- Chowdhury, T.; Mandal, S.M.; Kumari, R.; Ghosh, A.K. Purification and Characterization of a Novel Antimicrobial Peptide (QAK) from the Hemolymph of Antheraea Mylitta. Biochem. Biophys. Res. Commun. 2020, 527, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.C.; Silva, O.N.; Mundim, N.C.C.R.; de Carvalho, M.J.A.; Migliolo, L.; Leite, J.R.S.A.; Prates, M.V.; Bocca, A.L.; Franco, O.L.; Felipe, M.S.S. Predicting Antimicrobial Peptides from Eukaryotic Genomes: In Silico Strategies to Develop Antibiotics. Peptides 2012, 37, 301–308. [Google Scholar] [CrossRef]
- Prichula, J.; Primon-Barros, M.; Luz, R.C.Z.; Castro, Í.M.S.; Paim, T.G.S.; Tavares, M.; Ligabue-Braun, R.; d’Azevedo, P.A.; Frazzon, J.; Frazzon, A.P.G.; et al. Genome Mining for Antimicrobial Compounds in Wild Marine Animals-Associated Enterococci. Mar. Drugs 2021, 19, 328. [Google Scholar] [CrossRef]
- De la Lastra, J.M.P.; Garrido-Orduña, C.; Borges, A.A.; Jiménez-Arias, D.; García-Machado, F.J.; Hernández, M.; González, C.; Boto, A. Bioinformatics discovery of vertebrate cathelicidins from the mining of available genomes. In Drug Discovery—Concepts to Market; Bobbarala, V., Ed.; InTech: London, UK, 2018; ISBN 978-1-78923-696-5. [Google Scholar]
- Tomazou, M.; Oulas, A.; Anagnostopoulos, A.K.; Tsangaris, G.T.; Spyrou, G.M. In Silico Identification of Antimicrobial Peptides in the Proteomes of Goat and Sheep Milk and Feta Cheese. Proteomes 2019, 7, 32. [Google Scholar] [CrossRef]
- Mhade, S.; Panse, S.; Tendulkar, G.; Awate, R.; Kadam, S.; Kaushik, K.S. AMPing Up the Search: A Structural and Functional Repository of Antimicrobial Peptides for Biofilm Studies, and a Case Study of Its Application to Corynebacterium striatum, an Emerging Pathogen. Front. Cell. Infect. Microbiol. 2021, 11, 803774. [Google Scholar] [CrossRef]
- Li, C.; Sutherland, D.; Hammond, S.A.; Yang, C.; Taho, F.; Bergman, L.; Houston, S.; Warren, R.L.; Wong, T.; Hoang, L.M.N.; et al. AMPlify: Attentive Deep Learning Model for Discovery of Novel Antimicrobial Peptides Effective against WHO Priority Pathogens. BMC Genom. 2022, 23, 77. [Google Scholar] [CrossRef]
- Muir, P.; Li, S.; Lou, S.; Wang, D.; Spakowicz, D.J.; Salichos, L.; Zhang, J.; Weinstock, G.M.; Isaacs, F.; Rozowsky, J.; et al. The Real Cost of Sequencing: Scaling Computation to Keep Pace with Data Generation. Genome Biol. 2016, 17, 53. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef]
- Guo, R.; Chen, D.; Diao, Q.; Xiong, C.; Zheng, Y.; Hou, C. Transcriptomic Investigation of Immune Responses of the Apis Cerana Cerana Larval Gut Infected by Ascosphaera Apis. J. Invertebr. Pathol. 2019, 166, 107210. [Google Scholar] [CrossRef]
- Li, J.; Xu, X.; Xu, C.; Zhou, W.; Zhang, K.; Yu, H.; Zhang, Y.; Zheng, Y.; Rees, H.H.; Lai, R.; et al. Anti-Infection Peptidomics of Amphibian Skin. Mol. Cell. Proteom. 2007, 6, 882–894. [Google Scholar] [CrossRef]
- Song, Y.; Ji, S.; Liu, W.; Yu, X.; Meng, Q.; Lai, R. Different Expression Profiles of Bioactive Peptides in Pelophylax Nigromaculatus from Distinct Regions. Biosci. Biotechnol. Biochem. 2013, 77, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, S.; Guo, C.; Zhang, W.; Zhang, X.; Zhang, B.; Li, S.; Ren, J.; Hu, Y.; Wang, H. Identification and Functional Analyses of Novel Antioxidant Peptides and Antimicrobial Peptides from Skin Secretions of Four East Asian Frog Species. Acta Biochim. Biophys. Sin. 2017, 49, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Rifflet, A.; Gavalda, S.; Téné, N.; Orivel, J.; Leprince, J.; Guilhaudis, L.; Génin, E.; Vétillard, A.; Treilhou, M. Identification and Characterization of a Novel Antimicrobial Peptide from the Venom of the Ant Tetramorium Bicarinatum. Peptides 2012, 38, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Téné, N.; Bonnafé, E.; Berger, F.; Rifflet, A.; Guilhaudis, L.; Ségalas-Milazzo, I.; Pipy, B.; Coste, A.; Leprince, J.; Treilhou, M. Biochemical and Biophysical Combined Study of Bicarinalin, an Ant Venom Antimicrobial Peptide. Peptides 2016, 79, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Stoldt, M.; Jongepier, E.; Feldmeyer, B.; Menzel, F.; Bornberg-Bauer, E.; Foitzik, S. Ant Behaviour and Brain Gene Expression of Defending Hosts Depend on the Ecological Success of the Intruding Social Parasite. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180192. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The Protein Families Database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Sarkar, N. Polyadenylation of MRNA in Prokaryotes. Annu. Rev. Biochem. 1997, 66, 173–197. [Google Scholar] [CrossRef]
- Wangsanuwat, C.; Heom, K.A.; Liu, E.; O’Malley, M.A.; Dey, S.S. Efficient and Cost-Effective Bacterial MRNA Sequencing from Low Input Samples through Ribosomal RNA Depletion. BMC Genom. 2020, 21, 717. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting Antimicrobial Peptides with Improved Accuracy by Incorporating the Compositional, Physico-Chemical and Structural Features into Chou’s General PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, P.; Lin, W.-Z.; Jia, J.-H.; Chou, K.-C. IAMP-2L: A Two-Level Multi-Label Classifier for Identifying Antimicrobial Peptides and Their Functional Types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Veltri, D.; Kamath, U.; Shehu, A. Deep Learning Improves Antimicrobial Peptide Recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef]
- Das, P.; Wadhawan, K.; Chang, O.; Sercu, T.; Santos, C.D.; Riemer, M.; Chenthamarakshan, V.; Padhi, I.; Mojsilovic, A. PepCVAE: Semi-Supervised Targeted Design of Antimicrobial Peptide Sequences. arXiv 2018, arXiv:1810.07743. [Google Scholar] [CrossRef]
- Dean, S.N.; Alvarez, J.A.E.; Zabetakis, D.; Walper, S.A.; Malanoski, A.P. PepVAE: Variational Autoencoder Framework for Antimicrobial Peptide Generation and Activity Prediction. Front. Microbiol. 2021, 12, 725727. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, P.; Możejko, M.; Grzegorzek, T.; Bauer, M.; Neubauer, D.; Michalski, M.; Sroka, J.; Setny, P.; Kamysz, W.; Szczurek, E. HydrAMP: A Deep Generative Model for Antimicrobial Peptide Discovery. bioRxiv 2022. [Google Scholar] [CrossRef]
- Porto, W.F.; Pires, A.S.; Franco, O.L. Computational Tools for Exploring Sequence Databases as a Resource for Antimicrobial Peptides. Biotechnol. Adv. 2017, 35, 337–349. [Google Scholar] [CrossRef]
- Ramazi, S.; Mohammadi, N.; Allahverdi, A.; Khalili, E.; Abdolmaleki, P. A Review on Antimicrobial Peptides Databases and the Computational Tools. Database 2022, 2022, baac011. [Google Scholar] [CrossRef]
- Aronica, P.G.A.; Reid, L.M.; Desai, N.; Li, J.; Fox, S.J.; Yadahalli, S.; Essex, J.W.; Verma, C.S. Computational Methods and Tools in Antimicrobial Peptide Research. J. Chem. Inf. Model. 2021, 61, 3172–3196. [Google Scholar] [CrossRef]
- Cho, J.H.; Sung, B.H.; Kim, S.C. Buforins: Histone H2A-Derived Antimicrobial Peptides from Toad Stomach. Biochim. Biophys. Acta BBA-Biomembr. 2009, 1788, 1564–1569. [Google Scholar] [CrossRef]
- De Gregorio, E.; Spellman, P.T.; Tzou, P.; Rubin, G.M.; Lemaitre, B. The Toll and Imd Pathways Are the Major Regulators of the Immune Response in Drosophila. EMBO J. 2002, 21, 2568–2579. [Google Scholar] [CrossRef] [PubMed]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski, L.D.S.; Silva-Pereira, I.; Kyaw, C.M. Antibiotic Development Challenges: The Various Mechanisms of Action of Antimicrobial Peptides and of Bacterial Resistance. Front. Microbiol. 2013, 4, 353. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rojas, A.; Baeder, D.Y.; Johnston, P.; Regoes, R.R.; Rolff, J. Bacteria Primed by Antimicrobial Peptides Develop Tolerance and Persist. PLoS Pathog. 2021, 17, e1009443. [Google Scholar] [CrossRef]
- da Cunha, N.B.; Cobacho, N.B.; Viana, J.F.C.; Lima, L.A.; Sampaio, K.B.O.; Dohms, S.S.M.; Ferreira, A.C.R.; de la Fuente-Núñez, C.; Costa, F.F.; Franco, O.L.; et al. The next Generation of Antimicrobial Peptides (AMPs) as Molecular Therapeutic Tools for the Treatment of Diseases with Social and Economic Impacts. Drug Discov. Today 2017, 22, 234–248. [Google Scholar] [CrossRef]
- Cao, J.; de la Fuente-Nunez, C.; Ou, R.W.; Torres, M.D.T.; Pande, S.G.; Sinskey, A.J.; Lu, T.K. Yeast-Based Synthetic Biology Platform for Antimicrobial Peptide Production. ACS Synth. Biol. 2018, 7, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Hazam, P.K.; Goyal, R.; Ramakrishnan, V. Peptide Based Antimicrobials: Design Strategies and Therapeutic Potential. Prog. Biophys. Mol. Biol. 2019, 142, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Saito, C.; Goto, C.; Yokoo, H.; Kawano, R.; Misawa, T.; Demizu, Y. Rational Design of Helix-Stabilized Antimicrobial Peptide Foldamers Containing α,α-Disubstituted AAs or Side-Chain Stapling. ChemPlusChem 2020, 85, 2731–2736. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; On behalf of the International Nucleotide Sequence Database Collaboration. Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Nip, K.M.; Chiu, R.; Yang, C.; Chu, J.; Mohamadi, H.; Warren, R.L.; Birol, I. RNA-Bloom Enables Reference-Free and Reference-Guided Sequence Assembly for Single-Cell Transcriptomes. Genome Res. 2020, 30, 1191–1200. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov Model Speed Heuristic and Iterative HMM Search Procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Duckert, P.; Brunak, S.; Blom, N. Prediction of Proprotein Convertase Cleavage Sites. Protein Eng. Des. Sel. 2004, 17, 107–112. [Google Scholar] [CrossRef]
- Wang, X.; Song, Y.; Li, J.; Liu, H.; Xu, X.; Lai, R.; Zhang, K. A New Family of Antimicrobial Peptides from Skin Secretions of Rana Pleuraden. Peptides 2007, 28, 2069–2074. [Google Scholar] [CrossRef]
- Yi, H.-Y.; Chowdhury, M.; Huang, Y.-D.; Yu, X.-Q. Insect Antimicrobial Peptides and Their Applications. Appl. Microbiol. Biotechnol. 2014, 98, 5807–5822. [Google Scholar] [CrossRef]
- Jiang, Z.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Effects of Net Charge and the Number of Positively Charged Residues on the Biological Activity of Amphipathic Alpha-Helical Cationic Antimicrobial Peptides. Biopolymers 2008, 90, 369–383. [Google Scholar] [CrossRef]
- Hart, A.J.; Ginzburg, S.; Xu, M.; Fisher, C.R.; Rahmatpour, N.; Mitton, J.B.; Paul, R.; Wegrzyn, J.L. EnTAP: Bringing Faster and Smarter Functional Annotation to Non-model Eukaryotic Transcriptomes. Mol. Ecol. Resour. 2020, 20, 591–604. [Google Scholar] [CrossRef]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Slater, G.; Birney, E. Automated Generation of Heuristics for Biological Sequence Comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Adamczak, R.; Porollo, A.; Meller, J. Combining Prediction of Secondary Structure and Solvent Accessibility in Proteins. Proteins 2005, 59, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and Broth Dilution Methods to Determine the Minimal Inhibitory Concentration (MIC) of Antimicrobial Substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Rodríguez, A.; Grau, J.H.; Lötters, S.; Künzel, S.; Saporito, R.A.; Ringler, E.; Schulz, S.; Wollenberg Valero, K.C.; Vences, M. Transcriptomic Signatures of Experimental Alkaloid Consumption in a Poison Frog. Genes 2019, 10, 733. [Google Scholar] [CrossRef]
- Siu-Ting, K.; Torres-Sánchez, M.; San Mauro, D.; Wilcockson, D.; Wilkinson, M.; Pisani, D.; O’Connell, M.J.; Creevey, C.J. Inadvertent Paralog Inclusion Drives Artifactual Topologies and Timetree Estimates in Phylogenomics. Mol. Biol. Evol. 2019, 36, 1344–1356. [Google Scholar] [CrossRef]
- Xia, Y.; Luo, W.; Yuan, S.; Zheng, Y.; Zeng, X. Microsatellite Development from Genome Skimming and Transcriptome Sequencing: Comparison of Strategies and Lessons from Frog Species. BMC Genom. 2018, 19, 886. [Google Scholar] [CrossRef]
- Fan, W.; Jiang, Y.; Zhang, M.; Yang, D.; Chen, Z.; Sun, H.; Lan, X.; Yan, F.; Xu, J.; Yuan, W. Comparative Transcriptome Analyses Reveal the Genetic Basis Underlying the Immune Function of Three Amphibians’ Skin. PLoS ONE 2017, 12, e0190023. [Google Scholar] [CrossRef]
- Reilly, B.D.; Schlipalius, D.I.; Cramp, R.L.; Ebert, P.R.; Franklin, C.E. Frogs and Estivation: Transcriptional Insights into Metabolism and Cell Survival in a Natural Model of Extended Muscle Disuse. Physiol. Genom. 2013, 45, 377–388. [Google Scholar] [CrossRef]
- Liscano Martinez, Y.; Arenas Gómez, C.M.; Smith, J.; Delgado, J.P. A Tree Frog (Boana Pugnax) Dataset of Skin Transcriptome for the Identification of Biomolecules with Potential Antimicrobial Activities. Data Brief 2020, 32, 106084. [Google Scholar] [CrossRef] [PubMed]
- Grogan, L.F.; Mulvenna, J.; Gummer, J.P.A.; Scheele, B.C.; Berger, L.; Cashins, S.D.; McFadden, M.S.; Harlow, P.; Hunter, D.A.; Trengove, R.D.; et al. Survival, Gene and Metabolite Responses of Litoria Verreauxii Alpina Frogs to Fungal Disease Chytridiomycosis. Sci. Data 2018, 5, 180033. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Yang, W.; Fu, J.; Song, Z. Transcriptome Profile of the Green Odorous Frog (Odorrana Margaretae). PLoS ONE 2013, 8, e75211. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Zhu, W.; Shi, S.; Zhang, M.; Jiang, J.; Li, C.; Xie, F.; Wang, B. Plateau Grass and Greenhouse Flower? Distinct Genetic Basis of Closely Related Toad Tadpoles Respectively Adapted to High Altitude and Karst Caves. Genes 2020, 11, 123. [Google Scholar] [CrossRef]
- Caty, S.N.; Alvarez-Buylla, A.; Byrd, G.D.; Vidoudez, C.; Roland, A.B.; Tapia, E.E.; Budnik, B.; Trauger, S.A.; Coloma, L.A.; O’Connell, L.A. Molecular Physiology of Chemical Defenses in a Poison Frog. J. Exp. Biol. 2019, 222, jeb.204149. [Google Scholar] [CrossRef]
- Shu, Y.; Xia, J.; Yu, Q.; Wang, G.; Zhang, J.; He, J.; Wang, H.; Zhang, L.; Wu, H. Integrated Analysis of MRNA and MiRNA Expression Profiles Reveals Muscle Growth Differences between Adult Female and Male Chinese Concave-Eared Frogs (Odorrana Tormota). Gene 2018, 678, 241–251. [Google Scholar] [CrossRef]
- Yoshida, N.; Kaito, C. Dataset for de Novo Transcriptome Assembly of the African Bullfrog Pyxicephalus Adspersus. Data Brief 2020, 30, 105388. [Google Scholar] [CrossRef]
- Bossuyt, F.; Schulte, L.M.; Maex, M.; Janssenswillen, S.; Novikova, P.Y.; Biju, S.D.; Van de Peer, Y.; Matthijs, S.; Roelants, K.; Martel, A.; et al. Multiple Independent Recruitment of Sodefrin Precursor-Like Factors in Anuran Sexually Dimorphic Glands. Mol. Biol. Evol. 2019, 36, 1921–1930. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Qin, Z.; Wang, H.; Li, J. A Screening Assay for Thyroid Hormone Signaling Disruption Based on Thyroid Hormone-Response Gene Expression Analysis in the Frog Pelophylax Nigromaculatus. J. Environ. Sci. 2015, 34, 143–154. [Google Scholar] [CrossRef]
- Eskew, E.A.; Shock, B.C.; LaDouceur, E.E.B.; Keel, K.; Miller, M.R.; Foley, J.E.; Todd, B.D. Gene Expression Differs in Susceptible and Resistant Amphibians Exposed to Batrachochytrium Dendrobatidis. R. Soc. Open sci. 2018, 5, 170910. [Google Scholar] [CrossRef]
- Stuckert, A.M.M.; Chouteau, M.; McClure, M.; LaPolice, T.M.; Linderoth, T.; Nielsen, R.; Summers, K.; MacManes, M.D. The Genomics of Mimicry: Gene Expression throughout Development Provides Insights into Convergent and Divergent Phenotypes in a Müllerian Mimicry System. Mol. Ecol. 2021, 30, 4039–4061. [Google Scholar] [CrossRef] [PubMed]
- Christenson, M.K.; Trease, A.J.; Potluri, L.-P.; Jezewski, A.J.; Davis, V.M.; Knight, L.A.; Kolok, A.S.; Davis, P.H. De Novo Assembly and Analysis of the Northern Leopard Frog Rana Pipiens Transcriptome. J. Genom. 2014, 2, 141–149. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Price, S.J.; Garner, T.W.J.; Balloux, F.; Ruis, C.; Paszkiewicz, K.H.; Moore, K.; Griffiths, A.G.F. A de Novo Assembly of the Common Frog (Rana Temporaria) Transcriptome and Comparison of Transcription Following Exposure to Ranavirus and Batrachochytrium Dendrobatidis. PLoS ONE 2015, 10, e0130500. [Google Scholar] [CrossRef]
- Furman, B.L.S.; Evans, B.J. Sequential Turnovers of Sex Chromosomes in African Clawed Frogs (Xenopus) Suggest Some Genomic Regions Are Good at Sex Determination. G3 (Bethesda) 2016, 6, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Birol, I.; Behsaz, B.; Hammond, S.A.; Kucuk, E.; Veldhoen, N.; Helbing, C.C. De Novo Transcriptome Assemblies of Rana (Lithobates) Catesbeiana and Xenopus Laevis Tadpole Livers for Comparative Genomics without Reference Genomes. PLoS ONE 2015, 10, e0130720. [Google Scholar] [CrossRef]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Colak, R.; et al. The Evolutionary Landscape of Alternative Splicing in Vertebrate Species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef]
- Arvidson, R.; Kaiser, M.; Lee, S.S.; Urenda, J.-P.; Dail, C.; Mohammed, H.; Nolan, C.; Pan, S.; Stajich, J.E.; Libersat, F.; et al. Parasitoid Jewel Wasp Mounts Multipronged Neurochemical Attack to Hijack a Host Brain. Mol. Cell. Proteom. 2019, 18, 99–114. [Google Scholar] [CrossRef]
- Yek, S.H.; Boomsma, J.J.; Schiøtt, M. Differential Gene Expression in Acromyrmex Leaf-Cutting Ants after Challenges with Two Fungal Pathogens. Mol. Ecol. 2013, 22, 2173–2187. [Google Scholar] [CrossRef]
- Yoon, K.A.; Kim, K.; Kim, W.-J.; Bang, W.Y.; Ahn, N.-H.; Bae, C.-H.; Yeo, J.-H.; Lee, S.H. Characterization of Venom Components and Their Phylogenetic Properties in Some Aculeate Bumblebees and Wasps. Toxins 2020, 12, 47. [Google Scholar] [CrossRef]
- McNamara-Bordewick, N.K.; McKinstry, M.; Snow, J.W. Robust Transcriptional Response to Heat Shock Impacting Diverse Cellular Processes despite Lack of Heat Shock Factor in Microsporidia. mSphere 2019, 4, e00219-19. [Google Scholar] [CrossRef]
- Becchimanzi, A.; Avolio, M.; Bostan, H.; Colantuono, C.; Cozzolino, F.; Mancini, D.; Chiusano, M.L.; Pucci, P.; Caccia, S.; Pennacchio, F. Venomics of the Ectoparasitoid Wasp Bracon Nigricans. BMC Genom. 2020, 21, 34. [Google Scholar] [CrossRef]
- de Bekker, C.; Ohm, R.A.; Loreto, R.G.; Sebastian, A.; Albert, I.; Merrow, M.; Brachmann, A.; Hughes, D.P. Gene Expression during Zombie Ant Biting Behavior Reflects the Complexity Underlying Fungal Parasitic Behavioral Manipulation. BMC Genom. 2015, 16, 620. [Google Scholar] [CrossRef] [PubMed]
- von Wyschetzki, K.; Lowack, H.; Heinze, J. Transcriptomic Response to Injury Sheds Light on the Physiological Costs of Reproduction in Ant Queens. Mol. Ecol. 2016, 25, 1972–1985. [Google Scholar] [CrossRef]
- Zhao, W.; Shi, M.; Ye, X.; Li, F.; Wang, X.; Chen, X. Comparative Transcriptome Analysis of Venom Glands from Cotesia Vestalis and Diadromus Collaris, Two Endoparasitoids of the Host Plutella Xylostella. Sci. Rep. 2017, 7, 1298. [Google Scholar] [CrossRef]
- Coffman, K.A.; Harrell, T.C.; Burke, G.R. A Mutualistic Poxvirus Exhibits Convergent Evolution with Other Heritable Viruses in Parasitoid Wasps. J. Virol. 2020, 94, e02059-19. [Google Scholar] [CrossRef] [PubMed]
- Burke, G.R.; Strand, M.R. Systematic Analysis of a Wasp Parasitism Arsenal. Mol. Ecol. 2014, 23, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Mueller, A.; Clayton, D.; Starobova, H.; Hamilton, B.R.; Payne, R.J.; Vetter, I.; King, G.F.; Undheim, E.A.B. A Comprehensive Portrait of the Venom of the Giant Red Bull Ant, Myrmecia Gulosa, Reveals a Hyperdiverse Hymenopteran Toxin Gene Family. Sci. Adv. 2018, 4, eaau4640. [Google Scholar] [CrossRef]
- Martinson, E.O.; Mrinalini; Kelkar, Y.D.; Chang, C.-H.; Werren, J.H. The Evolution of Venom by Co-Option of Single-Copy Genes. Curr. Biol. 2017, 27, 2007–2013. [Google Scholar] [CrossRef]
- Cook, N.; Boulton, R.A.; Green, J.; Trivedi, U.; Tauber, E.; Pannebakker, B.A.; Ritchie, M.G.; Shuker, D.M. Differential Gene Expression Is Not Required for Facultative Sex Allocation: A Transcriptome Analysis of Brain Tissue in the Parasitoid Wasp Nasonia vitripennis. R. Soc. Open sci. 2018, 5, 171718. [Google Scholar] [CrossRef]
- Sim, A.D.; Wheeler, D. The Venom Gland Transcriptome of the Parasitoid Wasp Nasonia Vitripennis Highlights the Importance of Novel Genes in Venom Function. BMC Genom. 2016, 17, 571. [Google Scholar] [CrossRef]
- Kazuma, K.; Masuko, K.; Konno, K.; Inagaki, H. Combined Venom Gland Transcriptomic and Venom Peptidomic Analysis of the Predatory Ant Odontomachus Monticola. Toxins 2017, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.R.; Helms Cahan, S.; Kemena, C.; Brady, S.G.; Yang, W.; Bornberg-Bauer, E.; Eriksson, T.; Gadau, J.; Helmkampf, M.; Gotzek, D.; et al. How Do Genomes Create Novel Phenotypes? Insights from the Loss of the Worker Caste in Ant Social Parasites. Mol. Biol. Evol. 2015, 32, 2919–2931. [Google Scholar] [CrossRef] [PubMed]
- Özbek, R.; Wielsch, N.; Vogel, H.; Lochnit, G.; Foerster, F.; Vilcinskas, A.; von Reumont, B.M. Proteo-Transcriptomic Characterization of the Venom from the Endoparasitoid Wasp Pimpla Turionellae with Aspects on Its Biology and Evolution. Toxins 2019, 11, 721. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, Y.; Liu, M.-M.; Yan, Z.-C.; Qiu, L.-M.; Fang, Q.; Wang, F.; Werren, J.H.; Ye, G.-Y. Identification and Comparative Analysis of Venom Proteins in a Pupal Ectoparasitoid, Pachycrepoideus Vindemmiae. Front. Physiol. 2020, 11, 9. [Google Scholar] [CrossRef]
- Bouzid, W.; Verdenaud, M.; Klopp, C.; Ducancel, F.; Noirot, C.; Vétillard, A. De Novo Sequencing and Transcriptome Analysis for Tetramorium Bicarinatum: A Comprehensive Venom Gland Transcriptome Analysis from an Ant Species. BMC Genom. 2014, 15, 987. [Google Scholar] [CrossRef]
- Negroni, M.A.; Foitzik, S.; Feldmeyer, B. Long-Lived Temnothorax Ant Queens Switch from Investment in Immunity to Antioxidant Production with Age. Sci. Rep. 2019, 9, 7270. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Prepro-Sequence | Putative Mature Peptide | ||||||
|---|---|---|---|---|---|---|---|
| Sequence | Length | Charge | AMPlify Score | MIC (μg/mL) * | Peptide ID | ||
| E. coli † | S. aureus† | ||||||
| MFTMKKSLLVLFFLGIVSLSLCEEERNADEDDGEMTEEVKR | GILDTLKQLGKAAVQGLLSKAACKLAKTC | 29 | 4 | 80.0 | 2–4 | 4–8 | AmMa1 |
| LGIVSLSLCQEERSADDEEGEVIEEEVKR | GFMDTAKNVAKNVAVTLLYNLKCKITKAC | 29 | 4 | 69.2 | 4 | 64 | OdMa12 |
| MFTMKKSLLFFFLGTIALSLCEEERGADEEENGGEITDEEVKR | GLLLDTVKGAAKNVAGILLNKLKCKVTGDC | 30 | 3 | 61.8 | 8 | 16–32 | PeNi10 |
| MFTMKKSLLLVFFLGTIALSLCEEERGADDDNGGEITDEEIKR | GILTDTLKGAAKNVAGVLLDKLKCKITGGC | 30 | 3 | 61.8 | 8–16 | 32–128 | PeNi11 |
| MFTLRKSLLLLFFLGMVSLSLCEQERDADEDEGEVTEEVKR | GLWTTIKEGVKNFSVGVLDKIRCKITGGC | 29 | 3 | 67.5 | 4–8 | 16–64 | PeNi14 |
| MKLLALVLVLSCVVAYTTARKRGQYWPTNTKIFTTPYRFRREADQGSIVANLKNTPQLPFDDNENLRLVLFDNDPTVDLGEDDKEIPGPQSQPNALSNNLHLIDENDYFSSYTSQPGTYRSFPRNFGTSGRYRWRREAGGHVEPRLRFDAETQRGNSFFTDFADLQRRANGRGIEPTVSATAGIRFRQEADQINPLAVRRERR | SWLSKSVKKLVNKKNYTRLEKLAKKKLFNE | 30 | 8 | 25.5 | 1–2 | >128 | TeRu4 |
| IFLVGCKLFGNFILQRMQLLLALADAVA | KIKIPWGKVKDFLVGGMKAVGKK | 23 | 6 | 45.0 | 1–4 | 2–8 | TeBi1 |
| Peptide ID | Source Organism | Highest Scoring Blastp Match | Sequence Identity (%) | ||
|---|---|---|---|---|---|
| Precursor | Prepro | Mature | |||
| AmMa1 | Amolops mantzorum | Palustrin-2GN3 (ADM34231.1) [Amolops granulosus] | 97 | 100 | 93 |
| OdMa12 | Odorrana margaretae | Odorranain-F2 (ABG76517.1) [Odorrana grahami] | 98 | 100 | 97 |
| PeNi10 | Leptobrachium boringii Polypedates megacephalus Pelophylax nigromaculatus Rhacophorus dennysi Rhacophorus omeimontis | Pelophylaxin-1 (Q2WCN8.1) [Pelophylax fukienensis] Ranatuerin-2N (AEM68233.1) * [Pelophylax nigromaculatus] | 82 98 | 86 97 | 77 100 |
| PeNi11 | Leptobrachium boringii Polypedates megacephalus Pelophylax nigromaculatus Rhacophorus dennysi Rhacophorus omeimontis | Pelophylaxin-1 (Q2WCN8.1) [Pelophylax fukienensis] | 100 | 100 | 100 |
| PeNi14 | Bufo gargarizans Polypedates megacephalus Pelophylax nigromaculatus Rhacophorus omeimontis | Palustrin-2HB1 (AIU998997.1) [Pelophylax hubeiensis] | 90 | 93 | 86 |
| TeRu4 | Temnothorax rugatulus | Uncharacterized protein (XP_024884948.1) [Temnothorax curvispinosus] Uncharacterized protein (TGZ47385.1) * [Temnothorax longispinosus] | 94 91 | 93 90 | 97 97 |
| TeBi1 | Tetramorium bicarinatum | M-myrmicitoxin(01)-Tb1a (W8GNV3.1) [Tetramorium bicarinatum] | 100 | - | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, D.; Sutherland, D.; Aninta, S.I.; Louie, N.; Nip, K.M.; Li, C.; Yanai, A.; Coombe, L.; Warren, R.L.; Helbing, C.C.; et al. Mining Amphibian and Insect Transcriptomes for Antimicrobial Peptide Sequences with rAMPage. Antibiotics 2022, 11, 952. https://doi.org/10.3390/antibiotics11070952
Lin D, Sutherland D, Aninta SI, Louie N, Nip KM, Li C, Yanai A, Coombe L, Warren RL, Helbing CC, et al. Mining Amphibian and Insect Transcriptomes for Antimicrobial Peptide Sequences with rAMPage. Antibiotics. 2022; 11(7):952. https://doi.org/10.3390/antibiotics11070952
Chicago/Turabian StyleLin, Diana, Darcy Sutherland, Sambina Islam Aninta, Nathan Louie, Ka Ming Nip, Chenkai Li, Anat Yanai, Lauren Coombe, René L. Warren, Caren C. Helbing, and et al. 2022. "Mining Amphibian and Insect Transcriptomes for Antimicrobial Peptide Sequences with rAMPage" Antibiotics 11, no. 7: 952. https://doi.org/10.3390/antibiotics11070952
APA StyleLin, D., Sutherland, D., Aninta, S. I., Louie, N., Nip, K. M., Li, C., Yanai, A., Coombe, L., Warren, R. L., Helbing, C. C., Hoang, L. M. N., & Birol, I. (2022). Mining Amphibian and Insect Transcriptomes for Antimicrobial Peptide Sequences with rAMPage. Antibiotics, 11(7), 952. https://doi.org/10.3390/antibiotics11070952